


Spiro-Flavonoids in Nature: A Critical Review of Structural Diversity and Bioactivity





Abstract
1. Introduction
2. Occurrence in Nature

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Plant | Part | Compound | References |
|---|---|---|---|---|
| Asparagaceae | Bessera elegans Schult.f. | bulbs | 57 | [13] |
| Chionodoxa luciliae Boiss. | bulbs | 49–52 | [14] | |
| Drimiopsis barteri Baker | bulbs and leaves | 50, 56, 60, 61, 64 | [15] | |
| Drimiopsis burkei Baker | bulbs | 53, 62 | [15] | |
| Drimiopsis maculata Lindl. & Paxton | bulbs | 52, 56 | [16,17] | |
| Eucomis schijffii Reyneke | bulbs | 49 | [17,18] | |
| Furcraea bedinghausii K.Koch | roots | 38, 39 | [19] | |
| Ledebouria graminifolia (Baker) Jessop | bulbs | 58, 61 | [20] | |
| Ledebouria hyderabadensis M.V.Ramana, Prasanna & Venu | bulbs | 49 | [21] | |
| Ledebouria ovatifolia (Baker) Jessop | bulbs | 49, 50 | [22] | |
| Ledebouria socialis (Baker) Jessop | bulbs | 63, 64 | [22] | |
| Merwilla natalensis (Planch.) Speta | bulbs | 49 | [23] | |
| Muscari armeniacum H.J.Veitch | bulbs | 52, 54 | [24] | |
| Muscari botryoides (L.) Mill. | bulbs | 52, 54, 56 | [24] | |
| Muscari comosum (L.) Mill. | bulbs | 53 | [25] | |
| Muscari neglectum Guss. ex Ten. | bulbs | 53–56 | [26] | |
| Scilla scilloides (Lindl.) Druce | bulbs | 49, 51, 52, 59 | [27] | |
| Yucca gloriosa L. | roots | 38–48 | [11,28,29] | |
| Yucca schidigera Roezl ex Ortgies | bark | 1, 5–7, 29, 36–41, 44, 46–48 | [30,31,32,33] | |
| Cistaceae | Fumana procumbens (Dunal) Gren. & Godr. | whole plant | 23 | [34] |
| Cupressaceae | Glyptostrobus pensilis (D.Don) K.Koch | trunk bark | 1–4, 10, 11 | [35] |
| Fabaceae | Caesalpinia sappan L. | heartwood | 65 | [36] |
| Pentaphylacaceae | Anneslea fragrans Wall. | twigs | 2, 8, 9, 30 | [37,38] |
| Pinaceae | Abies chensiensis Tiegh. | aerial parts | 1–3 | [39] |
| Abies delavayi Franch. var. delavayi | aerial parts | 1 | [40] | |
| Abies georgei Orr | aerial parts | 1, 12 | [41] | |
| Abies sachalinensis (F.Schmidt) Mast. | bark | 1, 4, 12–17 | [42] | |
| Larix decidua Mill. | bark | 1–3 | [43] | |
| Larix gmelinii (Rupr.) Kuzen. | bark | 1, 14, 18, 31 | [6,44,45] | |
| Larix olgensis Henry var. koreana Nakai | bark | 14, 16, 19–21 | [46] | |
| Larix sibirica Ledeb. | bark | 1, 14, 31 | [44] | |
| Pinus massoniana Lamb. | bark | 35 | [47] | |
| Tsuga longibracteata W.C.Cheng | bark | 1, 2 | [48] | |
| Thymelaeaceae | Daphne aurantiaca Diels | stem bark | 22, 24 | [49] |
| Daphne feddei H.Lév. | stem bark | 22, 24, 26, 27 | [50] | |
| Daphne genkwa Siebold & Zucc. | roots | 23 | [51] | |
| Daphne kiusiana Miq. | stem | 22, 24 | [52] | |
| Daphne kiusiana var. atrocaulis (Rehder) F.Maek. | stem | 22 | [53] | |
| Daphne linearifolia Hart | aerial parts | 23–25, 28 | [54] | |
| Daphne mucronata Royle | shoots | 23 | [55] | |
| Daphne odora Thunb. | roots | 22, 24 | [56,57] | |
| Edgeworthia chrysantha Lindl. | stem and twigs | 22, 24, 32–34 | [58] | |
| Stellera chamaejasme L. | roots | 23 | [59] | |
| Thymelaea microphylla Coss. & Durieu | roots | 23 | [60] | |
| Wikstroemia indica (L.) C.A.Mey. | roots | 22, 23 | [61,62] | |
| Vitaceae | Vitis amurensis Rupr. | seeds | 16 | [63] |
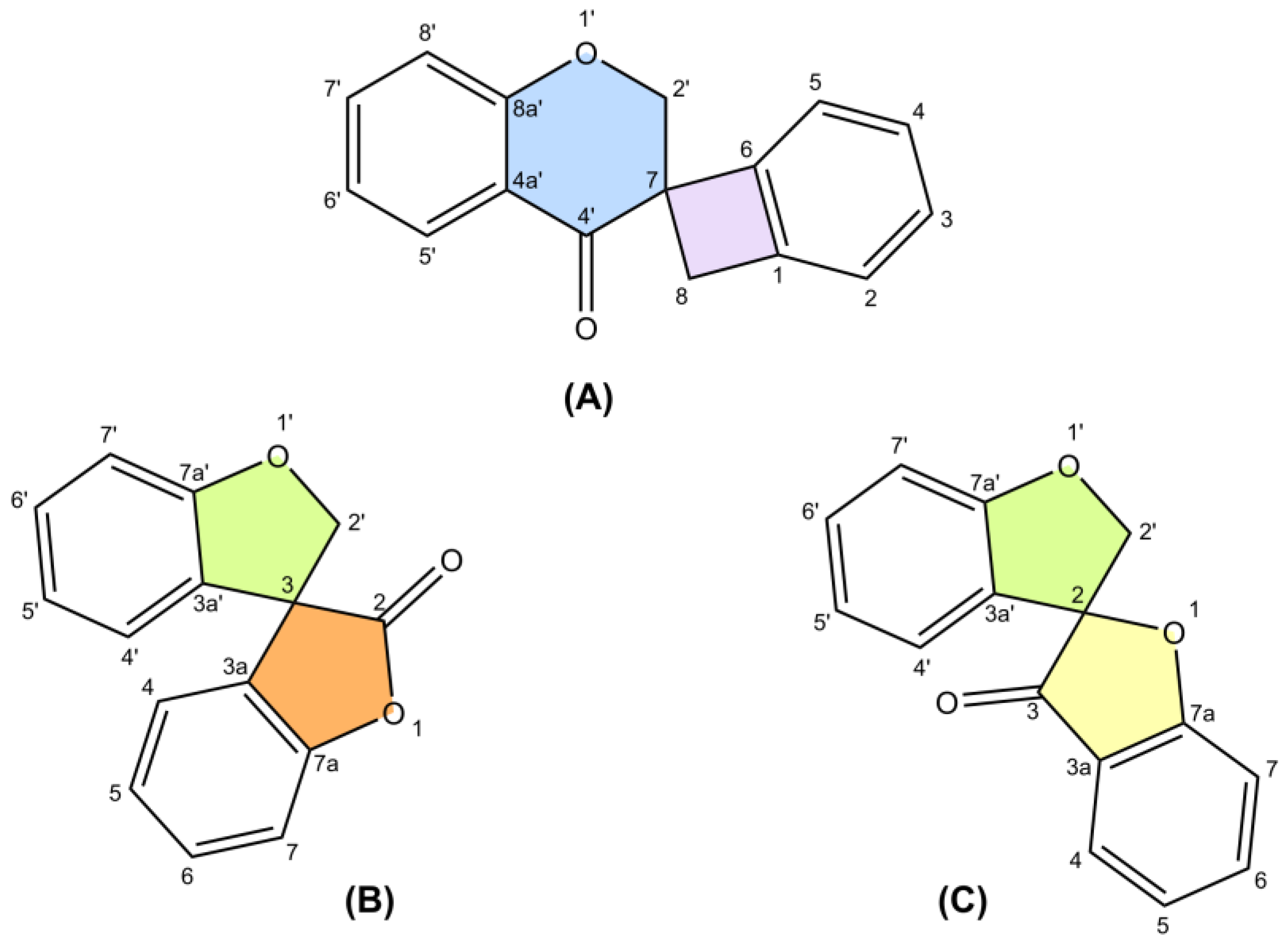
3. Methods of Extraction and Isolation
4. Stereochemistry of the Isolated Spiro-Flavonoids
4.1. Spiro-Biflavonoids
4.1.1. Larixinol Sub-Group (1–21) and Yuccaone A (29)
4.1.2. Daphnodorin C Sub-Group (22–28)
4.2. Spiro-Triflavonoids (30–31)
4.3. Spiro-Tetraflavonoids (32–35)
4.4. Spiro-Flavostilbenoids (36–48)
4.5. Scillascillin-Type Homoisoflavonoids (49–65)
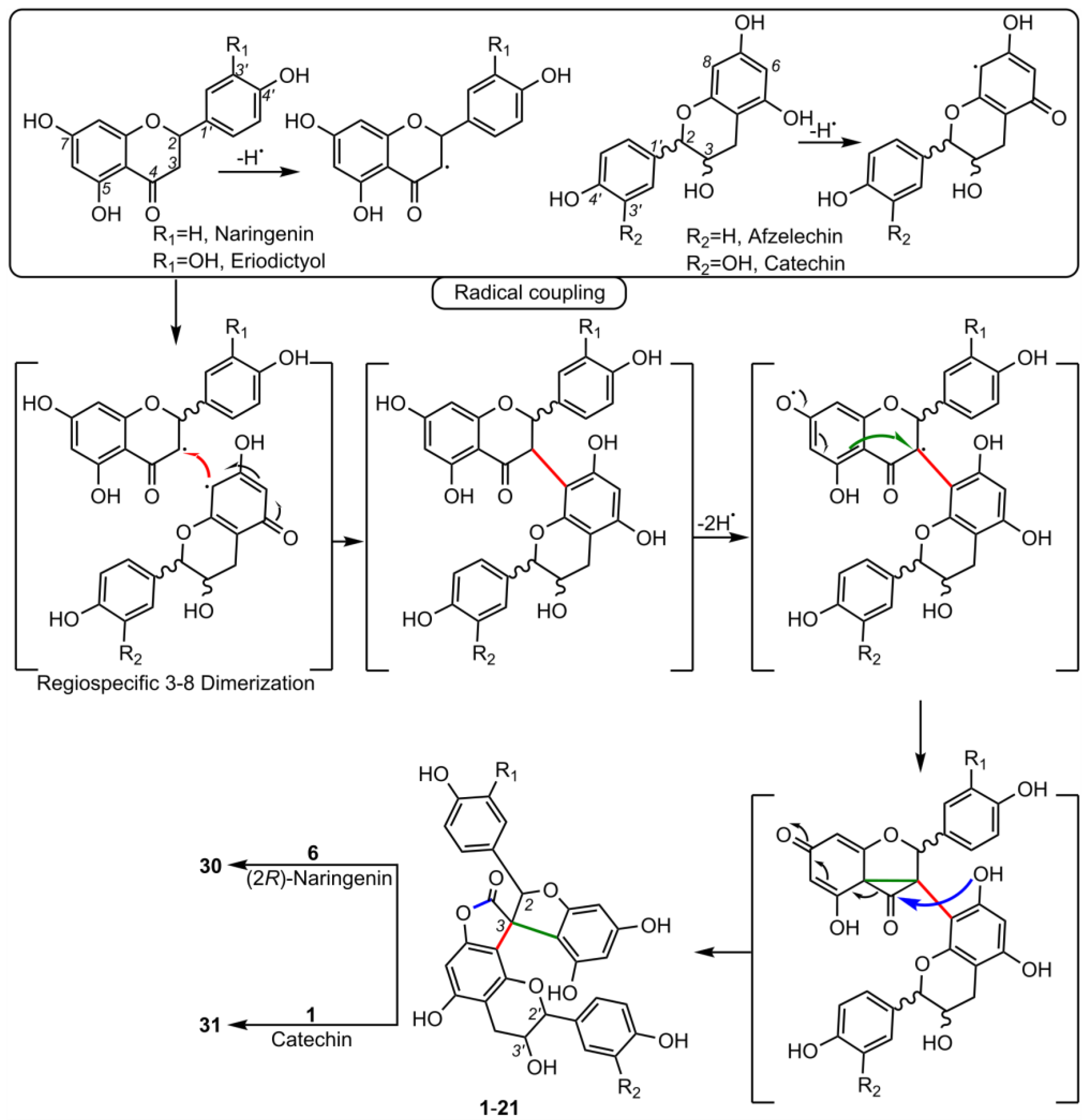
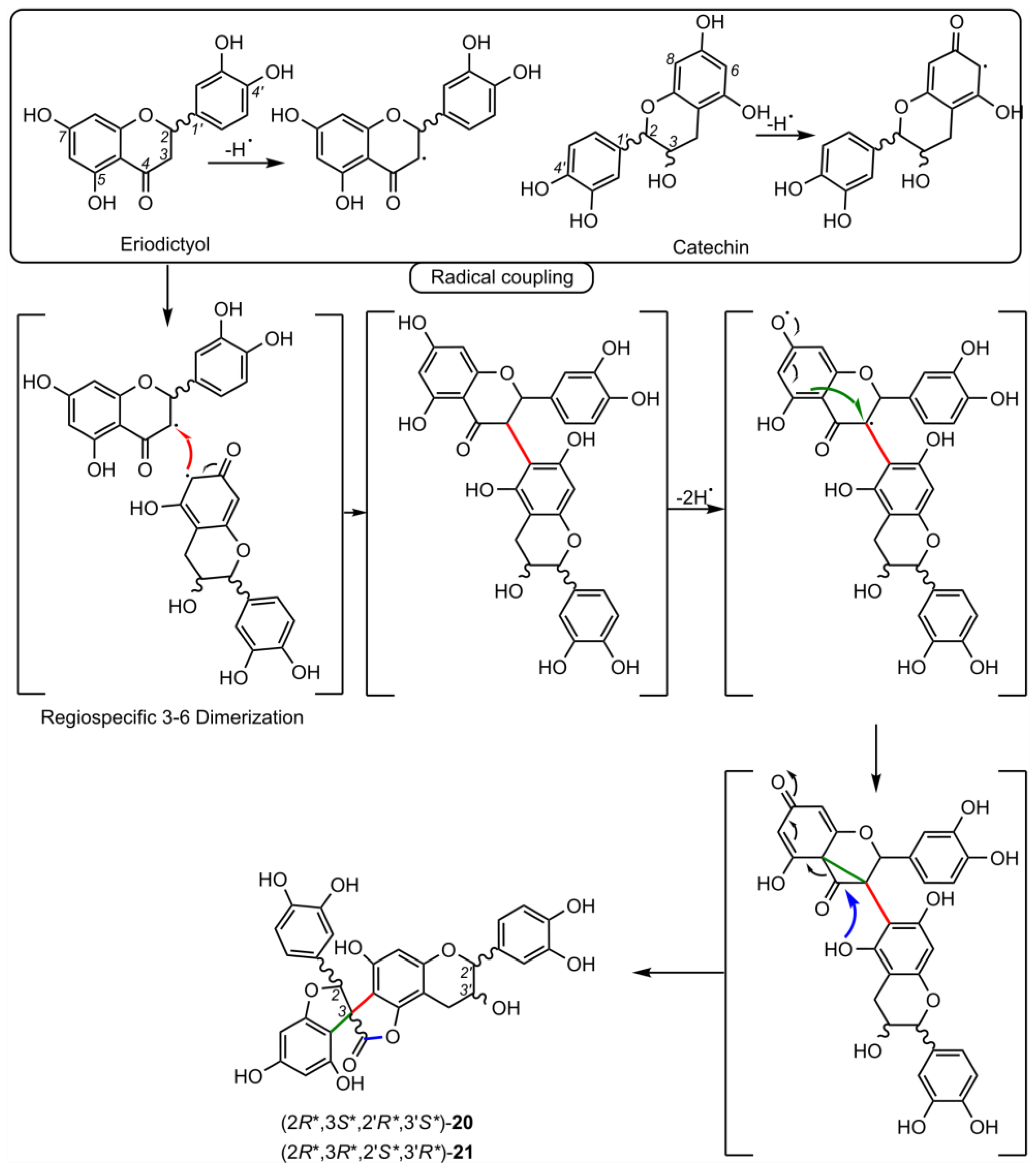
5. Biosynthesis of Spiro-Flavonoids
6. Biological Activities of Spiro-Flavonoids
6.1. Antioxidant Activity
6.2. Anti-Inflammatory Activity
6.3. Neuroprotective Activity
6.4. Anticancer and Antitumor Activity
6.5. Cytotoxicity/Mutagenicity
6.6. Antiplatelet Activity
6.7. Antidiabetic Activity
6.8. Antibacterial, Antifungal, and Antiviral Activity
6.9. Phytotoxic Activity
