

The Synthesis and Biological Evaluation of 2-(1H-Indol-3-yl)quinazolin-4(3H)-One Derivatives

 , , ,
, , , 
Abstract
:
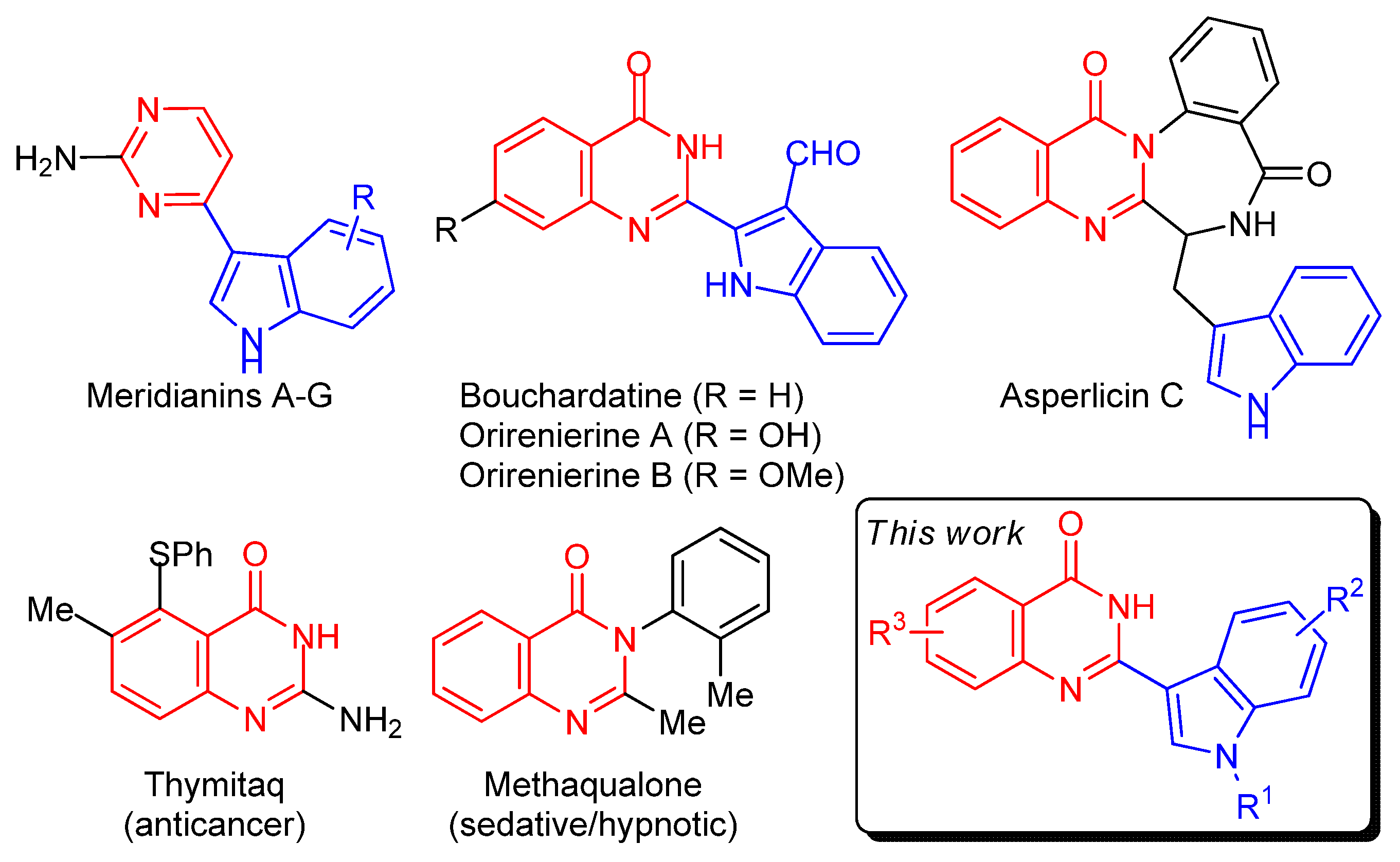
1. Introduction
2. Results and Discussion
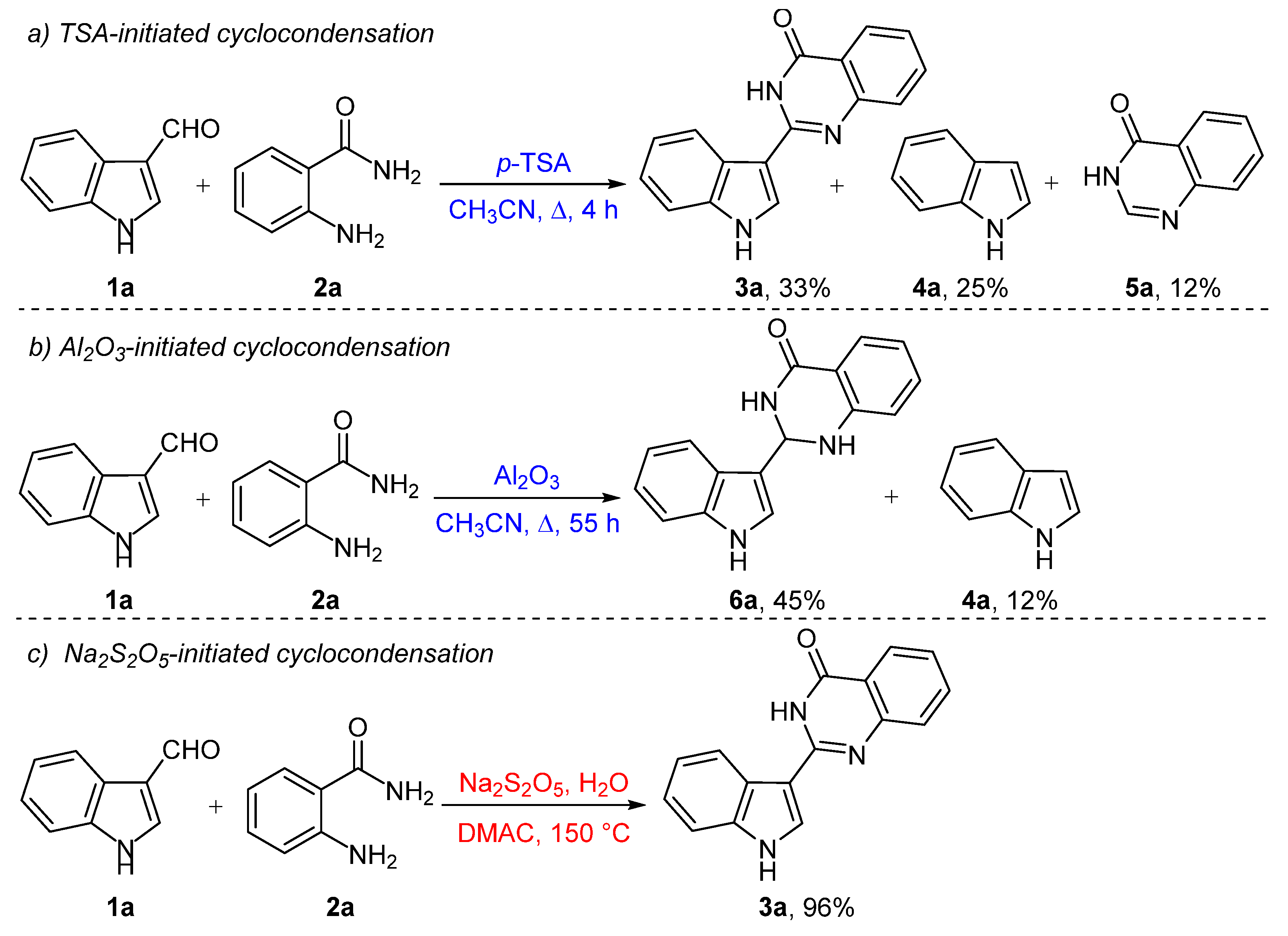
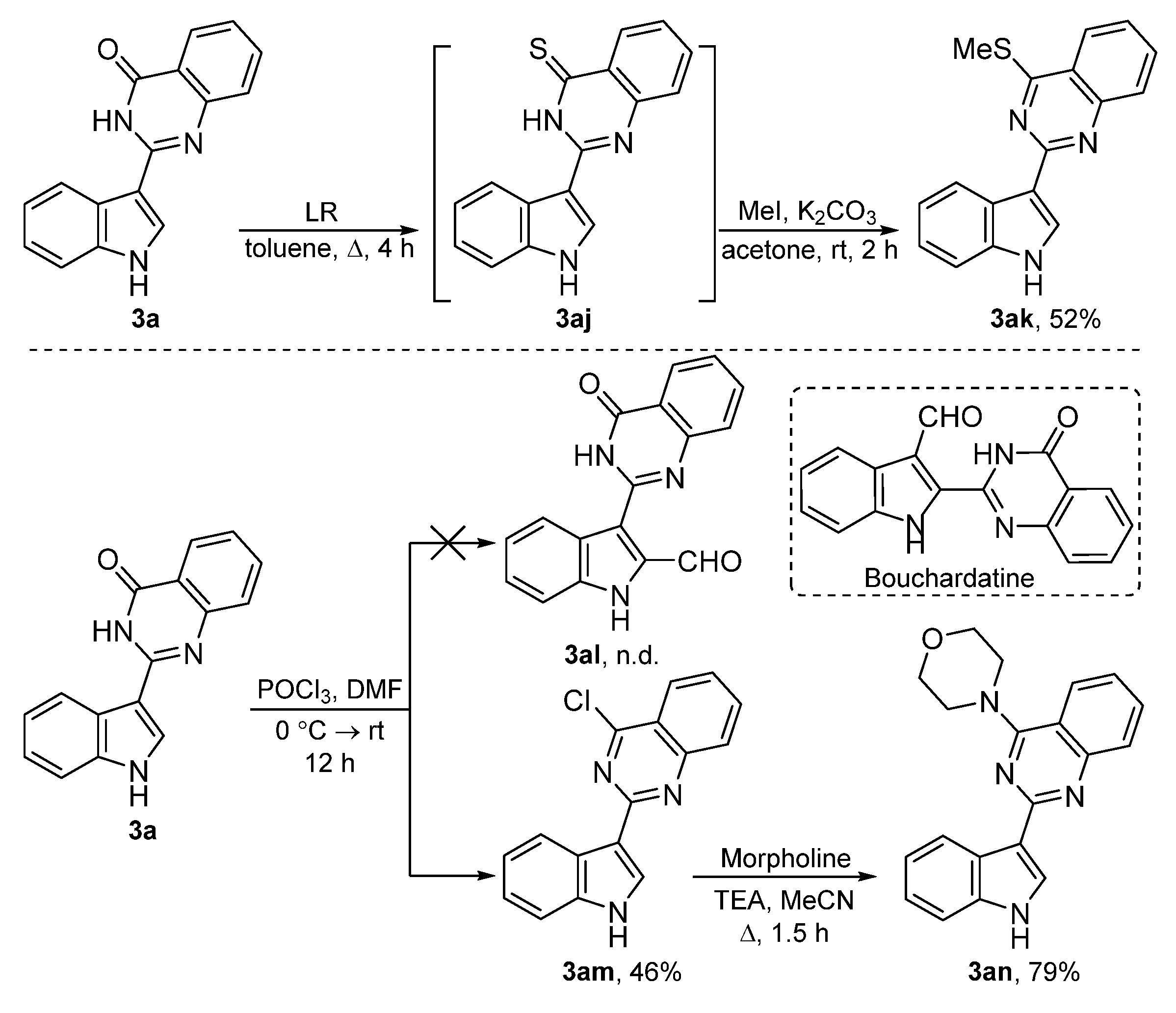
2.1. Chemistry
2.2. In Vitro Biological Evaluation
2.2.1. Antibacterial Activity
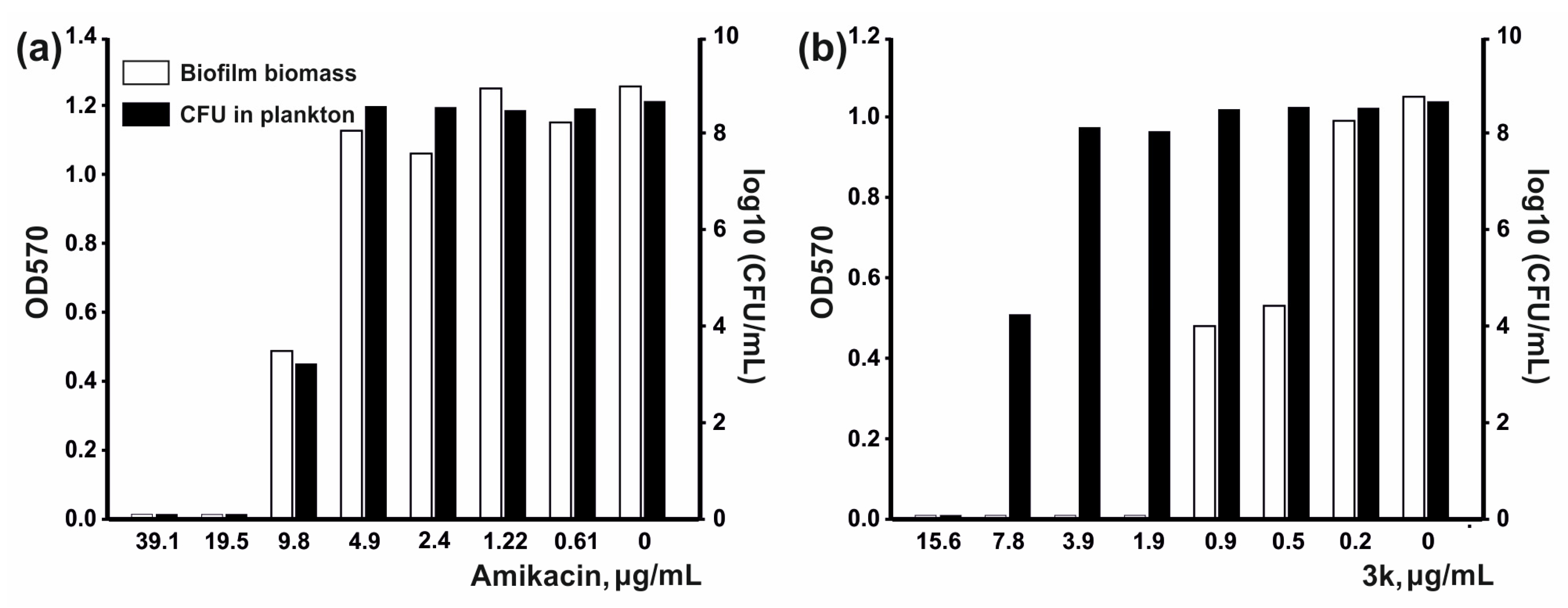
2.2.2. Antibiofilm Activity
2.2.3. Cytotoxicity Activity
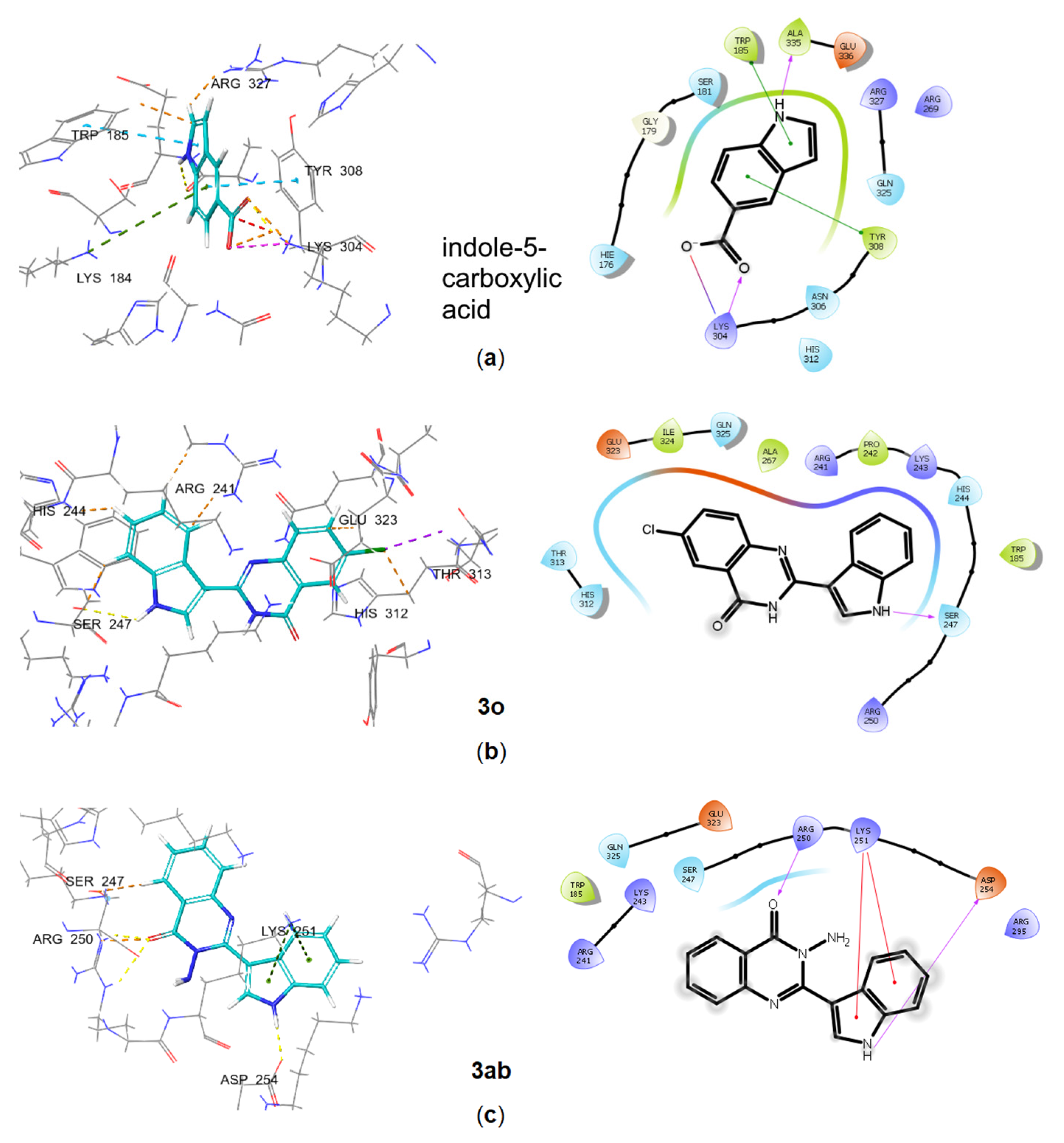
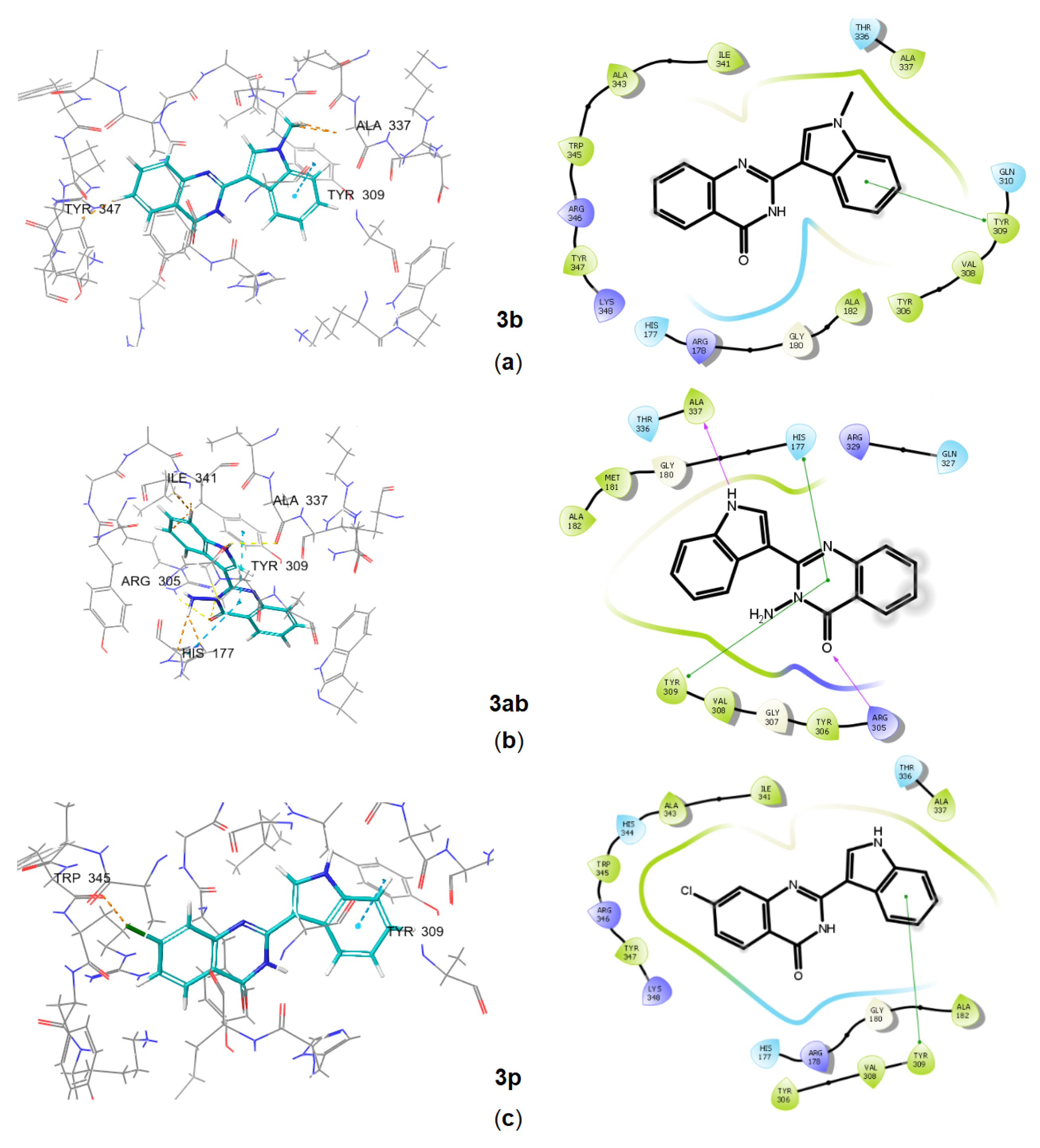
2.3. In Silico Characterization of Substituted Indoles Binding to Long RSH Proteins
Molecular Docking of Substituted Indoles
3. Materials and Methods
3.1. Instrumentation
3.2. Materials
3.3. Synthesis
3.3.1. Synthesis of the Starting Substrates
3.3.2. Synthesis of 2-(1H-indol-3-yl)quinazolin-4(3H)-one (3a) [80]
3.3.3. Synthesis of 2-(1H-indol-3-yl)-2,3-dihydroquinazolin-4(1H)-one (6a) [82]
3.3.4. General Procedure for the Synthesis of Quinazolin-4(3H)-Ones 3a–3g, 3k–3ab
3.3.5. Synthesis of 2-(5-methylfuran-2-yl)-2,3-dihydroquinazolin-4(1H)-one (6u) [88]
3.3.6. Synthesis of 2-(thiophen-2-yl)-2,3-dihydro-4H-benzo[e][1,3]oxazin-4-one (3ad) [89]
3.3.7. Synthesis of 2-(1-benzoyl-1H-indol-3-yl)quinazolin-4(3H)-one (3i)
3.3.8. Synthesis of Quinazolin-4(3H)-Ones 3b, 3ah, 3ai
3.3.9. Synthesis of 2-(1H-indol-3-yl)-4-(methylthio)quinazoline (3ak)
3.3.10. Synthesis of 4-Chloro-2-(1H-indol-3-yl)quinazoline (3am)
3.3.11. Synthesis of 4-[2-(1H-indol-3-yl)quinazolin-4-yl]morpholine (3an)
3.4. Antibacterial Activity
3.5. Biofilm Assay
3.6. Cytotoxicity Activity
3.6.1. Cell Lines and Culture Conditions
3.6.2. Cytotoxicity Measurements (Mosmann Assay)
3.7. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spellberg, B.; Bartlett, J.G.; Gilbert, D.N. The future of antibiotics and resistance. N. Engl. J. Med. 2013, 368, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, J. The Review on Antimicrobial Resistance; Government of the United States: London, UK, 2014. [Google Scholar]
- Farha, M.A.; Leung, A.; Sewell, E.W.; D’Elia, M.A.; Allison, S.E.; Ejim, L.; Pereira, P.M.; Pinho, M.G.; Wright, G.D.; Brown, E.D. Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to beta-lactams. ACS Chem. Biol. 2013, 8, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Report, G.T. Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Franco, L.H.; Joffe, E.B.; Puricelli, L.; Tatian, M.; Seldes, A.M.; Palermo, J.A. Indole alkaloids from the tunicate Aplidium meridianum. J. Nat. Prod. 1998, 61, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Seldes, A.M.; Brasco, M.F.; Franco, L.H.; Palermo, J.A. Identification of two meridianins from the crude extract of the tunicate Aplidium meridianum by tandem mass spectrometry. Nat. Prod. Res. 2007, 21, 555–563. [Google Scholar] [CrossRef]
- Yadav, R.R.; Khan, S.I.; Singh, S.; Khan, I.A.; Vishwakarma, R.A.; Bharate, S.B. Synthesis, antimalarial and antitubercular activities of meridianin derivatives. Eur. J. Med. Chem. 2015, 98, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Yadav, R.R.; Khan, S.I.; Tekwani, B.L.; Jacob, M.R.; Khan, I.A.; Vishwakarma, R.A. Meridianin G and its analogs as antimalarial agents. MedChemComm 2013, 4, 1042–1048. [Google Scholar] [CrossRef]
- Lebar, M.D.; Hahn, K.N.; Mutka, T.; Maignan, P.; McClintock, J.B.; Amsler, C.D.; van Olphen, A.; Kyle, D.E.; Baker, B.J. CNS and antimalarial activity of synthetic meridianin and psammopemmin analogs. Bioorg. Med. Chem. 2011, 19, 5756–5762. [Google Scholar] [CrossRef]
- Radwan, M.A.; El-Sherbiny, M. Synthesis and antitumor activity of indolylpyrimidines: Marine natural product meridianin D analogues. Bioorg. Med. Chem. 2007, 15, 1206–1211. [Google Scholar] [CrossRef]
