
New Acetamide-Sulfonamide-Containing Scaffolds: Antiurease Activity Screening, Structure-Activity Relationship, Kinetics Mechanism, Molecular Docking, and MD Simulation Studies


 , ,
, ,  , , ,
, , ,
Abstract
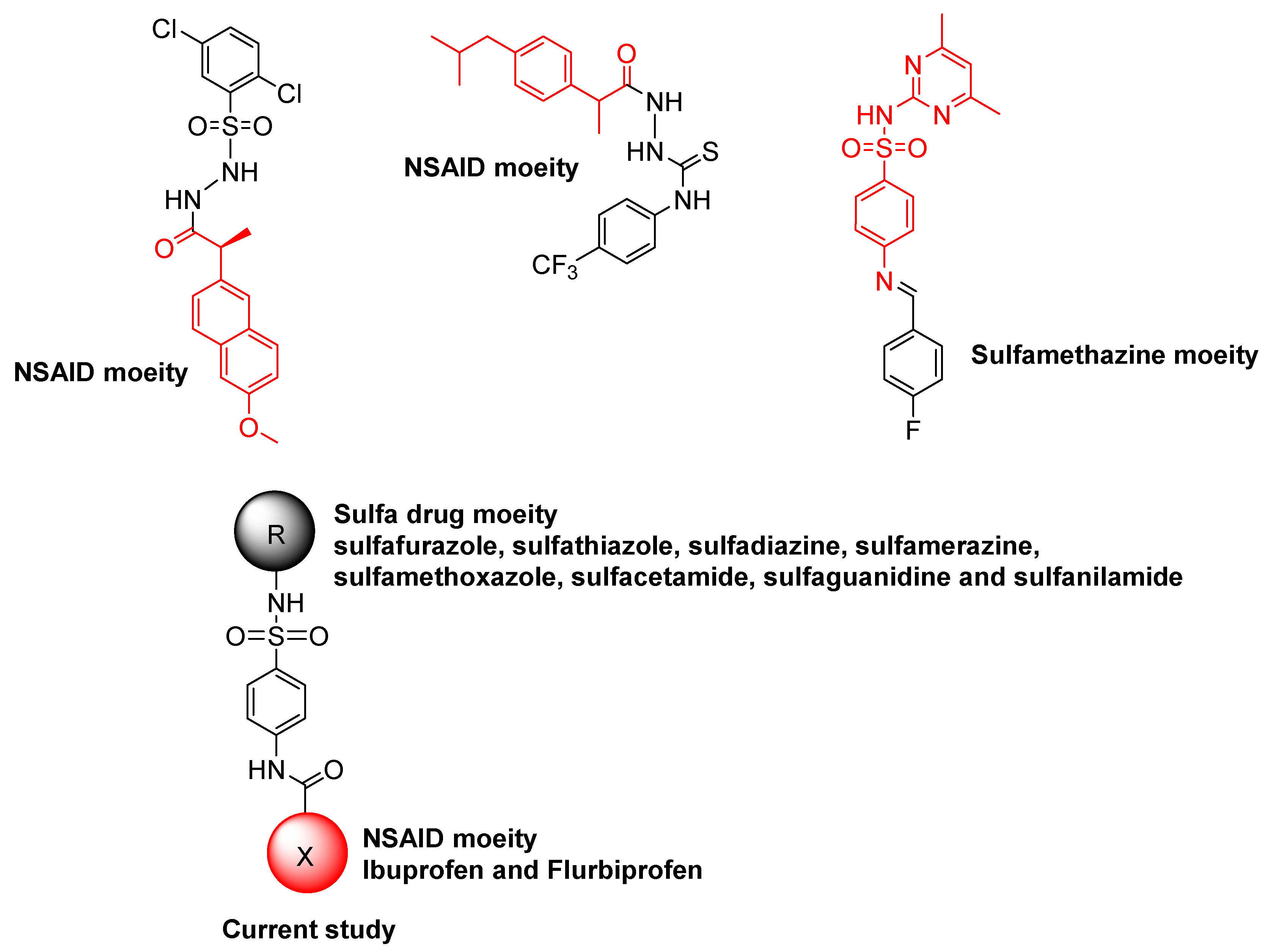
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Pharmacological Activity
2.2.1. Urease Inhibition and Structure–Activity Relationship (SAR)
2.2.2. Enzyme Kinetics Studies
2.3. Molecular Docking and Dynamics Simulation Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis Protocol for New Conjugates
3.1.3. Analytical Details of Ibuprofen–Sulfa Drug Conjugates
2-(4-Isobutylphenyl)-N-(4-sulfamoylphenyl)propanamide (4) [29]
N-(4-(N-(3,4-Dimethylisoxazol-5-yl)sulfamoyl)phenyl)-2-(4-isobutylphenyl)propanamide (5)
2-(4-Isobutylphenyl)-N-(4-(N-(thiazol-2-yl)sulfamoyl)phenyl)propanamide (6)
2-(4-Isobutylphenyl)-N-(4-(N-(pyrimidin-2-yl) sulfamoyl)phenyl)propanamide (7)
2-(4-Isobutylphenyl)-N-(4-(N-(4-methylpyrimidin-2yl)sulfamoyl)phenyl)propanamide (8)
2-(4-Isobutylphenyl)-N-(4-(N-(5-methylisoxazol-3-yl)sulfamoyl)phenyl)propanamide (9) [30]
N-(4-(N-Acetylsulfamoyl)phenyl)-2-(4-isobutylphenyl)propanamide (10)
N-(4-(N-Carbamimidoylsulfamoyl)phenyl)-2-(4-isobutylphenyl)propanamide (11)
3.1.4. Analytical Details of Flurbiprofen-Sulfa Drugs Conjugates
2-(2-Fluoro-[1,1′-biphenyl]-4-yl)-N-(4-sulfamoylphenyl)propanamide (12) [29]
N-(4-(N-(3,4-Dimethylisoxazol-5-yl)sulfamoyl)phenyl)-2-(2-fluoro-[1,1′-biphenyl]-4-yl)propanamide (13)
2-(2-Fluoro-[1,1′-biphenyl]-4-yl)-N-(4-(N-(thiazol-2-yl)sulfamoyl)phenyl)propanamide (14)
2-(2-Fluoro-[1,1′-biphenyl]-4-yl)-N-(4-(N-(pyrimidin-2-yl)sulfamoyl)phenyl)propanamide (15)
2-(2-Fluoro-[1,1′-biphenyl]-4-yl)-N-(4-(N-(4-methylpyrimidin-2-yl)sulfamoyl)phenyl)propanamide (16)
2-(2-Fluoro-[1,1′-biphenyl]-4-yl)-N-(4-(N-(5-methylisoxazol-3-yl)sulfamoyl)phenyl)propanamide (17)
N-(4-(N-Acetylsulfamoyl)phenyl)-2-(2-fluoro-[1,1′-biphenyl]-4-yl)propanamide (18)
N-(4-(N-Carbamimidoylsulfamoyl)phenyl)-2-(2-fluoro-[1,1′-biphenyl]-4-yl)propanamide (19)
3.2. Antiurease Assay
3.3. Molecular Docking and Dynamics Simulation Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tirmazi, S.A.A.S.; Qadir, M.A.; Ahmed, M.; Imran, M.; Hussain, R.; Sharif, M.; Yousaf, M.; Muddassar, M. Levofloxacin and sulfa drugs linked via schiff bases: Exploring their urease inhibition, enzyme kinetics and in silico studies. J. Mol. Struct. 2021, 1235, 130226. [Google Scholar] [CrossRef]
- Kappaun, K.; Piovesan, A.R.; Carlini, C.R.; Ligabue-Braun, R. Ureases: Historical aspects, catalytic, and non-catalytic properties–a review. J. Adv. Res. 2018, 13, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Imran, M.; Muddassar, M.; Hussain, R.; Khan, M.U.; Ahmad, S.; Mehboob, M.Y.; Ashfaq, S. Benzenesulfonohydrazides inhibiting urease: Design, synthesis, their in vitro and in silico studies. J. Mol. Struct. 2020, 1220, 128740. [Google Scholar] [CrossRef]