6.10. Other Activities
7. Conclusions and Further Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 12-HETE | 12-Hydroxy-5,8,10,14-eicosatetraenoic acid |
| AbsC | Absolute configuration |
| ABTS | 2,2′-Azino-bis(3-ethylbenzothiozoline-6-sulfonic acid) diammonium salt |
| AChE | Acetylcholinesterase |
| BChE | Butyrylcholinesterase |
| CC | Column chromatography |
| CDA | Chiral derivatizing agent |
| CHS | Chalcone synthase |
| COSY | Correlation spectroscopy |
| COX | Cyclooxygenase |
| DEPT | Distortion enhancement by polarization transfer |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl |
| EC50 | Half maximal effective concentration |
| ECD | Electronic circular dichroism |
| GIAO | Gauge-independent atomic orbital |
| GPC | Gel permeation chromatography |
| HHT | 12-Hydroxy-5,8,10-heptadecatrienoic acid |
| HIV-1 | Human immunodeficiency virus type 1 |
| HMBC | Heteronuclear multiple bond correlation |
| IC50 | Half maximal inhibitory concentration |
| iNOS | Inducible nitric oxide synthase |
| KS | Kaposi’s sarcoma |
| LLE | Liquid–liquid extraction |
| LOX | Lipoxygenase |
| LPS | Lipopolysaccharide |
| LTB4 | Leukotriene B4 |
| MAPK | Mitogen-activated protein kinases |
| MIC | Minimum inhibitory concentration |
| MPLC | Medium-pressure liquid chromatography |
| MTPA | α-methoxy-α-trifluoromethylphenylacetic acid |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NMR | Nuclear magnetic resonance |
| NO | Nitric oxide |
| NOESY | Nuclear Overhauser effect spectroscopy |
| NOR 1 | (±)-(E)-Methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-hex-3-enamide |
| NP | Normal phase |
| PAF | Platelet-activating factor |
| PARP-1 | Poly(ADP-ribose) polymerase 1 |
| PGE2 | Prostaglandin E2 |
| PLC | Preparative thin-layer chromatography |
| PTP1B | Human protein tyrosine phosphatase 1B |
| QM | Quantum mechanical |
| RelC | Relative configuration |
| ROS | Reactive oxygen species |
| RP | Reversed phase |
| STS | Stilbene synthase |
| TDDFT | Time-Dependent Density-Functional Theory |
| TEAC | Trolox Equivalent Antioxidant Capacity |
| THMS | trans-3,3′,5,5′-Tetrahydroxy-4′-methoxystilbene |
| TLC | Thin-layer chromatography |
| TNF-α | Tumor necrosis factor-α |
| UV/Vis | Ultraviolet/Visible |
| VCD | Vibrational circular dichroism |
| VEGF | Vascular endothelial growth factor |
| XRD | X-ray diffraction |
References
- Moss, G.P. Extension and Revision of the Nomenclature for Spiro Compounds. Pure Appl. Chem. 1999, 71, 531–558. [Google Scholar] [CrossRef]
- Ding, A.; Meazza, M.; Guo, H.; Yang, J.W.; Rios, R. New Development in the Enantioselective Synthesis of Spiro Compounds. Chem. Soc. Rev. 2018, 47, 5946–5996. [Google Scholar] [CrossRef]
- Rios, R. Enantioselective Methodologies for the Synthesis of Spiro Compounds. Chem. Soc. Rev. 2012, 41, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, K.; Dar’in, D.; Proschak, E.; Krasavin, M. Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. [Google Scholar] [CrossRef] [PubMed]
- Kouno, I.; Komori, T.; Kawasaki, T. Zur Struktur Der Neuen Typen Homo-Isoflavanone Aus Bulben von Scilla scilloides Druce. Tetrahedron Lett. 1973, 14, 4569–4572. [Google Scholar] [CrossRef]
- Fedorova, T.E.; Ivanova, S.Z.; Fedorov, S.V.; Babkin, V.A. Larisinol, a New Spirobiflavonoid from Larix gmelinii Bark. Chem. Nat. Compd. 2007, 43, 208–209. [Google Scholar] [CrossRef]
- Pecio, Ł.; Alilou, M.; Kozachok, S.; Orhan, I.E.; Eren, G.; Şenol Deniz, F.S.; Stuppner, H.; Oleszek, W. Absolute Configuration of Spiro-Flavostilbenoids from Yucca schidigera Roezl Ex Ortgies: First Indication of (2R)-Naringenin as the Key Building Block. Phytochemistry 2023, 207, 113584. [Google Scholar] [CrossRef]
- Fedorova, T.E.; Ivanova, S.Z.; Babkin, V.A. Spiroflavonoid Compounds: Structure and Distribution in Nature Review. Russ. J. Bioorg. Chem. 2010, 36, 793–801. [Google Scholar] [CrossRef]
- POWO. Plants of the World Online. Facilitated by the Royal Botanic Gaedens, Kew. 2022. Available online: http://www.plantsoftheworldonline.org/ (accessed on 29 August 2022).