- Huggins, W.M.; Barker, W.T.; Baker, J.T.; Hahn, N.A.; Melander, R.J.; Melander, C. Meridianin D Analogues Display Antibiofilm Activity against MRSA and Increase Colistin Efficacy in Gram-Negative Bacteria. ACS Med. Chem. Lett. 2018, 9, 702–707. [Google Scholar] [CrossRef]
- Zeiler, M.J.; Melander, R.J.; Melander, C. Second-Generation Meridianin Analogues Inhibit the Formation of Mycobacterium smegmatis Biofilms and Sensitize Polymyxin-Resistant Gram-Negative Bacteria to Colistin. ChemMedChem 2020, 15, 1672–1679. [Google Scholar] [CrossRef]
- Alagarsamy, V.; Raja Solomon, V.; Dhanabal, K. Synthesis and pharmacological evaluation of some 3-phenyl-2-substituted-3H-quinazolin-4-one as analgesic, anti-inflammatory agents. Bioorg. Med. Chem. 2007, 15, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Wansi, J.D.; Happi, E.N.; Bavoua, J.L.; Devkota, K.P.; Sewald, N. Bioactive beta-indoloquinazoline alkaloids from Oricia renieri. Planta Med. 2012, 78, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Ibrar, A.; Abbas, N.; Saeed, A. Recent advances in the structural library of functionalized quinazoline and quinazolinone scaffolds: Synthetic approaches and multifarious applications. Eur. J. Med. Chem. 2014, 76, 193–244. [Google Scholar] [CrossRef] [PubMed]
- Tseng, M.C.; Yang, H.Y.; Chu, Y.H. Total synthesis of asperlicin C, circumdatin F, demethylbenzomalvin A, demethoxycircumdatin H, sclerotigenin, and other fused quinazolinones. Org. Biomol. Chem. 2010, 8, 419–427. [Google Scholar] [CrossRef]
- Rao, Y.; Yu, H.; Gao, L.; Lu, Y.T.; Xu, Z.; Liu, H.; Gu, L.Q.; Ye, J.M.; Huang, Z.S. Natural alkaloid bouchardatine ameliorates metabolic disorders in high-fat diet-fed mice by stimulating the sirtuin 1/liver kinase B-1/AMPK axis. Br. J. Pharmacol. 2017, 174, 2457–2470. [Google Scholar] [CrossRef] [Green Version]
- Michael, J.P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 2008, 25, 166–187. [Google Scholar] [CrossRef]
- Vega, N.M.; Allison, K.R.; Khalil, A.S.; Collins, J.J. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 2012, 8, 431–433. [Google Scholar] [CrossRef]
- Kim, J.; Park, W. Indole: A signaling molecule or a mere metabolic byproduct that alters bacterial physiology at a high concentration? J. Microbiol. 2015, 53, 421–428. [Google Scholar] [CrossRef]
- Kim, C.S.; Li, J.H.; Barco, B.; Park, H.B.; Gatsios, A.; Damania, A.; Wang, R.; Wyche, T.P.; Piizzi, G.; Clay, N.K.; et al. Cellular Stress Upregulates Indole Signaling Metabolites in Escherichia coli. Cell Chem. Biol. 2020, 27, 698–707.e7. [Google Scholar] [CrossRef]
- Wood, T.K.; Song, S. Forming and waking dormant cells: The ppGpp ribosome dimerization persister model. Biofilm 2020, 2, 100018. [Google Scholar] [CrossRef]
- Lang, M.; Krin, E.; Korlowski, C.; Sismeiro, O.; Varet, H.; Coppee, J.Y.; Mazel, D.; Baharoglu, Z. Sleeping ribosomes: Bacterial signaling triggers RaiA mediated persistence to aminoglycosides. iScience 2021, 24, 103128. [Google Scholar] [CrossRef]
- Kashevarova, N.M.; Akhova, A.V.; Khaova, E.A.; Tkachenko, A.G. Role of alarmone (p)ppGpp in the regulation of indole formation depending on glucose content in Escherichia coli. Acta Biomed. Sci. 2022, 7, 162–168. [Google Scholar] [CrossRef]
- Tkachenko, A.G.; Kashevarova, N.M.; Sidorov, R.Y.; Nesterova, L.Y.; Akhova, A.V.; Tsyganov, I.V.; Vaganov, V.Y.; Shipilovskikh, S.A.; Rubtsov, A.E.; Malkov, A.V. A synthetic diterpene analogue inhibits mycobacterial persistence and biofilm formation by targeting (p)ppGpp synthetases. Cell Chem. Biol. 2021, 28, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Coppa, C.; Sorrentino, L.; Civera, M.; Minneci, M.; Vasile, F.; Sattin, S. New Chemotypes for the Inhibition of (p)ppGpp Synthesis in the Quest for New Antimicrobial Compounds. Molecules 2022, 27, 3097. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Sarma, D. Recent advances in the synthesis of Quinazoline analogues as Anti-TB agents. Tuberculosis 2020, 124, 101986. [Google Scholar] [CrossRef] [PubMed]