- Imran, M.; Waqar, S.; Ogata, K.; Ahmed, M.; Noreen, Z.; Javed, S.; Bibi, N.; Bokhari, H.; Amjad, A.; Muddassar, M. Identification of novel bacterial urease inhibitors through molecular shape and structure based virtual screening approaches. RSC Adv. 2020, 10, 16061–16070. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Qadir, M.A.; Hameed, A.; Arshad, M.N.; Asiri, A.M.; Muddassar, M. Azomethines, isoxazole, n-substituted pyrazoles and pyrimidine containing curcumin derivatives: Urease inhibition and molecular modeling studies. Biochem. Biophys. Res. Commun. 2017, 490, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Seraj, F.; Khan, K.M.; Khan, A.; Ali, M.; Khalil, R.; Ul-Haq, Z.; Hameed, S.; Taha, M.; Salar, U.; Perveen, S. Biology-oriented drug synthesis (biods), in vitro urease inhibitory activity, and in silico studies on ibuprofen derivatives. Mol. Divers. 2021, 25, 143–157. [Google Scholar] [CrossRef]
- Mohiuddin, G.; Khan, K.M.; Salar, U.; Lodhi, M.A.; Wadood, A.; Riaz, M.; Perveen, S. Biology-oriented drug synthesis (biods), in vitro urease inhibitory activity, and in silico study of s-naproxen derivatives. Bioorg. Chem. 2019, 83, 29–46. [Google Scholar] [CrossRef]
- Yakan, H.; Muğlu, H.; Türkeş, C.; Demir, Y.; Erdoğan, M.; Çavuş, M.S.; Beydemir, Ş. A novel series of thiosemicarbazone hybrid scaffolds: Design, synthesis, dft studies, metabolic enzyme inhibition properties, and molecular docking calculations. J. Mol. Struct. 2023, 1280, 135077. [Google Scholar] [CrossRef]
- Kumar, D.; Aggarwal, N.; Deep, A.; Kumar, H.; Chopra, H.; Marwaha, R.K.; Cavalu, S. An understanding of mechanism-based approaches for 1, 3, 4-oxadiazole scaffolds as cytotoxic agents and enzyme inhibitors. Pharmaceuticals 2023, 16, 254. [Google Scholar] [CrossRef]
- Yu, Z.; Huang, J.-P.; Yang, J.; Liu, C.; Yan, Y.; Wang, L.; Zhao, J.; Chen, Y.; Xiang, W.; Huang, S.-X. Discovery and biosynthesis of karnamicins as angiotensin converting enzyme inhibitors. Nat. Commun. 2023, 14, 209. [Google Scholar] [CrossRef]
- Peerzada, M.N.; Hamel, E.; Bai, R.; Supuran, C.T.; Azam, A. Deciphering the key heterocyclic scaffolds in targeting microtubules, kinases and carbonic anhydrases for cancer drug development. Pharmacol. Ther. 2021, 225, 107860. [Google Scholar] [CrossRef] [PubMed]