- IPNI. International Plant Names Index. 2022. Available online: https://www.ipni.org (accessed on 29 August 2022).
- Nakashima, K.; Abe, N.; Oyama, M.; Inoue, M. Yuccalides A–C, Three New Phenolic Compounds with Spiro-Structures from the Roots of Yucca gloriosa. Fitoterapia 2016, 111, 154–159. [Google Scholar] [CrossRef]
- He, X.; Yang, F.; Huang, X. Proceedings of Chemistry, Pharmacology, Pharmacokinetics and Synthesis of Biflavonoids. Molecules 2021, 26, 6088. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kurihara, R.; Akagi, N.; Mimaki, Y. Two New Homoisoflavonoids from the Bulbs of Bessera elegans. Nat. Prod. Commun. 2014, 9, 1725–1727. [Google Scholar] [CrossRef]
- Corsaro, M.M.; Lanzetta, R.; Mancino, A.; Parrilli, M. Homoisoflavanones from Chionodoxa luciliae. Phytochemistry 1992, 31, 1395–1397. [Google Scholar] [CrossRef]
- Ngamga, D.; Bipa, J.; Lebatha, P.; Hiza, C.; Mutanyatta, J.; Bezabih, M.-T.; Tane, P.; Abegaz, B.M. Isoquinoline Alkaloids and Homoisoflavonoids from Drimiopsis barteri Bak and D. burkei Bak. Nat. Prod. Commun. 2008, 3, 769–777. [Google Scholar] [CrossRef]
- Koorbanally, C.; Crouch, N.R.; Mulholland, D.A. Scillascillin-Type Homoisoflavanones from Drimiopsis maculata (Hyacinthaceae). Biochem. Syst. Ecol. 2001, 29, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Fusi, F.; Ferrara, A.; Koorbanally, C.; Crouch, N.R.; Mulholland, D.A.; Sgaragli, G. Vascular Myorelaxing Activity of Isolates from South African Hyacinthaceae Partly Mediated by Activation of Soluble Guanylyl Cyclase in Rat Aortic Ring Preparations. J. Pharm. Pharmacol. 2010, 60, 489–497. [Google Scholar] [CrossRef]
- Koorbanally, C.; Crouch, N.R.; Langlois, A.; Du Toit, K.; Mulholland, D.A.; Drewes, S.E. Homoisoflavanones and Spirocyclic Nortriterpenoids from Three Eucomis Species: E. comosa, E. schijffii and E. pallidiflora Subsp. Pole-Evansii (Hyacinthaceae). S. Afr. J. Bot. 2006, 72, 428–433. [Google Scholar] [CrossRef]
- Teponno, R.B.; Ponou, B.K.; Fiorini, D.; Barboni, L.; Tapondjou, L.A. Chemical Constituents from the Roots of Furcraea bedinghausii Koch. Int. Lett. Chem. Phys. Astron. 2013, 16, 9–19. [Google Scholar] [CrossRef]
- Mutanyatta, J.; Matapa, B.G.; Shushu, D.D.; Abegaz, B.M. Homoisoflavonoids and Xanthones from the Tubers of Wild and in Vitro Regenerated Ledebouria graminifolia and Cytotoxic Activities of Some of the Homoisoflavonoids. Phytochemistry 2003, 62, 797–804. [Google Scholar] [CrossRef]
- Chinthala, Y.; Chinde, S.; Kumar, A.N.; Srinivas, K.V.N.S.; Kumar, J.K.; Sastry, K.P.; Grover, P.; Ramana, M.V. Anticancer Active Homoisoflavone from the Underground Bulbs of Ledebouria hyderabadensis. Pharmacogn. Res. 2014, 6, 303–305. [Google Scholar] [CrossRef]
- Waller, C.P.; Thumser, A.E.; Langat, M.K.; Crouch, N.R.; Mulholland, D.A. COX-2 Inhibitory Activity of Homoisoflavanones and Xanthones from the Bulbs of the Southern African Ledebouria socialis and Ledebouria ovatifolia (Hyacinthaceae: Hyacinthoideae). Phytochemistry 2013, 95, 284–290. [Google Scholar] [CrossRef]
- Moodley, N.; Mulholland, D.A.; Crouch, N.R. Eucosterol-Type Nortriterpenoids from Merwilla natalensis. J. Nat. Prod. 2004, 67, 918–920. [Google Scholar] [CrossRef]
- Adinolfi, M.; Corsaro, M.M.; Lanzetta, R.; Laonigro, G.; Mangoni, L.; Parrilli, M. Ten Homoisoflavanones from Two Muscari Species. Phytochemistry 1986, 26, 285–290. [Google Scholar] [CrossRef]
- Adinolfi, M.; Barone, G.; Belardini, M.; Lanzetta, R.; Laonigro, G.; Parrilli, M. Homoisoflavanones from Muscari comosum Bulbs. Phytochemistry 1985, 24, 2423–2426. [Google Scholar] [CrossRef]
- Barone, G.; Corsaro, M.M.; Lanzetta, R.; Parrilli, M. Homoisoflavanones from Muscari neglectum. Phytochemistry 1988, 27, 921–923. [Google Scholar] [CrossRef]
- Nishida, Y.; Eto, M.; Miyashita, H.; Ikeda, T.; Yamaguchi, K.; Yoshimitsu, H.; Nohara, T.; Ono, M. A New Homostilbene and Two New Homoisoflavones from the Bulbs of Scilla scilloides. Chem. Pharm. Bull. 2008, 56, 1022–1025. [Google Scholar] [CrossRef]
- Bassarello, C.; Bifulco, G.; Montoro, P.; Skhirtladze, A.; Benidze, M.; Kemertelidze, E.; Pizza, C.; Piacente, S. Yucca gloriosa: A Source of Phenolic Derivatives with Strong Antioxidant Activity. J. Agric. Food Chem. 2007, 55, 6636–6642. [Google Scholar] [CrossRef] [PubMed]
- Bassarello, C.; Bifulco, G.; Montoro, P.; Skhirtladze, A.; Kemertelidze, E.; Piacente, S. Gloriosaols A and B, Two Novel Phenolics from Yucca gloriosa: Structural Characterization and Configurational Assignment by a Combined NMR-Quantum Mechanical Strategy. Tetrahedron 2007, 63, 148–154. [Google Scholar] [CrossRef]
- Oleszek, W.; Sitek, M.; Stochmal, A.; Piacente, S.; Pizza, C.; Cheeke, P. Resveratrol and Other Phenolics from the Bark of Yucca schidigera Roezl. J. Agric. Food Chem. 2001, 49, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Pecio, Ł.; Alilou, M.; Kozachok, S.; Orhan, I.E.; Eren, G.; Deniz, F.S.S.; Stuppner, H.; Oleszek, W. Yuccalechins A-C from the Yucca schidigera Roezl Ex Ortgies Bark: Elucidation of the Relative and Absolute Configurations of Three New Spirobiflavonoids and Their Cholinesterase Inhibitory Activities. Molecules 2019, 24, 4162. [Google Scholar] [CrossRef]
- Piacente, S.; Bifulco, G.; Pizza, C.; Stochmal, A.; Oleszek, W. A Novel Phenolic Spiro Derivative, Yuccaone A, from Yucca schidigera Bark. Tetrahedron Lett. 2002, 43, 9133–9136. [Google Scholar] [CrossRef]
- Piacente, S.; Montoro, P.; Oleszek, W.; Pizza, C. Yucca schidigera Bark: Phenolic Constituents and Antioxidant Activity. J. Nat. Prod. 2004, 67, 882–885. [Google Scholar] [CrossRef] [PubMed]
- Emerce, E.; Gürbüz, P.; Doğan, Ş.D.; Kadioglu, E.; Süntar, I. Cytotoxic Activity-Guided Isolation Studies on Fumana procumbens (Dunal) Gren. & Godr. Rec. Nat. Prod. 2019, 13, 189–198. [Google Scholar] [CrossRef]
- Xiong, J.; Hu, C.L.; Wang, P.P.; Gao, D.D.; Huang, F.; Li, J.; Hu, J.F. Spirobiflavonoid Stereoisomers from the Endangered Conifer Glyptostrobus pensilis and Their Protein Tyrosine Phosphatase 1B Inhibitory Activity. Bioorg. Med. Chem. Lett. 2020, 30, 126943. [Google Scholar] [CrossRef] [PubMed]
- Washiyama, M.; Sasaki, Y.; Hosokawa, T.; Nagumo, S. Anti-Inflammatory Constituents of Sappan Lignum. Biol. Pharm. Bull. 2009, 32, 941–944. [Google Scholar] [CrossRef]
- Omar, A.M.; Sun, S.; Kim, M.J.; Tawila, A.M.; Dibwe, D.F.; Phrutivorapongkul, A.; Toyooka, N.; Awale, S. Fragranol A: A New Class of Spiro-Triflavanoid Hybrid with an Unprecedented Carbon Skeleton from Anneslea fragrans. Tetrahedron Lett. 2020, 61, 152099. [Google Scholar] [CrossRef]