- Kausar, N.; Roy, I.; Chattopadhyay, D.; Das, A.R. Synthesis of 2,3-dihydroquinazolinones and quinazolin-4(3H)-ones catalyzed by graphene oxide nanosheets in an aqueous medium: “On-water” synthesis accompanied by carbocatalysis and selective C–C bond cleavage. RSC Adv. 2016, 6, 22320–22330. [Google Scholar] [CrossRef]
- Latha, G.; Devarajan, N.; Suresh, P. Framework Copper Catalyzed Oxidative Synthesis of Quinazolinones: A Benign Approach Using Cu3(BTC)2 MOF as an Efficient and Reusable Catalyst. ChemistrySelect 2020, 5, 10041–10047. [Google Scholar] [CrossRef]
- Safari, J.; Gandomi-Ravandi, S. Efficient synthesis of 2-aryl-2,3-dihydroquinazolin-4(1H)-ones in the presence of nanocomposites under microwave irradiation. J. Mol. Catal. A-Chem. 2014, 390, 1–6. [Google Scholar] [CrossRef]
- Karhale, S.; Survase, D.; Bhat, R.; Ubale, P.; Helavi, V. A practical and green protocol for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones using oxalic acid as organocatalyst. Res. Chem. Intermed. 2017, 43, 3915–3924. [Google Scholar] [CrossRef]
- Sharma, M.; Pandey, S.; Chauhan, K.; Sharma, D.; Kumar, B.; Chauhan, P.M. Cyanuric chloride catalyzed mild protocol for synthesis of biologically active dihydro/spiro quinazolinones and quinazolinone-glycoconjugates. J. Org. Chem. 2012, 77, 929–937. [Google Scholar] [CrossRef]
- Khan, D.; Ahmed, N.; Alsharif, M.A.; Alahmdi, M.I.; Mukhtar, S. SeO2 Mediated Synthesis of Selected Heterocycles by Oxidative C−C Bond Cleavage of AcetophenoneDerivatives. ChemistrySelect 2019, 4, 7585–7590. [Google Scholar] [CrossRef]
- Mohammed, S.; Vishwakarma, R.A.; Bharate, S.B. Iodine Catalyzed Oxidative Synthesis of Quinazolin-4(3H)-ones and Pyrazolo[4,3-d]pyrimidin-7(6H)-ones via Amination of sp(3) C-H Bond. J. Org. Chem. 2015, 80, 6915–6921. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, G.; Shen, J.; Kuai, C.; Cui, X. Cleavage of the C–C triple bond of ketoalkynes: Synthesis of 4(3H)-quinazolinones. Org. Chem. Front. 2015, 2, 366–368. [Google Scholar] [CrossRef]
- Laha, J.K.; Gulati, U.; Saima; Gupta, A.; Indurthi, H.K. Improved, gram-scale synthesis of sildenafil in water using arylacetic acid as the acyl source in the pyrazolo[4,3-d]pyrimidin-7-one ring formation. New J. Chem. 2021, 45, 2643–2648. [Google Scholar] [CrossRef]
- Kabri, Y.; Gellis, A.; Vanelle, P. Microwave-assisted synthesis in aqueous medium of new quinazoline derivatives as anticancer agent precursors. Green Chem. 2009, 11, 201–208. [Google Scholar] [CrossRef]
- Das, S.; Mondal, R.; Chakraborty, G.; Guin, A.K.; Das, A.; Paul, N.D. Zinc Stabilized Azo-anion Radical in Dehydrogenative Synthesis of N-Heterocycles. An Exclusively Ligand Centered Redox Controlled Approach. ACS Catal. 2021, 11, 7498–7512. [Google Scholar] [CrossRef]
- Wu, X.F.; He, L.; Neumann, H.; Beller, M. Palladium-catalyzed carbonylative synthesis of quinazolinones from 2-aminobenzamide and aryl bromides. Chemistry 2013, 19, 12635–12638. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, X.; Chen, G.; Zhao, P. Direct synthesis of quinazolinones by heterogeneous Cu(OH)X/OMS-2 catalyst under oxygen. Cat. Commun. 2018, 104, 106–111. [Google Scholar] [CrossRef]
- Steinchen, W.; Bange, G. The magic dance of the alarmones (p)ppGpp. Mol. Microbiol. 2016, 101, 531–544. [Google Scholar] [CrossRef]
- Yadav, R.R.; Battini, N.; Mudududdla, R.; Bharate, J.B.; Muparappu, N.; Bharate, S.B.; Vishwakarma, R.A. Deformylation of indole and azaindole-3-carboxaldehydes using anthranilamide and solid acid heterogeneous catalyst via quinazolinone intermediate. Tetrahedron Lett. 2012, 53, 2222–2225. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ishizaka, T.; Kawanami, H. Accelerated decarbonylation of 5-hydroxymethylfurfural in compressed carbon dioxide: A facile approach. Green Chem. 2018, 20, 2345–2355. [Google Scholar] [CrossRef]