- Elbadawi, M.M.; Eldehna, W.M.; Nocentini, A.; Somaa, W.R.; Al-Rashood, S.T.; Elkaeed, E.B.; El Hassab, M.A.; Abdel-Aziz, H.A.; Supuran, C.T.; Fares, M. Development of 4-((3-oxo-3-phenylpropyl) amino) benzenesulfonamide derivatives utilizing tail/dual-tail approaches as novel carbonic anhydrase inhibitors. Eur. J. Med. Chem. 2022, 238, 114412. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Qadir, M.A.; Hameed, A.; Arshad, M.N.; Asiri, A.M.; Muddassar, M. Sulfonamides containing curcumin scaffold: Synthesis, characterization, carbonic anhydrase inhibition and molecular docking studies. Bioorg. Chem. 2018, 76, 218–227. [Google Scholar] [CrossRef]
- Abdul Qadir, M.; Ahmed, M.; Iqbal, M. Synthesis, characterization, and antibacterial activities of novel sulfonamides derived through condensation of amino group containing drugs, amino acids, and their analogs. BioMed Res. Int. 2015, 2015, 938486. [Google Scholar] [CrossRef] [PubMed]
- Abdul Qadir, M.; Ahmed, M.; Aslam, H.; Waseem, S.; Shafiq, M.I. Amidine sulfonamides and benzene sulfonamides: Synthesis and their biological evaluation. J. Chem. 2015, 2015, 524056. [Google Scholar] [CrossRef]
- Qadir, M.A.; Ahmed, M.; Khaleeq, A. Synthesis and biological evaluation of amino terminal modified new sulfonamides of contemporary drugs. Lat. Am. J. Pharm. 2015, 34, 719–724. [Google Scholar]
- Shahzad, S.; Qadir, M.A.; Ahmed, M.; Ahmad, S.; Khan, M.J.; Gulzar, A.; Muddassar, M. Folic acid-sulfonamide conjugates as antibacterial agents: Design, synthesis and molecular docking studies. RSC Adv. 2020, 10, 42983–42992. [Google Scholar] [CrossRef]
- Nadeem, R.A.; Abdul Qadir, M.; Ahmed, M.; Sajid, I. Cephalosporin conjugated sulfonamides: Synthesis, characterization and anticancer activities. Lett. Drug Des. Discov. 2020, 17, 264–270. [Google Scholar] [CrossRef]
- Lane, S.S.; Modi, S.S.; Lehmann, R.P.; Holland, E.J. Nepafenac ophthalmic suspension 0.1% for the prevention and treatment of ocular inflammation associated with cataract surgery. J. Cataract Refract. Surg. 2007, 33, 53–58. [Google Scholar] [CrossRef]
- Orzalesi, G.; Selleri, R.; Caldini, O.; Volpato, I.; Innocenti, F.; Colome, J.; Sacristan, A.; Varez, G. Ibuproxam and ibuprofen. A pharmacological comparison. Arzneimittel-Forschung 1977, 27, 1006–1012. [Google Scholar]
- Agrawal, R.; Rewatkar, P.V.; Kokil, G.R.; Verma, A.; Kalra, A. Oseltamivir: A first line defense against swine flu. Med. Chem. 2010, 6, 247–251. [Google Scholar] [CrossRef]
- Khan, S.; Iqbal, S.; Shah, M.; Rehman, W.; Hussain, R.; Rasheed, L.; Alrbyawi, H.; Dera, A.A.; Alahmdi, M.I.; Pashameah, R.A. Synthesis, in vitro anti-microbial analysis and molecular docking study of aliphatic hydrazide-based benzene sulphonamide derivatives as potent inhibitors of α-glucosidase and urease. Molecules 2022, 27, 7129. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Alam, A.; Khan, K.M.; Salar, U.; Chigurupati, S.; Wadood, A.; Ali, F.; Mohammad, J.I.; Riaz, M.; Perveen, S. Flurbiprofen derivatives as novel α-amylase inhibitors: Biology-oriented drug synthesis (biods), in vitro, and in silico evaluation. Bioorg. Chem. 2018, 81, 157–167. [Google Scholar] [CrossRef]