- Omar, A.M.; Sun, S.; Kim, M.J.; Tawila, A.M.; Dibwe, D.F.; Phrutivorapongkul, A.; Toyooka, N.; Awale, S. Highly Oxygenated Spiro-Biflavanoids from Anneslea fragrans Twigs. Phytochem. Lett. 2020, 40, 21–25. [Google Scholar] [CrossRef]
- Li, Y.-L.; Yang, X.-W.; Li, S.-M.; Tang, J.; Tian, J.-M.; Peng, X.-Y.; Huang, D.-S.; Zhang, W.-D. Two New Spirobiflavonoids from Abies Chensiensis with Moderate NO Production Inhibitory Activity. Planta Med. 2009, 75, 1534–1537. [Google Scholar] [CrossRef]
- Yang, X.-W.; Li, S.M.; Li, Y.L.; Feng, L.; Shen, Y.H.; Lin, S.; Tian, J.M.; Zeng, H.W.; Wang, N.; Steinmetz, A.; et al. Chemical Constituents of Abies delavayi. Phytochemistry 2014, 105, 164–170. [Google Scholar] [CrossRef]
- Yang, X.-W.; Li, Y.L.; Li, S.M.; Shen, Y.H.; Tian, J.M.; Zhu, Z.J.; Feng, L.; Wu, L.; Lin, S.; Wang, N.; et al. Mono- and Sesquiterpenoids, Flavonoids, Lignans, and Other Miscellaneous Compounds of Abies georgei. Planta Med. 2011, 77, 742–748. [Google Scholar] [CrossRef]
- Wada, S.; Hitomi, T.; Tanaka, R. Phenolic Compounds Isolated from the Bark of Abies sachalinensis. Helv. Chim. Acta 2009, 92, 1610–1620. [Google Scholar] [CrossRef]
- Baldan, V.; Sut, S.; Faggian, M.; Dalla Gassa, E.; Ferrari, S.; De Nadai, G.; Francescato, S.; Baratto, G.; Dall’Acqua, S. Larix decidua Bark as a Source of Phytoconstituents: An LC-MS Study. Molecules 2017, 22, 1974. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, S.Z.; Fedorova, T.E.; Ivanova, N.V.; Fedorov, S.V.; Babkin, V.A. Triflarixinol—A New Spiroflavonoid from the Bark of the Larch Tree. Khimiya Rastit. Syr’ya 2006, 1, 37–40. [Google Scholar]
- Shen, Z.; Haslam, E.; Falshaw, C.P.; Begley, M.J. Procyanidins and Polyphenols of Larix gmelini Bark. Phytochemistry 1986, 25, 2629–2635. [Google Scholar] [CrossRef]
- Yang, B.-H.; Zhang, W.-D.; Liu, R.-H.; Tan, C.-H.; Li, T.-Z.; Zhang, C.; Xu, X.-K.; Su, J. Spiro-Biflavonoids from Larix olgensis Henry Var. Koreana Nakai. Helv. Chim. Acta 2005, 88, 2892–2896. [Google Scholar] [CrossRef]
- Zhou, B.; Alania, Y.; Reis, M.C.; McAlpine, J.B.; Bedran-Russo, A.K.; Pauli, G.F.; Chen, S.-N. Rare A-Type, Spiro-Type, and Highly Oligomeric Proanthocyanidins from Pinus massoniana. Org. Lett. 2020, 22, 5304–5308. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Nie, Q.; Weng, Q.; Zhang, M.; Ding, L.; Zhou, Y.; Chen, F.; Xiao, S. Chemical Constituents from Tsuga longibracteata and Its Chemotaxonomic Study. Biochem. Syst. Ecol. 2018, 77, 4–6. [Google Scholar] [CrossRef]
- Liang, S.; Tian, J.-M.; Feng, Y.; Liu, X.-H.; Xiong, Z.; Zhang, W.-D. Flavonoids from Daphne aurantiaca and Their Inhibitory Activities against Nitric Oxide Production. Chem. Pharm. Bull. 2011, 59, 653–656. [Google Scholar] [CrossRef]
- Liang, S.; Tang, J.; Shen, Y.-H.; Jin, H.-Z.; Tian, J.-M.; Wu, Z.-J.; Zhang, W.-D.; Yan, S.-K. Biflavonoids from Daphne feddei and Their Inhibitory Activities against Nitric Oxide Production. Chem. Pharm. Bull. 2008, 56, 1729–1731. [Google Scholar] [CrossRef]
- Baba, K.; Takeuchi, K.; Tabata, Y.; Taniguchi, M.; Kozawa, M. Chemical Studies on the Constituents of the Thymelaeaceous Plants. IV. Structure of a New Spiro Biflavonoid, Genkwanol A, from the Root of Daphne genkwa Sieb. et Zucc. Yakugaku Zasshi 1987, 107, 525–529. [Google Scholar] [CrossRef]
- Ryu, H.W.; Lee, J.W.; Kim, M.O.; Lee, R.W.; Kang, M.J.; Kim, S.M.; Min, J.H.; Oh, E.S.; Song, Y.N.; Jung, S.; et al. Daphnodorin C Isolated from the Stems of Daphne kiusiana Miquel Attenuates Airway Inflammation in a Mouse Model of Chronic Obstructive Pulmonary Disease. Phytomedicine 2022, 96, 153848. [Google Scholar] [CrossRef]
- Bai, Z.; Zhou, D.; Meng, Q.; Fang, M.; Chen, G.; Hou, Y.; Li, N. Characteristic Biflavonoids from Daphne Kiusiana Var. Atrocaulis (Rehd.) F. Maekawa. Nat. Prod. Res. 2022, 37, 1557–1564. [Google Scholar] [CrossRef]
- Malafronte, N.; Vassallo, A.; Dal Piaz, F.; Bader, A.; Braca, A.; De Tommasi, N. Biflavonoids from Daphne linearifolia Hart. Phytochem. Lett. 2012, 5, 621–625. [Google Scholar] [CrossRef]
- Ghanadian, M.; Ali, Z.; Khan, I.A.; Balachandran, P.; Nikahd, M.; Aghaei, M.; Mirzaei, M.; Sajjadi, S.E. A New Sesquiterpenoid from the Shoots of Iranian Daphne mucronata Royle with Selective Inhibition of STAT3 and Smad3/4 Cancer-Related Signaling Pathways. DARU J. Pharm. Sci. 2020, 28, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Baba, K.; Takeuchi, K.; Hamasaki, F.; Kozawa, M. Chemical Studies on the Constituents of the Thymelaeaceous Plants. I. Structures of Two New Flavans from Daphne odora Thunb. Chem. Pharm. Bull. 1986, 34, 595–602. [Google Scholar] [CrossRef]
- Taniguchi, M.; Baba, K. Three Biflavonoids from Daphne odora. Phytochemistry 1996, 42, 1447–1453. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, S.W.; Liu, S.S.; Cong, H.J.; Xuan, L.J. Daphnodorin Dimers from Edgeworthia chrysantha with α-Glucosidase Inhibitory Activity. Phytochem. Lett. 2010, 3, 242–247. [Google Scholar] [CrossRef]
- Yan, Z.; Guo, H.; Yang, J.; Liu, Q.; Jin, H.; Xu, R.; Cui, H.; Qin, B. Phytotoxic Flavonoids from Roots of Stellera chamaejasme L. (Thymelaeaceae). Phytochemistry 2014, 106, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, H.; Haba, H.; Marcourt, L.; Benkhaled, M.; Wolfender, J.L. Microphynolides A and B, New Spiro-γ-Lactone Glycosides from Thymelaea microphylla. Nat. Prod. Res. 2014, 28, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Kobayashi, H.; Dong, A.; Iwasaki, S.; Yao, X. Antifungal, Antimitotic and Anti-HIV-1 Agents from the Roots of Wikstroemia indica. Planta Med. 2000, 66, 564–567. [Google Scholar] [CrossRef]
- Shao, M.; Huang, X.J.; Liu, J.S.; Han, W.L.; Cai, H.B.; Tang, Q.F.; Fan, Q. A New Cytotoxic Biflavonoid from the Rhizome of Wikstroemia indica. Nat. Prod. Res. 2016, 30, 1417–1422. [Google Scholar] [CrossRef]
- Wang, J.-N.; Hano, Y.; Nomura, T.; Chen, Y.-J. Procyanidins from the Seeds of Vitis amurensis. Phytochemistry 2000, 53, 1097–1102. [Google Scholar] [CrossRef]
- Johnson, J.L.; Raghavan, V.; Cimmino, A.; Moeini, A.; Petrovic, A.G.; Santoro, E.; Superchi, S.; Berova, N.; Evidente, A.; Polavarapu, P.L. Absolute Configurations of Chiral Molecules with Multiple Stereogenic Centers without Prior Knowledge of the Relative Configurations: A Case Study of Inuloxin C. Chirality 2018, 30, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, X.-C.; Ferreira, D. Theoretical Calculation of Electronic Circular Dichroism of a Hexahydroxydiphenoyl-Containing Flavanone Glycoside. J. Nat. Prod. 2009, 72, 327–335. [Google Scholar] [CrossRef]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of Relative Configuration in Organic Compounds by NMR Spectroscopy and Computational Methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V. Probing Stereoelectronic Effects with Spectroscopic Methods. In Stereoelectronic Effects: A Bridge between Structure and Reactivity; John Wiley & Sons, Ltd.: Chichester, UK; Hoboken, NJ, USA, 2016; pp. 1–7. ISBN 9781118906378. [Google Scholar]