- Modak, A.; Rana, S.; Phukan, A.K.; Maiti, D. Palladium-Catalyzed Deformylation Reactions with Detailed Experimental and in Silico Mechanistic Studies. Eur. J. Org. Chem. 2017, 2017, 4168–4174. [Google Scholar] [CrossRef]
- Butin, A.V.; Uchuskin, M.G.; Pilipenko, A.S.; Serdyuk, O.V.; Trushkov, I.V. Unusual reactivity of β-(3-indolyl)-α,β-unsaturated ketones. 2-Acetylvinyl group removal by phenylhydrazine hydrochloride. Tetrahedron Lett. 2011, 52, 5255–5258. [Google Scholar] [CrossRef]
- Hour, M.J.; Huang, L.J.; Kuo, S.C.; Xia, Y.; Bastow, K.; Nakanishi, Y.; Hamel, E.; Lee, K.H. 6-Alkylamino- and 2,3-dihydro-3′-methoxy-2-phenyl-4-quinazolinones and related compounds: Their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 2000, 43, 4479–4487. [Google Scholar] [CrossRef]
- Gupta, A.; Vankar, J.K.; Jadav, J.P.; Gururaja, G.N. Water Mediated Direct Thioamidation of Aldehydes at Room Temperature. J. Org. Chem. 2022, 87, 2410–2420. [Google Scholar] [CrossRef]
- Kubicová, L.; Šustr, M.; Kráľová, K.; Chobot, V.; Vytlačilová, J.; Jahodář, L.; Vuorela, P.; Macháček, M.; Kaustová, J. Synthesis and Biological Evaluation of Quinazoline-4-thiones. Molecules 2003, 8, 756–769. [Google Scholar] [CrossRef]
- Jones, P.B.; Parrish, N.M.; Houston, T.A.; Stapon, A.; Bansal, N.P.; Dick, J.D.; Townsend, C.A. A new class of antituberculosis agents. J. Med. Chem. 2000, 43, 3304–3314. [Google Scholar] [CrossRef]
- Tortoli, E.; Cichero, P.; Piersimoni, C.; Simonetti, M.T.; Gesu, G.; Nista, D. Use of BACTEC MGIT 960 for recovery of mycobacteria from clinical specimens: Multicenter study. J. Clin. Microbiol. 1999, 37, 3578–3582. [Google Scholar] [CrossRef] [Green Version]
- Romero, A.H.; Rodriguez, N.; Oviedo, H. 2-Aryl-quinazolin-4(3H)-ones as an inhibitor of leishmania folate pathway: In Vitro biological evaluation, mechanism studies and molecular docking. Bioorg. Chem. 2019, 83, 145–153. [Google Scholar] [CrossRef]
- Hrast, M.; Rozman, K.; Jukic, M.; Patin, D.; Gobec, S.; Sova, M. Synthesis and structure-activity relationship study of novel quinazolinone-based inhibitors of MurA. Bioorg. Med. Chem. Lett. 2017, 27, 3529–3533. [Google Scholar] [CrossRef]
- Liao, B.L.; Pan, Y.J.; Zhang, W.; Pan, L.W. Four Natural Compounds Separated from Folium Isatidis: Crystal Structures and Antibacterial Activity. Chem. Biodivers. 2018, 15, e1800152. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.M.; Bessa, L.J.; Buttachon, S.; Costa, P.M.; Buaruang, J.; Dethoup, T.; Silva, A.M.; Kijjoa, A. Antibacterial and antibiofilm activities of tryptoquivalines and meroditerpenes isolated from the marine-derived fungi Neosartorya paulistensis, N. laciniosa, N. tsunodae, and the soil fungi N. fischeri and N. siamensis. Mar. Drugs 2014, 12, 822–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, R.; Tiwari, M.; Donelli, G.; Tiwari, V. Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action. Virulence 2018, 9, 522–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.R.; Arora, G.; Mattoo, A.; Sajid, A. Stringent Response in Mycobacteria: From Biology to Therapeutic Potential. Pathogens 2021, 10, 1417. [Google Scholar] [CrossRef]
- Singal, B.; Balakrishna, A.M.; Nartey, W.; Manimekalai, M.S.S.; Jeyakanthan, J.; Gruber, G. Crystallographic and solution structure of the N-terminal domain of the Rel protein from Mycobacterium tuberculosis. FEBS Lett. 2017, 591, 2323–2337. [Google Scholar] [CrossRef] [Green Version]