- Ullah, S.; Saeed, M.; Halimi, S.M.A.; Fakhri, M.I.; Khan, K.M.; Khan, I.; Perveen, S. Piroxicam sulfonates biology-oriented drug synthesis (biods), characterization and anti-nociceptive screening. Med. Chem. Res. 2016, 25, 1468–1475. [Google Scholar] [CrossRef]
- Ertl, P.; Altmann, E.; Racine, S. The most common linkers in bioactive molecules and their bioisosteric replacement network. Bioorg. Med. Chem. 2023, 81, 117194. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How molecular size impacts rmsd applications in molecular dynamics simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O.J.M.B. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Tahir, A.; Alharthy, R.D.; Naseem, S.; Mahmood, N.; Ahmed, M.; Shahzad, K.; Akhtar, M.N.; Hameed, A.; Sadiq, I.; Nawaz, H.; et al. Investigations of structural requirements for brd4 inhibitors through ligand- and structure-based 3d qsar approaches. Molecules 2018, 23, 1527. [Google Scholar] [CrossRef]
- Akgul, O.; Di Cesare Mannelli, L.; Vullo, D.; Angeli, A.; Ghelardini, C.; Bartolucci, G.; Alfawaz Altamimi, A.S.; Scozzafava, A.; Supuran, C.T.; Carta, F. Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (nsaids–cais) for the management of rheumatoid arthritis. J. Med. Chem. 2018, 61, 4961–4977. [Google Scholar] [CrossRef]
- Tatheer, A.; Murtaza, S.; Kausar, N.; Altaf, A.A.; Kausar, S.; Ahmed, S.; Muhammad, S.; Hussain, A. Synthesis, theoretical investigations and biological evaluation of ibuprofen drug hybrids. Med. Chem. Res. 2022, 31, 2032–2044. [Google Scholar] [CrossRef]
- Schrödinger, L.J.S.S. Protein Preparation Wizard; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the prediction of absolute solvation free energies using the next generation opls force field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on cpu and gpu architectures with namd. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E. Amber 2021: Reference Manual; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. RW Impey, and ML Klein. J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Grant, B.J.; Skjærven, L.; Yao, X.Q. The bio3d packages for structural bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conjugate | IC50 (µM); Mean ± SEM (% Inhibition) | a Vmax (app) (µM/min) | b Km (app) (mM) | c Ki (µM) | Mode of Inhibition |
|---|---|---|---|---|---|
| 4 | 14.26 ± 0.14 (84.2) | 3.64 | 1.66 | 8.20 | Mixed |
| 5 | 23.27 ± 0.41 (84.0) | - | - | - | - |
| 6 | 9.95 ± 0.14 (90.6) | 1.39 | 2.30 | 0.17 | Competitive |
| 7 | 50.18 ± 1.24 (71.6) | - | - | - | - |
| 8 | 12.43 ± 0.54 (87.7) | 3.0 | 0.384 | 3.01 | Mixed |
| 9 | 39.89 ± 1.17 (76.7) | - | - | - | - |
| 10 | 10.27 ± 0.11 (88.9) | 1.04 | 1.92 | 2.62 | Mixed |