- Hameed, R.; Van Mourik, T.; Khan, A. 13C-1H Coupling Constants as a Conformational Tool for Structural Assignment of Quinic and Octulosonic Acid. J. Mol. Model. 2018, 24, 324. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds Using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Kong, L.Y.; Wang, P. Determination of the Absolute Configuration of Natural Products. Chin. J. Nat. Med. 2013, 11, 193–198. [Google Scholar] [CrossRef]
- Sullivan, G.R.; Dale, J.A.; Mosher, H.S. Correlation of Configuration and 19F Chemical Shifts of α-Methoxy-α-trifluoromethylphenylacetate Derivatives. J. Org. Chem. 1973, 38, 2143–2147. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear Magnetic Resonance Enantiomer Regents. Configurational Correlations via Nuclear Magnetic Resonance Chemical Shifts of Diastereomeric Mandelate, O-Methylmandelate, and α-Methoxy-α-trifluoromethylphenylacetate (MTPA) Esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Petrovic, A.G.; Navarro-Vázquez, A.; Alonso-Gómez, J.L. From Relative to Absolute Configuration of Complex Natural Products: Interplay between NMR, ECD, VCD, and ORD Assisted by Ab Initio Calculations. Curr. Org. Chem. 2010, 14, 1612–1628. [Google Scholar] [CrossRef]
- Valentín-Pérez, Á.; Rosa, P.; Hillard, E.A.; Giorgi, M. Chirality Determination in Crystals. Chirality 2022, 34, 163–181. [Google Scholar] [CrossRef]
- Shen, Z.; Falshaw, C.P.; Haslam, E.; Begleyc, M.J. A Novel Sprio-Biflavonoid from Larix gmelini. J. Chem. Soc. Chem. Commun. 1985, 1135–1137. [Google Scholar] [CrossRef]
- Baba, K.; Takeuchi, K.; Doi, M.; Kozawa, M. Chemical Studies on the Constituents of the Thymelaeaceous Plants. III. Structure of a Novel Spiro Biflavonoid, Daphnodorin C, from Daphne odora THUNB. Chem. Pharm. Bull. 1987, 35, 1853–1859. [Google Scholar] [CrossRef]
- Baba, K.; Takeuchi, K.; Doi, M.; Inoue, M.; Kozawa, M. Chemical Studies on the Constituents of the Thymelaeaceous Plants. II. Stereochemistry of Daphnodorin A and Daphnodorin B. Chem. Pharm. Bull. 1986, 34, 1540–1545. [Google Scholar] [CrossRef]
- Adinolfi, M.; Barone, G.; Giordano, F.; Lanzetta Michelangelo Parrilli, R. Absolute Configuration of Benzocyclobutene Homoisoflavanones from Muscari Species. Tetrahedron 1990, 46, 6565–6574. [Google Scholar] [CrossRef]
- Grotewold, E. The Genetics and Biochemistry of Floral Pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef]
- Tropf, S.; Lanz, T.; Rensing, S.A.; Schröder, J.; Schröder, G. Evidence That Stilbene Synthases Have Developed from Chalcone Synthases Several Times in the Course of Evolution. J. Mol. Evol. 1994, 38, 610–618. [Google Scholar] [CrossRef]
- Forkmann, G.; Heller, W. Confirm. In Comprehensive Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 1999; pp. 713–748. [Google Scholar]
- Bednar, R.A.; Hadcock, J.R. Purification and Characterization of Chalcone Isomerase from Soybeans. J. Biol. Chem. 1988, 263, 9582–9588. [Google Scholar] [CrossRef]
- Shirley, B.W.; Kubasek, W.L.; Storz, G.; Bruggemann, E.; Koornneef, M.; Ausubel, F.M.; Goodman, H.M. Analysis of Arabidopsis Mutants Deficient in Flavonoid Biosynthesis. Plant J. 1995, 8, 659–671. [Google Scholar] [CrossRef]
- Cahn, R.S.; Ingold, C.K.; Prelog, V. The Specification of Asymmetric Configuration in Organic Chemistry. Experientia 1956, 12, 81–94. [Google Scholar] [CrossRef]
- Zeb, N.; Rashid, M.H.; Mubarak, M.Q.E.; Ghafoor, S.; de Visser, S.P. Flavonol Biosynthesis by Nonheme Iron Dioxygenases: A Computational Study into the Structure and Mechanism. J. Inorg. Biochem. 2019, 198, 110728. [Google Scholar] [CrossRef] [PubMed]
- Lukačin, R.; Wellmann, F.; Britsch, L.; Martens, S.; Matern, U. Flavonol Synthase from Citrus unshiu Is a Bifunctional Dioxygenase. Phytochemistry 2003, 62, 287–292. [Google Scholar] [CrossRef]
- Britsch, L.; Grisebach, H. Purification and Characterization of (2S)-Flavanone 3-hydroxylase from Petunia hybrida. Eur. J. Biochem. 1986, 156, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, J.-L.; Austin, M.B.; Stewart, C.; Noel, J.P. Structure and Function of Enzymes Involved in the Biosynthesis of Phenylpropanoids. Plant Physiol. Biochem. 2008, 46, 356–370. [Google Scholar] [CrossRef]
- Hanhineva, K.; Kokko, H.; Siljanen, H.; Rogachev, I.; Aharoni, A.; Kärenlampi, S.O. Stilbene Synthase Gene Transfer Caused Alterations in the Phenylpropanoid Metabolism of Transgenic Strawberry (Fragaria×ananassa). J. Exp. Bot. 2009, 60, 2093–2106. [Google Scholar] [CrossRef]
- Jeandet, P.; Vannozzi, A.; Sobarzo-Sánchez, E.; Uddin, M.S.; Bru, R.; Martínez-Márquez, A.; Clément, C.; Cordelier, S.; Manayi, A.; Nabavi, S.F.; et al. Phytostilbenes as Agrochemicals: Biosynthesis, Bioactivity, Metabolic Engineering and Biotechnology. Nat. Prod. Rep. 2021, 38, 1282–1329. [Google Scholar] [CrossRef]
- Liu, Z.; Zhuang, C.; Sheng, S.; Shao, L.; Zhao, W.; Zhao, S. Overexpression of a Resveratrol Synthase Gene (PcRS) from Polygonum Cuspidatum in Transgenic Arabidopsis Causes the Accumulation of Trans-Piceid with Antifungal Activity. Plant Cell Rep. 2011, 30, 2027–2036. [Google Scholar] [CrossRef]
- Nicoletti, I.; De Rossi, A.; Giovinazzo, G.; Corradini, D. Identification and Quantification of Stilbenes in Fruits of Transgenic Tomato Plants (Lycopersicon esculentum Mill.) by Reversed Phase HPLC with Photodiode Array and Mass Spectrometry Detection. J. Agric. Food Chem. 2007, 55, 3304–3311. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The Grapevine Genome Sequence Suggests Ancestral Hexaploidization in Major Angiosperm Phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef]
- González-Barrio, R.; Beltrán, D.; Cantos, E.; Gil, M.I.; Espín, J.C.; Tomás-Barberán, F.A. Comparison of Ozone and UV-C Treatments on the Postharvest Stilbenoid Monomer, Dimer, and Trimer Induction in Var. ‘Superior′ White Table Grapes. J. Agric. Food Chem. 2006, 54, 4222–4228. [Google Scholar] [CrossRef]
- Pezet, R.; Perret, C.; Jean-Denis, J.B.; Tabacchi, R.; Gindro, K.; Viret, O. δ-Viniferin, a Resveratrol Dehydrodimer: One of the Major Stilbenes Synthesized by Stressed Grapevine Leaves. J. Agric. Food Chem. 2003, 51, 5488–5492. [Google Scholar] [CrossRef] [PubMed]
- Timperio, A.M.; D’Alessandro, A.; Fagioni, M.; Magro, P.; Zolla, L. Production of the Phytoalexins Trans-Resveratrol and Delta-Viniferin in Two Economy-Relevant Grape Cultivars upon Infection with Botrytis cinerea in Field Conditions. Plant Physiol. Biochem. 2012, 50, 65–71. [Google Scholar] [CrossRef]