- Hogg, T.; Mechold, U.; Malke, H.; Cashel, M.; Hilgenfeld, R. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response [corrected]. Cell 2004, 117, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Bag, S.; Das, B.; Dasgupta, S.; Bhadra, R.K. Mutational analysis of the (p)ppGpp synthetase activity of the Rel enzyme of Mycobacterium tuberculosis. Arch. Microbiol. 2014, 196, 575–588. [Google Scholar] [CrossRef]
- Ann, J.; Czikora, A.; Saini, A.S.; Zhou, X.; Mitchell, G.A.; Lewin, N.E.; Peach, M.L.; Blumberg, P.M.; Lee, J. α-Arylidene Diacylglycerol-Lactones (DAG-Lactones) as Selective Ras Guanine-Releasing Protein 3 (RasGRP3) Ligands. J. Med. Chem. 2018, 61, 6261. [Google Scholar] [CrossRef]
- Liu, K.; Ding, Y.; Kang, C. Synthesis and Antiproliferative Activity of New N-Acylhydrazone Derivatives Containing Benzothiazole and Indole Based Moiety. Pharm. Chem. J. 2020, 54, 345–352. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Nguyen, T.S.; May, J.A. Bronsted Acid Catalyzed Homoconjugate Addition of Organotrifluoroborates to Arylated Cyclopropyl Ketones. Org. Lett. 2016, 18, 3786–3789. [Google Scholar] [CrossRef]
- van Baar, J.F.; Horton, A.D.; de Kloe, K.P.; Kragtwijk, E.; Mkoyan, S.G.; Nifant’ev, I.E.; Schut, P.A.; Taidakov, I.V. ansa-Zirconocenes Based on N-Substituted 2-Methylcyclopenta[b]Indoles: Synthesis and Catalyst Evaluation in Liquid Propylene Polymerization. Organometallics 2003, 22, 2711–2722. [Google Scholar] [CrossRef]
- Durgadevi, G.; Arjunan, V.; Thirunarayanan, S.; Marchewka, M.K.; Mohan, S. Structure, electronic, spectroscopic and reactivity investigations of pharmacologically active compound 1–acetyl–3–indolecarboxaldehyde—An experimental and theoretical approach. J. Mol. Struct. 2018, 1164, 57–69. [Google Scholar] [CrossRef]
- Wang, J.; Sun, G.; Li, Z.; Mai, W.; Xie, J. Synthesis and Biological Evaluation of Curcumin Analogues having a Piperidone Core as Potential Antioxidant Agents. J. Chem. Res. 2012, 36, 63–65. [Google Scholar] [CrossRef]
- Spoehrle, S.S.M.; West, T.H.; Taylor, J.E.; Slawin, A.M.Z.; Smith, A.D. Tandem Palladium and Isothiourea Relay Catalysis: Enantioselective Synthesis of alpha-Amino Acid Derivatives via Allylic Amination and [2,3]-Sigmatropic Rearrangement. J. Am. Chem. Soc. 2017, 139, 11895–11902. [Google Scholar] [CrossRef] [Green Version]
- Canche Chay, C.I.; Gomez Cansino, R.; Espitia Pinzon, C.I.; Torres-Ochoa, R.O.; Martinez, R. Synthesis and anti-tuberculosis activity of the marine natural product caulerpin and its analogues. Mar. Drugs. 2014, 12, 1757–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, I.; Luxami, V.; Choudhury, D.; Paul, K. Synthesis and photobiological applications of naphthalimide-benzothiazole conjugates: Cytotoxicity and topoisomerase IIalpha inhibition. RSC Adv. 2021, 12, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Guo, X.; Zhang, X.; Li, B.; Huang, Q. Access to 5H-benzo[a]carbazol-6-ols and benzo[6,7]cyclohepta[1,2-b]indol-6-ols via rhodium-catalyzed C–H activation/carbenoid insertion/aldol-type cyclization. Org. Chem. Front. 2020, 7, 3146–3159. [Google Scholar] [CrossRef]
- Christiansen, L.B.; Tagmoste, T.M.; Olesen, P.H.; Hansen, T.K. Indole Derivatives for Use as Chemical Uncoupler. WO Patent 2005051908, 9 June 2005. [Google Scholar]
- Senaweera, S.; Weaver, J.D. S(N)Ar catalysis enhanced by an aromatic donor-acceptor interaction; facile access to chlorinated polyfluoroarenes. Chem. Commun. 2017, 53, 7545–7548. [Google Scholar] [CrossRef]
- Kuo, Y.-H.; Shih, K.-S. A new method for preparation of 3-hydroxypyridines from furfurylamines by photooxygenation. Chem. Pharm. Bull. 1991, 39, 181–183. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Luo, M.; Cai, B.; Haroon Ur, R.; Huang, M.; Jiang, J.; Wang, L.; Wu, L. Design, synthesis, biological evaluation and structure-activity relationship of sophoridine derivatives bearing pyrrole or indole scaffold as potential antitumor agents. Eur. J. Med. Chem. 2018, 157, 665–682. [Google Scholar] [CrossRef]
