
Computational Modeling of Human Serum Albumin Binding of Per- and Polyfluoroalkyl Substances Employing QSAR, Read-Across, and Docking



Abstract
:
1. Introduction
2. Results and Discussion
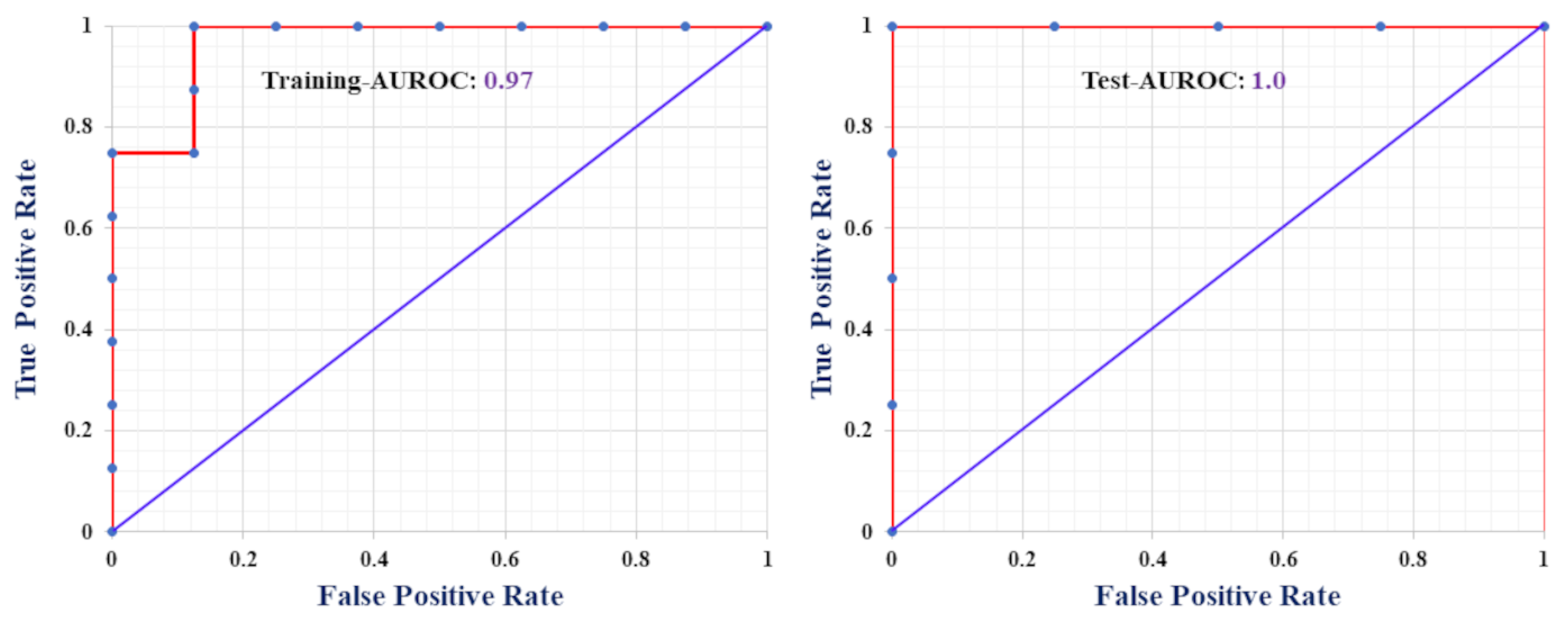
2.1. Classification-Based QSAR Model
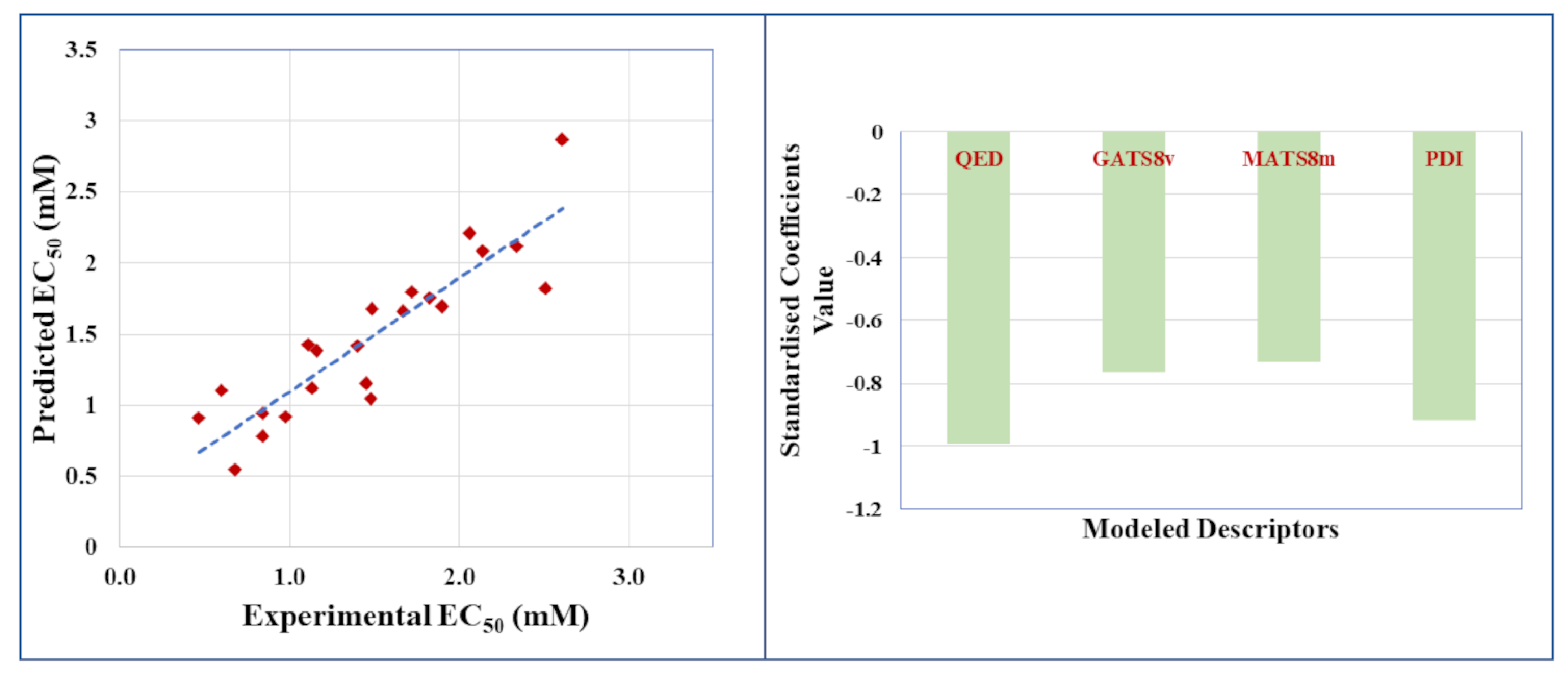
2.2. Regression-Based ‘Small Dataset QSAR’ Model for Undivided Dataset
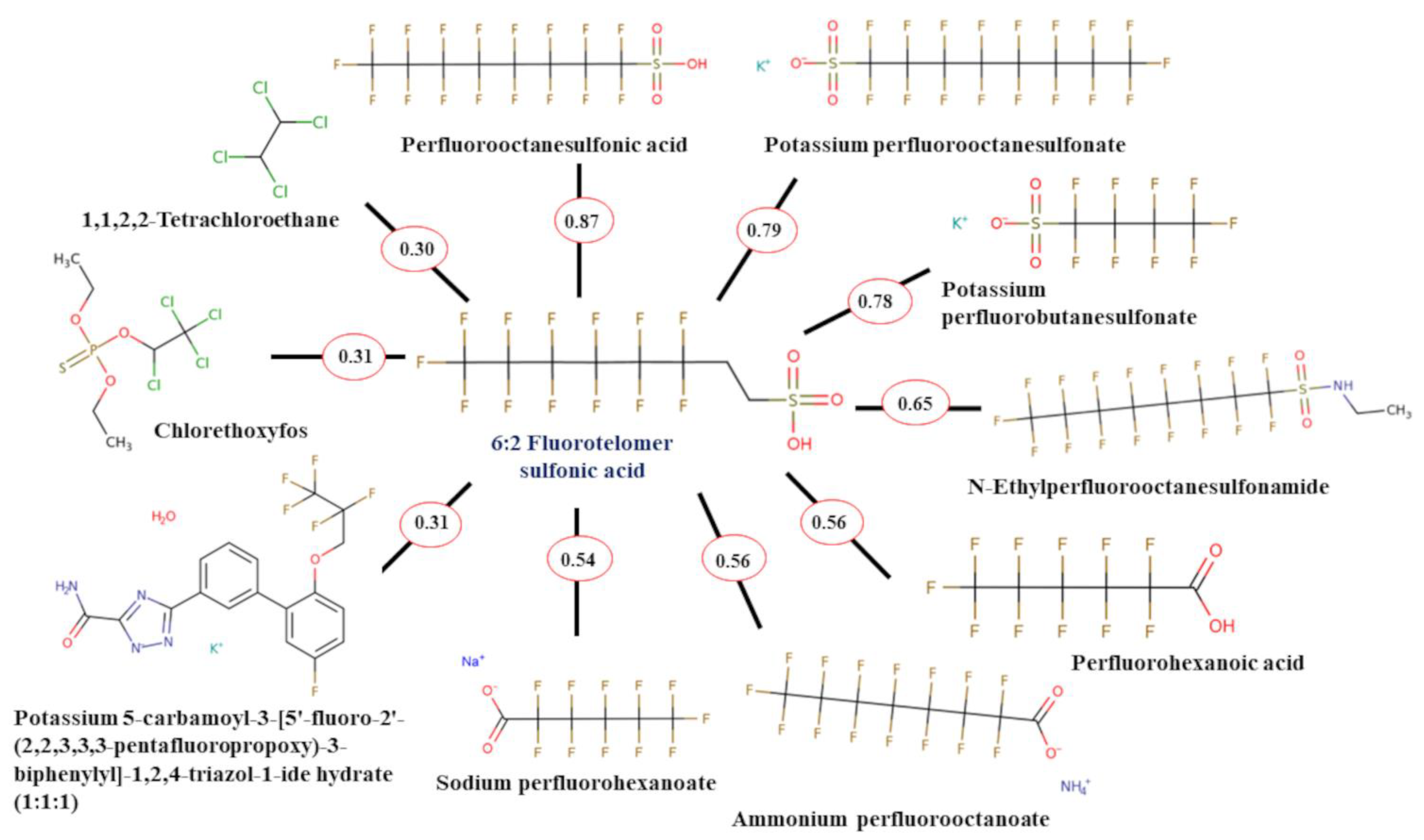
2.3. Read-Across Results
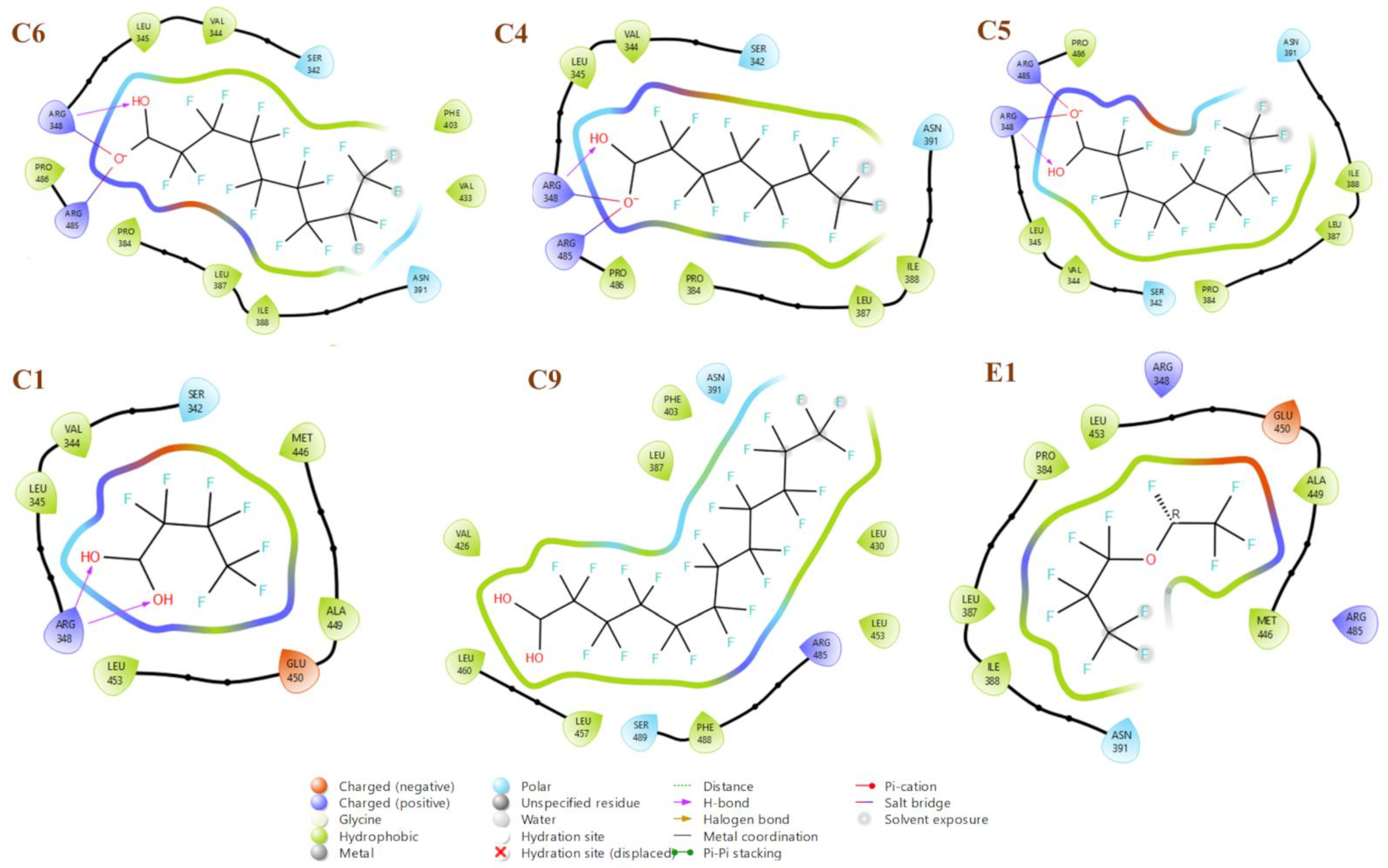
2.4. Docking Results
3. Materials and Methods
3.1. Dataset
3.2. Descriptor Calculation
3.3. QSAR Modeling
3.4. Validation, Applicability Domain, and Randomization
3.5. Docking Study
3.6. Read-Across
4. Overview and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- EPA. Our Current Understanding of the Human Health and Environmental Risks of PFAS; EPA: Washington, DC, USA, 2022. [Google Scholar]