- Calderón, A.A.; Zapata, J.M.; Pedreño, M.A.; Muñoz, R.; Barceló, A.R. Levels of 4-Hydroxystilbene-Oxidizing Isoperoxidases Related to Constitutive Disease Resistance in in Vitro-Cultured Grapevine. Plant Cell. Tissue Organ Cult. 1992, 29, 63–70. [Google Scholar] [CrossRef]
- Langcake, P.; Pryce, R.J. A New Class of Phytoalexins from Grapevines. Experientia 1977, 33, 151–152. [Google Scholar] [CrossRef]
- Gorham, J.; Coughlan, S.J. Inhibition of Photosynthesis by Stilbenoids. Phytochemistry 1980, 19, 2059–2064. [Google Scholar] [CrossRef]
- Sangha, A.K.; Parks, J.M.; Standaert, R.F.; Ziebell, A.; Davis, M.; Smith, J.C. Radical Coupling Reactions in Lignin Synthesis: A Density Functional Theory Study. J. Phys. Chem. B 2012, 116, 4760–4768. [Google Scholar] [CrossRef]
- Elder, T.; Rencoret, J.; del Río, J.C.; Kim, H.; Ralph, J. Radical Coupling Reactions of Hydroxystilbene Glucosides and Coniferyl Alcohol: A Density Functional Theory Study. Front. Plant Sci. 2021, 12, 319. [Google Scholar] [CrossRef]
- Givens, R.S.; Heger, D.; Hellrung, B.; Kamdzhilov, Y.; Mac, M.; Conrad, P.G.; Cope, E.; Lee, J.I.; Mata-Segreda, J.F.; Schowen, R.L.; et al. The Photo-Favorskii Reaction of p-Hydroxyphenacyl Compounds Is Initiated by Water-Assisted, Adiabatic Extrusion of a Triplet Biradical. J. Am. Chem. Soc. 2008, 130, 3307–3309. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E. Rules for Ring Closure. J. Chem. Soc. Chem. Commun. 1976, 734–736. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Thomas, R.C.; Kruse, L.I.; Silberman, L. Rules for Ring Closure: Ring Formation by Conjugate Addition of Oxygen Nucleophiles. J. Org. Chem. 1977, 42, 3846–3852. [Google Scholar] [CrossRef]
- Pecio, Ł.; Kozachok, S.; Brinza, I.; Stefan Boiangiu, R.; Hritcu, L.; Mircea, C.; Flavia Burlec, A.; Cioanca, O.; Hancianu, M.; Wronikowska-Denysiuk, O.; et al. Neuroprotective Effect of Yucca schidigera Roezl Ex Ortgies Bark Phenolic Fractions, Yuccaol B and Gloriosaol A on Scopolamine-Induced Memory Deficits in Zebrafish. Molecules 2022, 27, 3692. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.V.; López, S.N. Homoisoflavonoids: Occurrence, Biosynthesis, and Biological Activity. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; Volume 54, pp. 315–354. [Google Scholar]
- Dewick, P.M. Biosynthesis of the 3-Benzylchroman-4-One Eucomin. J. Chem. Soc. Chem. Commun. 1973, 438–439. [Google Scholar] [CrossRef]
- Dewick, P.M. Biosynthesis of the 3-Benzylchroman-4-One Eucomin in Eucomis bicolor. Phytochemistry 1975, 14, 983–988. [Google Scholar] [CrossRef]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The Use of Spirocyclic Scaffolds in Drug Discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.J.; Tice, C.M. The Utilization of Spirocyclic Scaffolds in Novel Drug Discovery. Expert Opin. Drug Discov. 2016, 11, 831–834. [Google Scholar] [CrossRef]
- Benabdallah, M.; Talhi, O.; Nouali, F.; Choukchou-Braham, N.; Bachari, K.; Silva, A.M.S. Advances in Spirocyclic Hybrids: Chemistry and Medicinal Actions. Curr. Med. Chem. 2018, 25, 3748–3767. [Google Scholar] [CrossRef]
- Acosta-Quiroga, K.; Rojas-Peña, C.; Nerio, L.S.; Gutiérrez, M.; Polo-Cuadrado, E. Spirocyclic Derivatives as Antioxidants: A Review. RSC Adv. 2021, 11, 21926–21954. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Guan, W.; Su, W.; Li, G.; Zhang, S.; Yao, H. Spiral Molecules with Antimalarial Activities: A Review. Eur. J. Med. Chem. 2022, 237, 114361. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Nishida, Y.; Wada, K.; Toyohisa, D.; Tanaka, T.; Ono, M.; Yasuda, S. Homoisoflavones as the Antioxidants Responsible from Bulbs of Scilla scilloides. Nat. Prod. Res. 2013, 27, 2360–2362. [Google Scholar] [CrossRef] [PubMed]
- Wenzig, E.M.; Oleszek, W.; Stochmal, A.; Kunert, O.; Bauer, R. Influence of Phenolic Constituents from Yucca schidigera Bark on Arachidonate Metabolism in Vitro. J. Agric. Food Chem. 2008, 56, 8885–8890. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Piacente, S.; Pizza, C.; Oleszek, W.; Stochmal, A.; Pinto, A.; Sorrentino, R.; Autore, G. Inhibition of Inducible Nitric Oxide Synthase Expression by Yuccaol C from Yucca schidigera Roezl. Life Sci. 2004, 75, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Sugahara, S.; Wada, K.; Toyohisa, D.; Tanaka, T.; Ono, M.; Yasuda, S. Inhibitory Effects of the Ethyl Acetate Extract from Bulbs of Scilla scilloides on Lipoxygenase and Hyaluronidase Activities. Pharm. Biol. 2014, 52, 1351–1357. [Google Scholar] [CrossRef]
- Du Toit, K.; Elgorashi, E.E.; Malan, S.F.; Drewes, S.E.; Van Staden, J.; Crouch, N.R.; Mulholland, D.A. Anti-Inflammatory Activity and QSAR Studies of Compounds Isolated from Hyacinthaceae Species and Tachiadenus longiflorus Griseb. (Gentianaceae). Bioorg. Med. Chem. 2005, 13, 2561–2568. [Google Scholar] [CrossRef]
- Wada, S.-I.; Hitomi, T.; Tokuda, H.; Tanaka, R. Anti-Tumor-Initiating Effects of Spiro-Biflavonoids from Abies sachalinensis. Chem. Biodivers. 2010, 7, 2303–2308. [Google Scholar] [CrossRef]
- Balestrieri, C.; Felice, F.; Piacente, S.; Pizza, C.; Montoro, P.; Oleszek, W.; Visciano, V.; Balestrieri, M.L. Relative Effects of Phenolic Constituents from Yucca schidigera Roezl. Bark on Kaposi’s Sarcoma Cell Proliferation, Migration, and PAF Synthesis. Biochem. Pharmacol. 2006, 71, 1479–1487. [Google Scholar] [CrossRef]
- Nigro, P.; Bloise, E.; Turco, M.C.; Skhirtladze, A.; Montoro, P.; Pizza, C.; Piacente, S.; Belisario, M.A. Antiproliferative and Pro-Apoptotic Activity of Novel Phenolic Derivatives of Resveratrol. Life Sci. 2007, 81, 873–883. [Google Scholar] [CrossRef]
- Schwikkard, S.; Whitmore, H.; Sishtla, K.; Sulaiman, R.S.; Shetty, T.; Basavarajappa, H.D.; Waller, C.; Alqahtani, A.; Frankemoelle, L.; Chapman, A.; et al. The Antiangiogenic Activity of Naturally Occurring and Synthetic Homoisoflavonoids from the Hyacinthaceae (Sensu APGII). J. Nat. Prod. 2019, 82, 1227–1239. [Google Scholar] [CrossRef]
- Czeczot, H.; Podsiad, M.; Skrzycki, M.; Stochmal, A.; Oleszek, W. Evaluation of the Mutagenic Activity of Phenolics from the Bark of Yucca schidigera Roezl. Acta Pol. Pharm. 2003, 60, 357–362. [Google Scholar]
- Sakuma, S.; Fujimoto, Y.; Tsunomori, M.; Tagano, S.; Nishida, H.; Baba, K.; Fujita, T. Effects of Daphnodorin A, B and C, New Flavans Isolated from Traditional Chinese Medicine, on the 12. Lipoxygenase and Cyclooxygenase Metabolism of Arachidonic Acid in Rabbit Platelets. Prostaglandins, Leukot. Essent. Fat. Acids 1998, 58, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Olas, B.; Wachowicz, B.; Stochmal, A.; Oleszek, W. Anti-Platelet Effects of Different Phenolic Compounds from Yucca schidigera Roezl. Bark. Platelets 2002, 13, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Inamori, Y.; Takeuchi, K.; Baba, K.; Kozawa, M. Antifungal and Insecticidal Activities of Daphnodorins A, B and C. Chem. Pharm. Bull. 1987, 35, 3931–3934. [Google Scholar] [CrossRef]