- Mahiwal, K.; Kumar, P.; Narasimhan, B. Synthesis, antimicrobial evaluation, ot-QSAR and mt-QSAR studies of 2-amino benzoic acid derivatives. Med. Chem. Res. 2010, 21, 293–307. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, H.; Luo, X.; Wang, Y.; Liu, Y.; Jin, H.; Liu, Z.; Yang, W.; Yu, P.; Zhang, L.; et al. Design, synthesis and biological activities of 2,3-dihydroquinazolin-4(1H)-one derivatives as TRPM2 inhibitors. Eur. J. Med. Chem. 2018, 152, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hao, X.-C.; Chen, Y.; Ding, S.; Liu, H.-S.; Wang, D. Synthesis of New Substituted 5-amino-8H-phthalazino[1,2-b]quinazolin-8-one Derivatives. J. Chem. Res. 2017, 41, 18–20. [Google Scholar] [CrossRef]
- Woods, M.; Sherry, A.D. Synthesis and luminescence studies of aryl substituted tetraamide complexes of europium(III): A new approach to pH responsive luminescent europium probes. Inorg. Chem. 2003, 42, 4401–4408. [Google Scholar] [CrossRef]
- Lepri, S.; Nannetti, G.; Muratore, G.; Cruciani, G.; Ruzziconi, R.; Mercorelli, B.; Palu, G.; Loregian, A.; Goracci, L. Optimization of small-molecule inhibitors of influenza virus polymerase: From thiophene-3-carboxamide to polyamido scaffolds. J. Med. Chem 2014, 57, 4337–4350. [Google Scholar] [CrossRef]
- Wannaporn, D.; Ishikawa, T. Polymer-supported and polymeric chiral guanidines: Preparation and application to the asymmetric Michael reaction of iminoacetate with methyl vinyl ketone. Mol. Divers. 2005, 9, 321–331. [Google Scholar] [CrossRef]
- Chen, X.; Chen, T.; Ji, F.; Zhou, Y.; Yin, S.-F. Iron-catalyzed aerobic oxidative functionalization of sp3 C–H bonds: A versatile strategy for the construction of N-heterocycles. Catal. Sci. Technol. 2015, 5, 2197–2202. [Google Scholar] [CrossRef]
- Mertens, M.D.; Pietsch, M.; Schnakenburg, G.; Gutschow, M. Regioselective sulfonylation and N- to O-sulfonyl migration of quinazolin-4(3H)-ones and analogous thienopyrimidin-4(3H)-ones. J. Org. Chem. 2013, 78, 8966–8979. [Google Scholar] [CrossRef]
- Shinde, A.R.; Mane, Y.D.; Muley, D.B. One-pot B(C6F5)3 catalyzed cascade synthesis of 2-substituted-2,3-dihydroquinazolin-4(1H)-ones. Synth. Commun. 2019, 50, 33–40. [Google Scholar] [CrossRef]
- Zhang, F.L.; Wang, Y.F.; Chiba, S. Orthogonal aerobic conversion of N-benzyl amidoximes to 1,2,4-oxadiazoles or quinazolinones. Org. Biomol. Chem. 2013, 11, 6003–6007. [Google Scholar] [CrossRef]
- Rao, K.R.; Mekala, R.; Raghunadh, A.; Meruva, S.B.; Kumar, S.P.; Kalita, D.; Laxminarayana, E.; Prasad, B.; Pal, M. A catalyst-free rapid, practical and general synthesis of 2-substituted quinazolin-4(3H)-ones leading to luotonin B and E, bouchardatine and 8-norrutaecarpine. RSC Adv. 2015, 5, 61575–61579. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Hwang, I.Y.; Marshall, E.S.; Lindsay, B.S.; Denny, W.A.; Gilchrist, C.; Joseph, W.R.; Greenhalgh, D.; Richardson, E.; Kestell, P.; et al. Therapeutic reactivation of mutant p53 protein by quinazoline derivatives. Investig. New Drugs 2012, 30, 2035–2045. [Google Scholar] [CrossRef] [PubMed]
- Memarian, H.R.; Ebrahimi, S. Light induced oxidation of 2,3-dihydroquinazolin-4(1H)-ones. J. Photochem. Photobiol. A Chem. 2013, 271, 8–15. [Google Scholar] [CrossRef]
- Hao, S.; Yang, J.; Liu, P.; Xu, J.; Yang, C.; Li, F. Linear-Organic-Polymer-Supported Iridium Complex as a Recyclable Auto-Tandem Catalyst for the Synthesis of Quinazolinones via Selective Hydration/Acceptorless Dehydrogenative Coupling from o-Aminobenzonitriles. Org. Lett. 2021, 23, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Zaytsev, V.P.; Revutskaya, E.L.; Kuz´menko, M.G.; Novikov, R.A.; Zubkov, F.I.; Sorokina, E.A.; Nikitina, E.V.; Toze, F.A.A.; Varlamov, A.V. Synthesis of furyl-, furylvinyl-, thienyl-, pyrrolinylquinazolines and isoindolo[2,1-a]quinazolines. Russ. Chem. Bull. 2016, 64, 1345–1353. [Google Scholar] [CrossRef]