- Ahrens, L. Polyfluoroalkyl compounds in the aquatic environment: A review of their occurrence and fate. J. Environ. Monit. 2011, 13, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Lassen, C.; Jensen, A.A.; Potrykus, A.; Christensen, F.; Kjølholt, J.; Jeppesen, C.N.; Mikkelsen, S.H.; Innanen, S. Survey of PFOS, PFOA and Other Perfluoroalkyl and Polyfluoroalkyl Substances. 2013. Available online: https://www2.mst.dk/udgiv/publications/2013/04/978-87-93026-03-2.pdf (accessed on 6 June 2023).
- National Toxicology Program (NTP). Immunotoxicity Associated with Exposure to Perfluorooctanoic Acid (PFOA) or Perfluorooctane Sulfonate (PFOS). 2016. Available online: https://ntp.niehs.nih.gov/go/749926 (accessed on 6 June 2023).
- Fenton, S.E.; Ducatman, A.; Boobis, A.; DeWitt, J.C.; Lau, C.; Ng, C.; Smith, J.S.; Roberts, S.M. Per- and polyfluoroalkyl substance toxicity and human health review: Current state of knowledge and strategies for informing future research. Environ. Toxicol. Chem. 2021, 40, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.; Berger, U.; Goss, K.-U.; Ulrich, N. Partition coefficients of four perfluoroalkyl acid alternatives between bovine serum albumin (BSA) and water in comparison to ten classical perfluoroalkyl acids. Environ. Sci. Process. Impacts 2019, 21, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.W.; Scheibly, C.M.; Polera, M.E.; Belcher, S.M. Rapid Characterization of human serum albumin binding for per- and polyfluoroalkyl substances using differential scanning fluorimetry. Environ. Sci. Technol. 2021, 55, 12291–12301. [Google Scholar] [CrossRef] [PubMed]
- Chi, Q.; Li, Z.; Huang, J.; Ma, J.; Wang, X. Interactions of perfluorooctanoic acid and perfluorooctanesulfonic acid with serum albumins by native mass spectrometry, fluorescence and molecular docking. Chemosphere 2018, 198, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Delva-Wiley, J.; Jahan, I.; Newman, R.H.; Zhang, L.; Dong, M. Computational analysis of the binding mechanism of GenX and HSA. ACS Omega 2021, 6, 29166–29170. [Google Scholar] [CrossRef] [PubMed]
- Alesio, J.L.; Slitt, A.; Bothun, G.D. Critical new insights into the binding of poly- and perfluoroalkyl substances (PFAS) to albumin protein. Chemosphere 2022, 287, 131979. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Sepúlveda, M.S.; Roy, K.; Leszczynski, J. Endocrine-disrupting activity of per- and polyfluoroalkyl substances: Exploring combined approaches of ligand and structure based modeling. Chemosphere 2017, 184, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Hoover, G.; Kar, S.; Guffey, S.; Leszczynski, J.; Sepúlveda, M.S. In vitro and in silico modeling of perfluoroalkyl substances mixture toxicity in an amphibian fibroblast cell line. Chemosphere 2019, 233, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Bertola, M. Alvascience: A new software suite for the QSAR workflow applied to the blood brain barrier permeability. Int. J. Mol. Sci. 2022, 23, 12882. [Google Scholar] [CrossRef] [PubMed]
- Ambure, P.; Roy, K. DTC Lab Software Tools. Available online: http://teqip.jdvu.ac.in/QSAR_Tools/ (accessed on 6 June 2023).