- Yusa, K.; Oh-hara, T.; Tsukahara, S.; Baba, K.; Taniguchi, M.; Kozawa, M.; Takeuchi, S.; Hara, H.; Tsuruo, T. Inhibition of Human Immunodeficiency Virus Type 1 (HIV-1) Replication by Daphnodorins. Antivir. Res. 1994, 25, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, K.; Elgorashi, E.E.; Malan, S.F.; Mulholland, D.A.; Drewes, S.E.; Van Staden, J. Antibacterial Activity and QSAR of Homoisoflavanones Isolated from Six Hyacinthaceae Species. S. Afr. J. Bot. 2007, 73, 236–241. [Google Scholar] [CrossRef]
- Takai, S.; Sakaguchi, M.; Jin, D.; Baba, K.; Miyazaki, M. Effects of Daphnodorin A, Daphnodorin B and Daphnodorin C on Human Chymase-Dependent Angiotensin II Formation. Life Sci. 1999, 64, 1889–1896. [Google Scholar] [CrossRef]
- Dal Piaz, F.; Ferro, P.; Vassallo, A.; Vasaturo, M.; Forte, G.; Chini, M.G.; Bifulco, G.; Tosco, A.; De Tommasi, N. Identification and Mechanism of Action Analysis of the New PARP-1 Inhibitor 2″-Hydroxygenkwanol A. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 1806–1814. [Google Scholar] [CrossRef]
- Sasaki, Y.; Suzuki, M.; Matsumoto, T.; Hosokawa, T.; Kobayashi, T.; Kamata, K.; Nagumo, S. Vasorelaxant Activity of Sappan Lignum Constituents and Extracts on Rat Aorta and Mesenteric Artery. Biol. Pharm. Bull. 2010, 33, 1555–1560. [Google Scholar] [CrossRef]






 | |||||
|---|---|---|---|---|---|
| Compound | Name | R1 | R2 | Stereochemistry | |
| C30H22O10 (MW = 542.49) | |||||
| 1 | Larixinol (=Abiesinol E) | H | H | (2R,3R,2′R,3′R) | |
| 2 a | 3-Epi-larixinol | H | H | (2R*,3R*,2′R*,3′S*) | |
| 3 a | 3,2′-Epi-larixinol | H | H | (2S*,3R*,2′R*,3′S*) | |
| 4 | Abiesinol F | H | H | (2R,3S,2′R,3′R) | |
| 5 | Yuccalechin A | H | H | (2S,3R,2′R,3′R) | |
| 6 | Yuccalechin B | H | H | (2S,3R,2′R,3′S) | |
| 7 | Yuccalechin C | H | H | (2S,3S,2′R,3′S) | |
| 8 | Fragranol B | H | H | (2S,3R,2′S,3′R) | |
| 9 | Fragranol C | H | H | (2R,3S,2′S,3′R) | |
| 10 | Spiropensilisol A | H | H | (2R*,3S*,2′R*,3′S*) | |
| 11 | Spiropensilisol B | H | H | (2S*,3S*,2′R*,3′S*) | |
| C30H22O11 (MW = 558.49) | |||||
| 12 a | Abiesinol A (=13-Hydroxylarixinol) | H | OH | (2R,3R,2′R,3′R) | |
| 13 | Abiesinol B | H | OH | (2R,3S,2′R,3′R) | |
| 14 | Abiesinol C (=Olgensisinol A) | OH | H | (2R,3R,2′R,3′R) | |
| 15 | Abiesinol D | OH | H | (2R,3S,2′R,3′R) | |
| C30H22O12 (MW = 574.49) | |||||
| 16 | Abiesinol G (=Vitisinol) | OH | OH | (2R,3R,2′R,3′R) | |
| 17 | Abiesinol H | OH | OH | (2R,3S,2′R,3′R) | |
| 18 | Larisinol | OH | OH | (2R*,3R*,2′R*,3′S*) | |
| 19 | Olgensisinol B | OH | OH | (2R,3S,2′S,3′S) | |
 | |||||
| Compound | Name | Stereochemistry | |||
| C30H22O12 (MW = 574.49) | |||||
| 20 | Olgensisinol C | (2R*,3S*,2′R*,3′S*) | |||
| 21 | Olgensisinol D | (2R*,3R*,2′S*,3′R*) | |||
 | |||||
| Compound | Name | R1 | R2 | R3 | Stereochemistry |
| C30H22O9 (MW = 526.49) | |||||
| 22 | Daphnodorin C | H | H | H | (2S,3S,2′S) |
| C30H22O10 (MW = 542.49) | |||||
| 23 | Genkwanol A | OH | H | H | (2R,3R,2′R,3′S) |
| 24 | Daphnodorin I | OH | H | H | (2S,3S,2′R,3′S) |
| C31H24O10 (MW = 556.52) | |||||
| 25 a | 4′-Methylgenkwanol A | OH | H | CH3 | (2R,3S,2′R,3′S) |
| 26 a | 2″-Methoxy-daphnodorin C | H | OCH3 | H | (2R*,3S*,2′R*) |
| 27 a | 2″-Methoxy-2-epi-daphnodorin C | H | OCH3 | H | (2R*,3S*,2′S*) |
| C30H22O11 (MW = 558.49) | |||||
| 28 a | 2″-Hydroxygenkwanol A | (2R,3S,2′R,3′S) | |||
 | |||||
| Compound | Name | Stereochemistry | |||
| C29H22O9 (MW = 514.48) | |||||
| 29 | Yuccaone A | (2R*,4R*,2′S*,3′R*) | |||
 | ||
|---|---|---|
| Compound | Name | Stereochemistry |
| C45H30O15 (MW = 810.71) | ||
| 30 | Fragranol A | (2S,3S,2′S,3′R,2″R,3″S) |
 | ||
| 31 | Triflarixinol | (2R*,3R*,2′R*,3′R*,2‴S*,3‴S*) |
 | |||
|---|---|---|---|
| Compound | Name | R | Stereochemistry |
| C60H42O18 (MW = 1050.97) | |||
| 32 | Edgechrin A | H | (2S*,3S*,2′S*,4′R*,2‴S*) |
| C60H42O19 (MW = 1066.97) | |||
| 34 | Edgechrin B | OH | (2S*,3S*,2′S*,4′R*,2‴R*,3‴S*) |
 | |||
| Compound | Name | Stereochemistry | |
| C60H42O18 (MW = 1050.97) | |||
| 33 | Edgechrin D | (2S*,3S*,2′S*,2‴S*,4‴R*) | |
 | |||
| Compound | Name | Stereochemistry | |
| C60H44O24 (MW = 1148.98) | |||
| 35 | Pinuspirotetrin | (2R,3S,2′R,3′S,2‴R,4‴R,2‴″S,3‴″R,4‴″R) | |
 | |||||
|---|---|---|---|---|---|
| Compound | Name | R1 | R2 | R3 | Stereochemistry |
| C29H20O8 (MW = 496.47) | |||||
| 36 | Yuccaol A | H | OH | H | (2S,3R) |
| 37 | Yuccaol B | H | OH | H | (2S,3S) |
| C30H22O10 (MW = 542.49) | |||||
| 38 | Yuccaol C | OH | OCH3 | H | (2S,3S) |
| 39 | Yuccaol D | OH | OCH3 | H | (2S,3R) |
| 40 | Yuccaol E | OH | H | OCH3 | (2S,3S) |
| 41 | Yuccalide A | OH | H | OCH3 | (2S,3R) |
 | |||||
| Compound | Name | R1 | R2 | R3 | Stereochemistry |
| C30H22O10 (MW = 542.49) | |||||
| 42 | Yuccalide B | OH | OCH3 | H | (2R*,3R*) |
| 43 | Yuccalide C | OH | OCH3 | H | (2R*,3S*) |
 | |||||
| Compound | Name | Stereochemistry | |||
| C45H30O15 (MW = 810.71) | |||||
| 44 | Gloriosaol A | (2S,3S,2′S,3′S) | |||
| 45 | Gloriosaol B | (2S*,3S*,2′R*,3′R*) | |||
| 46 | Gloriosaol C | (2S,3R,2′S,3′R) | |||
| 47 | Gloriosaol D | (2S,3S,2′S,3′R) | |||
| 48 | Gloriosaol E | (2S,3R,2′S,3′S) | |||
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Name | R1 | R2 | Stereochemistry | ||||
| C17H12O6 (MW = 312.27) | ||||||||
| 49 | Scillascillin | H | H | (3R) | ||||
| C18H14O7 (MW = 342.30) | ||||||||
| 50 | 2-Hydroxy-7-O-methyl-scillascillin | OH | CH3 | rac-(2R*,3R) | ||||
| C17H12O7 (MW = 328.27) | ||||||||
| 51 | 2-Hydroxy-scillascillin | OH | H | (3R*) | ||||
 | ||||||||
| Compound | Name | R1 | R2 | R3 | R4 | R5 | R6 | Stereochemistry |
| C17H14O6 (MW = 314.29) | ||||||||
| 52 | 3′,5,7-Trihydroxy-4′-methoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (=isomuscomosin) | H | OH | CH3 | H | H | H | (3R) |
| 53 | 4′,5,7-Trihydroxy-3′-methoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (=muscomosin) | H | OCH3 | H | H | H | H | (3R) |
| 54 | 3′,4′,5-Trihydroxy-7-methoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | H | OH | H | H | CH3 | H | (3R) |
| C18H16O6 (MW = 328.32) | ||||||||
| 55 | 5,5′-Dihydroxy-4′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | H | H | CH3 | OH | CH3 | H | (3R) |
| 56 | 3′,5-Dihydroxy-4′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | H | OH | CH3 | H | CH3 | H | (3R) |
| 57 | 5,5′,7-Trihydroxy-4′-methoxy-6-methylspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]-trien]-4-one | H | H | CH3 | OH | H | CH3 | (3R) |