- Kidwai, M.; Singhal, K.; Singhal, P. One-Pot Heterocondensation for Benzoxazinone Derivatives Using Sodium Perborate as Catalyst. Heterocycles 2007, 71, 1615–1622. [Google Scholar] [CrossRef]
- Garad, D.N.; Viveki, A.B.; Mhaske, S.B. Pd-Catalyzed Regioselective Mono-Arylation: Quinazolinone as the Inherent Directing Group for C(sp(2))-H Activation. J. Org. Chem. 2017, 82, 6366–6372. [Google Scholar] [CrossRef]
- O’Toole, G.A. Microtiter dish biofilm formation assay. J. Vis. Exp. 2011, 47, e2437. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Maestro. Schrödinger Release 2018-1; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | C. a. 10231 b | M. s. 70084 c | E. c. 25922 d | E. c. 8739 e | S. a. 25923 f | MRSA g | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC | MFC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| 3d | 125 | 125 | - h | - | - | - | - | - | - | - | - | - |
| 3g | - | - | - | - | - | - | - | - | - | - | - | - |
| 3k | 7.80 | 7.80 | - | - | - | - | - | - | 3.90 | 7.80 | 0.98 | 3.90 |
| 3p | 250 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 | >500 |
| 3t | 125 | 125 | - | - | - | - | - | - | - | - | - | - |
| 3u | 125 | 500 | - | - | - | - | - | - | - | - | - | - |
| 3z | >250 | >250 | >250 | >250 | >250 | >250 | >250 | >250 | >250 | >250 | >250 | >250 |
| 3ab | 500 | - | - | - | - | - | - | - | - | - | - | - |
| 3ai | 62.50 | - | - | - | - | - | - | - | - | - | - | - |
| 5a | 250 | 500 | - | - | - | - | - | - | - | - | - | - |
| 6u | 125 | - | - | - | - | - | - | - | - | - | - | - |
| Cefotaxime | n.d. i | n.d. | n.d. | n.d. | 0.038 | 0.038 | 0.038 | 0.038 | 0.31 | 0.61 | 19.53 | 39.06 |
| Cefazolin | n.d. | n.d. | n.d. | n.d. | 2.44 | 2.44 | 2.44 | 2.44 | 0.15 | 0.61 | 9.77 | 39.06 |
| Amikacin | n.d. | n.d. | n.d. | n.d. | 19.53 | 19.53 | 19.53 | 19.53 | 4.88 | 9.77 | 9.77 | 9.77 |
| Fluconazole | 1.94 | 7.8 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Isoniazid | n.d. | n.d. | 4.58 | 9.16 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Rifampicin | n.d. | n.d. | 1.22 | 19.53 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Conc., µg/mL | Fluorescence Change (BACTEC MGIT 960 Growth System), Days | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 18 | 25 | 30 | 35 | 41 | |
| Control | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 100.0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 50.0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 25.0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 10.0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 5.0 | - | - | - | - | - | - | - | - | - | - | - | - | + | + | + | + |
| 2.5 | - | - | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 1.0 | - | - | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Compounds | MCF7′ | VA13 | A549 | HEK293T | ||||
|---|---|---|---|---|---|---|---|---|
| IC50 | IC50abs | IC50 | IC50abs | IC50 | IC50abs | IC50 | IC50abs | |
| 3b | 2.1 ± 0.4 | 27.1 ± 4.7 | 11.4 ± 1.2 | 29.4 ± 4.3 | 2.1 ± 0.3 | 3.6 ± 0.4 | 6.2 ± 0.7 | 8.3 ± 0.7 |
| 3c | 4.1 ± 0.6 | 10.9 ± 1.1 | 3.7 ± 0.4 | 4.0 ± 0.2 | 3.7 ± 0.4 | 1.7 ± 0.2 | 1.8 ± 0.7 | 3.4 ± 0.6 |
| 3d | le b | x > 100 | 47.0 ± 11.8 | 69.3 ± 3.1 | 43.0 ± 5.1 | 40.5 ± 2.7 | 5.5 ± 0.9 | 56.8 ± 7.9 |
| 3e | x > 100 | x > 100 | x > 100 | x > 100 | ~1 | 8.0 ± 2.3 | ~1 | x > 100 |
| 3f | 4.9 ± 0.7 | 7.1 ± 0.8 | 3.4 ± 0.4 | 3.8 ± 0.3 | 0,96 ± 0,18 | 1.3 ± 0.09 | 0.95 ± 0.28 | 1.9 ± 0.16 |
| 3g | 0.57 ± 0.05 | 27.6 ± 7.4 | 0.84 ± 0.38 | 13.5 ± 3.6 | 0.75 ± 0.07 | 3.3 ± 0,6 | 0.73 ± 0.15 | 9.8 ± 2.5 |
| 3i | 11.8 ± 0.7 | 10.9 ± 0.6 | 6.6 ± 0.5 | 7.1 ± 0.4 | 11.7± 1.2 | 9.0 ± 0.7 | 3.4 ± 0.2 | 4.1 ± 0.3 |
| 3k | 5.9 ± 0.3 | 6.1 ± 0.3 | 7.9 ± 0.4 | 8.5 ± 0.3 | 6.5 ± 0.2 | 6.5± 0.2 | 3.4 ± 0.1 | 3.5 ± 0.1 |