| 11 | 44.46 ± 1.41 (78.7) | - | - | - | - |
| 12 | 24.92 ± 0.24 (82.1) | - | - | - | - |
| 13 | 35.30 ± 0.21 (84.6) | - | - | - | - |
| 14 | 63.42 ± 1.15 (60.4) | - | - | - | - |
| 15 | 16.74 ± 0.23 (84.1) | 1.89 | 1.80 | 7.95 | Competitive |
| 16 | 17.48 ± 0.76 (86.1) | 1.01 | 3.67 | 1.30 | Mixed |
| 17 | 13.39 ± 0.11 (86.1) | 1.56 | 5.61 | 10.40 | Competitive |
| 18 | 14.78 ± 0.16 (87.1) | 1.02 | 1.41 | 4.11 | Mixed |
| 19 | 33.83 ± 0.18 (83.8) | - | - | - | - |
| d Thiourea | 22.61 ± 0.23 (92.3) | 18.61 | 2.18 | 18.18 | Competitive |
| Conjugate | MW | HBD | HBA | QPlogPo/w | QPlogHERG | QPPCaco | QPlogBB | QPlogKhsa |
|---|---|---|---|---|---|---|---|---|
| 4 | 360.47 | 3 | 7 | 2.617 | −5.908 | 237.115 | −1.649 | 0.114 |
| 6 | 443.57 | 2 | 8 | 4.011 | −6.829 | 568.346 | −1.265 | 0.346 |
| 8 | 452.57 | 2 | 9 | 4.13 | −7.227 | 510.708 | −1.536 | 0.468 |
| 11 | 402.50 | 2 | 7 | 3.263 | −6.067 | 196.034 | −1.805 | 0.379 |
| 15 | 476.52 | 2 | 9 | 4.263 | −8.303 | 445.399 | −1.467 | 0.42 |
| 16 | 490.55 | 2 | 9 | 4.631 | −8.203 | 522.792 | −1.416 | 0.573 |
| 17 | 479.52 | 2 | 8 | 4.395 | −7.972 | 342.919 | −1.644 | 0.563 |
| 18 | 440.48 | 2 | 7 | 3.777 | −7.161 | 203.357 | −1.715 | 0.483 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.; Abdul Qadir, M.; Ahmed, M.; Imran, M.; Yousaf, N.; Wani, T.A.; Zargar, S.; Ali, I.; Muddassar, M. New Acetamide-Sulfonamide-Containing Scaffolds: Antiurease Activity Screening, Structure-Activity Relationship, Kinetics Mechanism, Molecular Docking, and MD Simulation Studies. Molecules 2023, 28, 5389. https://doi.org/10.3390/molecules28145389
Ahmad S, Abdul Qadir M, Ahmed M, Imran M, Yousaf N, Wani TA, Zargar S, Ali I, Muddassar M. New Acetamide-Sulfonamide-Containing Scaffolds: Antiurease Activity Screening, Structure-Activity Relationship, Kinetics Mechanism, Molecular Docking, and MD Simulation Studies. Molecules. 2023; 28(14):5389. https://doi.org/10.3390/molecules28145389
Chicago/Turabian StyleAhmad, Saghir, Muhammad Abdul Qadir, Mahmood Ahmed, Muhammad Imran, Numan Yousaf, Tanveer A. Wani, Seema Zargar, Ijaz Ali, and Muhammad Muddassar. 2023. "New Acetamide-Sulfonamide-Containing Scaffolds: Antiurease Activity Screening, Structure-Activity Relationship, Kinetics Mechanism, Molecular Docking, and MD Simulation Studies" Molecules 28, no. 14: 5389. https://doi.org/10.3390/molecules28145389
APA StyleAhmad, S., Abdul Qadir, M., Ahmed, M., Imran, M., Yousaf, N., Wani, T. A., Zargar, S., Ali, I., & Muddassar, M. (2023). New Acetamide-Sulfonamide-Containing Scaffolds: Antiurease Activity Screening, Structure-Activity Relationship, Kinetics Mechanism, Molecular Docking, and MD Simulation Studies. Molecules, 28(14), 5389. https://doi.org/10.3390/molecules28145389