- Chicco, D.; Jurman, G. The Matthews correlation coefficient (MCC) should replace the ROC AUC as the standard metric for assessing binary classification. BioData Min. 2023, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- LigPrep, Schrödinger Release 2023-2: Schrödinger, LLC, New York, NY. 2023. Available online: https://www.schrodinger.com/products/ligprep (accessed on 6 June 2023).
- Glide, Schrödinger Release 2023-2: Schrödinger, LLC, New York, NY. 2023. Available online: https://www.schrodinger.com/products/glide (accessed on 6 June 2023).
- Generalized Read-Across (GenRA). Available online: https://comptox.epa.gov/genra/ (accessed on 6 June 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | CAS | Chemical Name | Regression-Based QSAR | Classification-Based QSAR | Docking | ||
|---|---|---|---|---|---|---|---|
| Observed EC50 (mM) | Predicted EC50 (PLS Model) | Observed Classification | Predicted Classification (LDA Model) | Glide Energy (kcal/mol) | |||
| C1 | 375-22-4 | Perfluorobutanoic acid (PFBA) | 2.61 | 2.87 | L | L | −19.583 |
| C2 | 2706-90-3 | Perfluoropentanoic acid (PFPeA) | 2.14 | 2.09 | L | L | −21.171 |
| C3 | 307-24-4 | Perfluorohexanoic acid (PFHxA) | 1.40 | 1.41 | L | L | −20.473 |
| C4 | 375-85-9 | Perfluoroheptanoic acid (PFHpA) | 0.68 | 0.55 | H | H | −31.114 |
| C5 * | 335-67-1 | Perfluorooctanoic acid (PFOA) | 0.84 | 0.78 | H | H | −33.059 |
| C6 | 375-95-1 | Perfluorononanoic acid (PFNA) | 0.60 | 1.10 | H | H | −37.045 |
| C7 * | 335-76-2 | Perfluorodecanoic acid (PFDA) | 1.11 | 1.42 | H | H | −40.381 |
| C8 | 2058-94-8 | Perfluoroundecanoic acid (PFUnDA) | 1.49 | 1.68 | H | H | −35.144 |
| C9 | 307-55-1 | Perfluorododecanoic acid (PFDoA) | 2.51 | 1.83 | L | L | −25.948 |
| C10 | 356-02-5 | 3:3 Fluorotelomer carboxylic acid (3:3 FTCA) | 2.06 | 2.21 | L | L | −22.287 |
| C11 | 914637-49-3 | 5:3 Fluorotelomer carboxylic acid (5:3 FTCA) | 1.48 | 1.04 | H | H | −25.859 |
| C12 | 27854-30-4 | 6:3 Fluorotelomer carboxylic acid (6:3 FTCA) | 0.84 | 0.95 | H | H | −27.848 |
| C13 | 34598-33-9 | 8:3 Fluorotelomer carboxylic acid (8:3 FTCA) | 1.16 | 1.38 | H | H | −29.169 |
| E1 * | 3330-15-2 | Heptafluoropropyl 1,2,2,2-tetrafluoroethyl ether (E1) | 2.34 | 2.11 | L | L | −18.76 |
| E2 | 13252-13-6 | 2,3,3,3-Tetrafluoro-2-(heptafluoropropoxy)propanoic acid (HFPO-DA) | 1.83 | 1.75 | L | L | −24.764 |
| E3 | 749836-20-2 | 7H-Perfluoro-4-methyl-3,6-dioxaoctanesulfonic acid (Nafion BP2) | 1.90 | 1.70 | L | L | −27.534 |
| E4 * | 151772-59-7 | Perfluoro-3,6,9-trioxadecanoic acid (PFO3DoDA) | 1.67 | 1.66 | L | L | −34.337 |
| S1 * | 375-73-5 | Perfluorobutanesulfonic acid (PFBS) | 1.72 | 1.79 | L | L | −26.738 |
| S2 * | 355-46-4 | Perfluorohexanesulfonic acid (PFHxS) | 0.98 | 0.92 | H | H | −27.822 |
| S3 * | 1763-23-1 | Perfluorooctanesulfonic acid (PFOS) | 1.13 | 1.12 | H | H | −31.682 |