| 3o | n.a. c | 57.1 ± 6.2 | ~20 | 67.8 ± 21.1 | 9.1 ± 1.2 | 38.1 ± 6.4 | n.a. | 19.8 ± 3.4 |
| 3q | ~20 | 19.7 ± 2.5 | 16.5 ± 2.5 | 23.2 ± 2.9 | 6.1 ± 1.1 | 6.9 ± 0.6 | 8.2 ± 1.1 | 9.0 ± 0.7 |
| 3r | 5.4 ± 2.2 | x > 100 | 2.5 ± 0.7 | 60.1 ± 23.0 | 3.5 ± 03 | 27.6 ± 4.9 | 1.8 ± 0.4 | 48.9 ± 13.6 |
| 3s | 13.6 ± 1.2 | 10.1 ± 1.0 | 13.5 ± 1.8 | 12.9 ± 0.8 | 9.4 ± 03 | 9.3 ± 0.3 | 12.4 ± 0.5 | 12,7 ± 0.4 |
| 3t | x > 33.3 | x > 33.3 | x > 33.3 | x > 33.3 | x > 333 | x > 33.3 | x > 33.3 | x > 33.3 |
| 3u | 0.30 ± 0.14 (le) | x > 100 | x > 100 | x > 100 | n.a. | 107.4 ± 27.7 | n.a. | 66.8 ± 3.6 |
| 3v | n.a. | 93.0 ± 3.1 | n.a. | 80.6 ± 3.9 | n.a. | 57.0 ± 3.9 | n.a. | 62.9 ± 2.8 |
| 3w | 0.53 ± 0.44 (le) | x > 100 | x > 100 | 115.8 ± 0.2 | n.a. | 105.1 ± 15.8 | n.a. | 104.8 ± 2.1 |
| 3x | le | x > 100 | le | x > 100 | le | x > 100 | 57.3 ± 34.5 | 152.3 ± 30.3 |
| 3y | x > 100 | x > 100 | x > 100 | x > 100 | 61.4 ± 58.1 | x > 100 | x > 100 | x > 100 |
| 3z | 8.4 ± 0.70 | 16.1 ± 0.9 | 3.0 ± 0.4 | 4.2 ± 0.4 | 2.6 ± 0.3 | 3.2 ± 0.3 | 3.9 ± 0.1 | 4.0 ± 0.1 |
| 3aa | x > 100 | x > 100 | x > 100 | 163.4 ± 19.7 | x > 100 | 65.5 ± 5.8 | x > 100 | 195.8 ± 51.0 |
| 3ab | x > 100 | x > 100 | 44.9 ± 11.7 | 90.8 ± 8.7 | n.a. | 169.1 ± 47.2 | 30.5 ± 1.1 | 32.2 ± 1.6 |
| 6u | x > 100 | x > 100 | x > 100 | 129.3 ± 23.4 | x > 100 | 215.7 ± 35.8 | x > 100 | x > 100 |
| doxorubicin | 0.195 ± 0.01 | 0.195 ± 0.01 | 0.087 ± 0.04 | 0.087 ± 0.04 | 0.038 ± 0.01 | 0.038 ± 0.01 | 0.018 ± 0.001 | 0.018 ± 0.001 |
| RelSeq SYN Active Site | RelMtb SYN Active Site | ||
|---|---|---|---|
| Ligand | Docking Score (kcal mol−1) | Ligand | Docking Score (kcal mol−1) |
| Indole-5-carboxylic acid (COO−) | −6.42 | 3b | −7.61 |
| 3o | −4.76 | 3ab | −7.43 |
| 3ab | −4.72 | 3p | −6.71 |
| 3a | −4.64 | 3a | −6.70 |
| 3b | −4.53 | 3ah | −6.67 |
| 3d | −4.48 | 3r | −6.66 |
| 3e | −4.48 | 3am | −6.62 |
| 3ak | −4.45 | 3o | −6.56 |
| 7a | −4.39 | Indole-5-carboxylicacid (COO−) | −6.55 |
| 3ah | −4.34 | 7a | −6.52 |
| 3r | −4.33 | 3l | −6.51 |
| 3p | −4.32 | DMNP a (COO−) | −6.41 |
| DMNP a (COO−) | −4.28 | 3k | −6.17 |
| 3q | −4.28 | 3c | −6.10 |
| 3g | −4.15 | 3d | −6.07 |
| 3c | −4.11 | 3q | −6.05 |
| 3l | −4.05 | 3ai | −5.82 |
| 3i | −4.01 | 3f | −5.77 |
| 3ai | −3.97 | 3i | −5.57 |
| 3f | −3.84 | 3ak | −5.50 |
| 3am | −3.78 | 3g | −5.33 |
| 3k | −3.77 | 7b | −4.96 |
| 7b | −3.73 | 3e | −4.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendogralo, E.Y.; Nesterova, L.Y.; Nasibullina, E.R.; Shcherbakov, R.O.; Tkachenko, A.G.; Sidorov, R.Y.; Sukonnikov, M.A.; Skvortsov, D.A.; Uchuskin, M.G. The Synthesis and Biological Evaluation of 2-(1H-Indol-3-yl)quinazolin-4(3H)-One Derivatives. Molecules 2023, 28, 5348. https://doi.org/10.3390/molecules28145348
Mendogralo EY, Nesterova LY, Nasibullina ER, Shcherbakov RO, Tkachenko AG, Sidorov RY, Sukonnikov MA, Skvortsov DA, Uchuskin MG. The Synthesis and Biological Evaluation of 2-(1H-Indol-3-yl)quinazolin-4(3H)-One Derivatives. Molecules. 2023; 28(14):5348. https://doi.org/10.3390/molecules28145348
Chicago/Turabian StyleMendogralo, Elena Y., Larisa Y. Nesterova, Ekaterina R. Nasibullina, Roman O. Shcherbakov, Alexander G. Tkachenko, Roman Y. Sidorov, Maxim A. Sukonnikov, Dmitry A. Skvortsov, and Maxim G. Uchuskin. 2023. "The Synthesis and Biological Evaluation of 2-(1H-Indol-3-yl)quinazolin-4(3H)-One Derivatives" Molecules 28, no. 14: 5348. https://doi.org/10.3390/molecules28145348
APA StyleMendogralo, E. Y., Nesterova, L. Y., Nasibullina, E. R., Shcherbakov, R. O., Tkachenko, A. G., Sidorov, R. Y., Sukonnikov, M. A., Skvortsov, D. A., & Uchuskin, M. G. (2023). The Synthesis and Biological Evaluation of 2-(1H-Indol-3-yl)quinazolin-4(3H)-One Derivatives. Molecules, 28(14), 5348. https://doi.org/10.3390/molecules28145348