| S4 | 757124-72-4 | 4:2 Fluorotelomer sulfonic acid (4:2 FTSA) | 1.45 | 1.16 | H | L | −23.829 |
| S5 | 59587-38-1 | 6:2 Fluorotelomer sulfonic acid (6:2 FTSA) | 0.47 | 0.91 | H | H | −28.131 |
| O1 | 2043-47-2 | 4:2 Fluorotelomer alcohol (4:2 FTOH) | N/A | N/A | L | L | −16.373 |
| O2 * | 647-42-7 | 6:2 Fluorotelomer alcohol (6:2 FTOH) | N/A | N/A | L | L | −21.834 |
| Metrics | Training Set | Test Set |
|---|---|---|
| Sensitivity (%) | 87.5 | 100 |
| Specificity (%) | 100 | 100 |
| Precision (%) | 100 | 100 |
| Accuracy (%) | 93.75 | 100 |
| F-measure (%) | 93.33 | 100 |
| MCC | 0.88 | 1 |
| AUROC | 0.97 | 1 |
| Cohen’s κ | 0.88 | 1 |
| G-means | 93.5 | 100 |
| Model | Chemometric Tool | No. of Descriptors | LV | R2 | Q2(LOO) | MAE(95%) | |
|---|---|---|---|---|---|---|---|
| 1 | MLR | 4 | - | 0.805 | 0.677 | 0.588 | 0.221 |
| 2 | PLS | 4 | 3 | 0.802 | 0.691 | 0.594 | 0.205 |
| Metrics Defining Statistical Quality of the Classification-Based QSAR Models | ||
|---|---|---|
| Sl. No. | Mathematical Definition | |
| 1 | Goodness-of-fit and quality measures | |
| 2 | ||
| 3 | Internal and external validation metrics and parameters for ROC analysis | |
| 4 | ||
| 5 | ||
| 6 | ||
| 7 | ||
| 8 | ||
| 9 | ||
| 10 | ||
| Metrics defining statistical quality of the regression-based models | ||
| Sl. No. | Mathematical definition | |
| 11 | Goodness-of-fit and quality measures | |
| 12 | Internal parameters For robustness checking | |
| 13 | Mean absolute error | Prediction error |
| 14 | rm2 metric where ) The parameters r2 and r02 are defined as follows: & The terms k and k’ are defined as: & The Yobs and Ypred values have been scaled at the beginning using the following formula: | Scaled rm2 metrics for internal predictivity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallagher, A.; Kar, S.; Sepúlveda, M.S. Computational Modeling of Human Serum Albumin Binding of Per- and Polyfluoroalkyl Substances Employing QSAR, Read-Across, and Docking. Molecules 2023, 28, 5375. https://doi.org/10.3390/molecules28145375
Gallagher A, Kar S, Sepúlveda MS. Computational Modeling of Human Serum Albumin Binding of Per- and Polyfluoroalkyl Substances Employing QSAR, Read-Across, and Docking. Molecules. 2023; 28(14):5375. https://doi.org/10.3390/molecules28145375
Chicago/Turabian StyleGallagher, Andrea, Supratik Kar, and Maria S. Sepúlveda. 2023. "Computational Modeling of Human Serum Albumin Binding of Per- and Polyfluoroalkyl Substances Employing QSAR, Read-Across, and Docking" Molecules 28, no. 14: 5375. https://doi.org/10.3390/molecules28145375
APA StyleGallagher, A., Kar, S., & Sepúlveda, M. S. (2023). Computational Modeling of Human Serum Albumin Binding of Per- and Polyfluoroalkyl Substances Employing QSAR, Read-Across, and Docking. Molecules, 28(14), 5375. https://doi.org/10.3390/molecules28145375