| 58 | 5,7-Dihydroxy-3′,4′-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | H | OCH3 | CH3 | H | H | H | (3R*) |
| C18H16O7 (MW = 344.32) | ||||||||
| 59 | 2′,5,7-Trihydroxy-3′,4′-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (=scillavone A) | OH | OCH3 | CH3 | H | H | H | (3R) |
| 60 | 2′,4′,5-Trihydroxy-3′,7-dimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | OH | OCH3 | H | H | CH3 | H | (3R*) |
| C19H18O6 (MW = 342.34) | ||||||||
| 61 | 5-Hydroxy-3′,4′,7-trimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | H | OCH3 | CH3 | H | CH3 | H | (3R*) |
| C19H18O7 (MW = 358.34) | ||||||||
| 62 | 5,7-Dihydroxy-2′,3′,4′-trimethoxyspiro[2H-1-benzopyran-3(4H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | OCH3 | OCH3 | CH3 | H | H | H | (3R*) |
| 63 | 2′,5-Dihydroxy-3′,4′,7-trimethoxyspiro[4H-1-benzopyran-3(2H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one (=socialinone) | OH | OCH3 | CH3 | H | CH3 | H | (3R*) |
| C20H20O7 (MW = 372.37) | ||||||||
| 64 | 5-Hydroxy-2′,3′,4′,7-tetramethoxyspiro[4H-1-benzopyran-3(2H),7′-bicyclo[4.2.0]octa[1,3,5]trien]-4-one | OCH3 | OCH3 | CH3 | H | CH3 | H | (3R*) |
 | ||||||||
| Compound | Name | Stereochemistry | ||||||
| C32H28O12 (MW = 604.56) | ||||||||
| 65 | Protosappanin D | Unknown | ||||||
| Group | Compound | Extraction Solvent | Isolation 1st Step | Isolation 2nd Step | Isolation Final Steps | References |
|---|---|---|---|---|---|---|
| Spiro-biflavonoids | 1 | Acetone (maceration) | LLE H2O-CHCl3-EtOAc * | CC GPC LH-20 (EtOH) | CC NP Silica gel (CHCl3-MeOH) | [45] |
| 22, 24 | Acetone-H2O (7:3, v/v) (maceration, r.t.) | CC RP HP20 (gr. MeOH-H2O) | CC GPC LH-20 (gr. MeOH-H2O) for 22 or CC RP MCI (gr. MeOH-H2O) for 24 | CC RP MCI (MeOH-H2O) → CC RP C8 (MeOH-H2O) for 22 or CC RP HW-40F (MeOH-H2O) for 24 | [58] | |
| 1, 4, 12–17 | CHCl3 → MeOH (maceration, r.t., successively) | CC RP Diaion HP-20 (MeOH-H2O) | CC NP Silica gel (gr. CHCl3-MeOH) | MPLC NP Silica gel → CC GPC LH-20 (MeOH) → HPLC RP C18 (MeOH-H2O) | [42] | |
| 23 | CHCl3-MeOH (2:1, v/v) (reflux) | CC RP C18 (gr. MeOH-H2O) | FLASH CC NP Silica gel (gr. CHCl3-MeOH-Acetone) | FLASH CC NP Silica gel (Hexanes-EtOAc-MeOH) → CC NP Silica gel (EtOAc-CHCl3-MeOH-H2O) → HPLC RP C18 (CH3CN-H2O) | [55] | |
| 1, 14, 18, 22, 24 | EtOAc (reflux) | CC NP Silica gel (gr. CHCl3-MeOH) for 1, 14, 18 or CC NP Silica gel (gr. n-Hexane-EtOAc) for 22, 24 | CC NP Silica gel (CHCl3-MeOH) or CC GPC LH-20 (MeOH) for 22 | CC GPC LH-20 (MeOH) for 24 | [6,44,56,57] | |
| 23 | EtOH (r.t.) | LLE H2O-Petroleum ether-CHCl3-EtOAc | CC NP Silica gel (gr. CHCl3-MeOH) | [59] | ||
| 1–3 | EtOH-H2O (40%, v/v) (stirring, r.t.) | HPLC RP C18 (CH3CN-H2O w/FA) | [43] | |||
| 23 | EtOH-H2O (7:3, v/v) (maceration, r.t.) | LLE H2O-Petroleum ether-DCM-EtOAc-n-BuOH | CC NP Silica gel (gr. Petroleum Ether-EtOAc-MeOH) | CC NP Silica gel (gr. DCM-MeOH) → CC GPC LH-20 (MeOH) → TLC RP (MeOH-H2O) | [60] | |
| 23 | EtOH-H2O (75%, v/v) (reflux) | LLE H2O-EtOAc-n-BuOH | CC NP Silica gel (gr. CHCl3-MeOH) | HPLC RP C18 (MeOH-H2O) | [61] | |
| 1–3 | EtOH-H2O (80%, v/v) | LLE H2O-CHCl3-EtOAc-n-BuOH | CC NP Silica gel (gr. CHCl3-MeOH) | CC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) | [39] | |
| 14, 16, 19–21 | EtOH-H2O (80%, v/v) | LLE H2O-CHCl3-EtOAc | CC RP XAD-7 HP (gr. MeOH-H2O) | CC GPC LH-20 (gr. MeOH-H2O) → CC RP MCI gel CHP-20P (EtOH-H2O) for 14, 16, 19 or CC GPC LH-20 (gr. MeOH-H2O) → CC GPC LH-20 (MeOH-H2O) → CC RP C18 (MeOH-H2O) for 20, 21 | [46] | |
| 16 | EtOH-H2O (80%, v/v) | LLE H2O-CHCl3-EtOAc-n-BuOH | CC RP Diaion HP-20 (MeOH-H2O) | CC GPC LH-20 (gr. MeOH-H2O) → CC RP MCI CHP-20P (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O w/AcOH) | [63] | |
| 1, 12 | EtOH-H2O (80%, v/v) (reflux) | LLE H2O-CHCl3-EtOAc-n-BuOH | CC NP Silica gel (gr. CHCl3-Acetone) | MPLC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) | [41] | |
| 1 | EtOH-H2O (85:15, v/v) (maceration, r.t.) | LLE H2O-CHCl3-EtOAc-n-BuOH | CC NP Silica gel (gr. CHCl3-Acetone) | MPLC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) | [40] | |
| 2, 8, 9 | EtOH-H2O (95%, v/v) (maceration, r.t.) | LLE H2O-EtOAc | MPLC NP Silica gel (DCM-EtOAc) | TLC NP Silica gel (DCM-EtOAc) | [38] | |
| 22 | EtOH-H2O (95%, v/v) (reflux) | LLE H2O-Petroleum ether-EtOAc | CC NP Silica gel (gr. DCM-MeOH) | CC NP Silica gel (unknown solvent) → HPLC RP C18 (MeOH-H2O) | [53] | |
| 1, 2, 22 | EtOH-H2O (95%, v/v) (r.t.) | LLE H2O-Petroleum ether-EtOAc-n-BuOH | CC NP Silica gel (gr. CHCl3-MeOH) for 22 or CC NP Silica gel (Petroleum ether-Acetone) for 1, 2 | CC GPC LH-20 (CHCl3-MeOH) for 22 or HPLC RP C18 (MeOH-H2O) → CC GPC LH-20 (MeOH) for 1, 2 | [48,62] | |
| 22, 24, 26, 27 | MeOH | LLE H2O-Petroleum ether-EtOAc-n-BuOH | CC NP Silica gel (gr. CHCl3-MeOH) | CC RP C18 (gr. MeOH-H2O) → CC GPC LH-20 (MeOH) or CC GPC LH-20 (MeOH) for 22, 24 | [49,50] | |
| 22, 24 | MeOH(maceration, r.t.) | LLE H2O-n-Hexane-EtOAc-n-BuOH | CC RP C18 (MeOH-H2O) | HPLC RP C18 (CH3CN-H2O) | [52] | |
| 29 | MeOH (maceration, r.t.) | CC GPC LH-20 (MeOH) | CC RP C18 (CH3CN-H2O w/H3PO4) | [32] | ||
| 23 | MeOH (reflux) | CC NP Silica gel (gr. n-Hexane-EtOAc → CHCl3-MeOH) | CC GPC LH-20 (MeOH-H2O) | CC RP MCI CHP 20P (MeOH-H2O) | [51] | |
| 1 | MeOH (r.t.) | CC RP C18 (gr. MeOH-H2O) | CC RP C18 (gr. CH3CN-H2O) | [33] | ||
| 5–7 | MeOH (r.t.) | LLE H2O-n-Hexane- EtOAc | CC GPC LH-20 (MeOH) | CC NP Silica gel (gr. CHCl3-Acetone-AcOH) → HPLC RP C18 (CH3CN/MeOH-H2O) | [31] | |
| 23 | MeOH-H2O (80%, v/v) (37 °C) | LLE H2O-n-BuOH | CC GPC LH-20 (MeOH) | CC NP Silica gel (gr. cyclohexane-EtOAc-MeOH) → CC MPLC RP (CH3CN-H2O) | [34] | |
| 1–4, 10, 11 | MeOH-H2O (90%, v/v) (r.t.) | CC GPC LH-20 | HPLC RP C18 | [35] | ||
| 23–25, 28 | n-Hexane → CHCl3 → CHCl3-MeOH (9:1, v/v) → MeOH (maceration, r.t., successively) | CC GPC LH-20 (MeOH) | HPLC RP C18 (MeOH-H2O) | [54] | ||
| Spiro-triflavonoids | 31 | EtOAc (reflux) | CC NP Silica gel (gr. CHCl3-MeOH) | CC NP Silica gel (CHCl3-MeOH) | [44] | |
| 30 | EtOH-H2O (95%, v/v) (maceration, r.t.) | LLE H2O-EtOAc | MPLC NP Silica gel (gr. DCM-EtOAc-MeOH) | MPLC NP Silica gel (gr. DCM-MeOH) | [37] | |
| Spiro-tetraflavonoids | 32–34 | Acetone-H2O (7:3, v/v) (maceration, r.t.) | CC RP HP20 (gr. MeOH-H2O) | CC GPC LH-20 (gr. MeOH-H2O) | CC NP Silica gel (DCM-MeOH) → CC RP HW-40F (MeOH-H2O) → CC RP C8 (MeOH-H2O) → CC RP C18 (MeOH-H2O) for 32 or CC RP HW-40F (MeOH-H2O) → HPLC RP C18 (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O) for 33 or CC NP Silica gel (DCM-MeOH) → CC RP C18 (MeOH-H2O) for 34 | [58] |
| 35 | Pine bark extract ** | LLE H2O-EtOAc | CPC Hexane-EtOAc-MeOH-H2O (2-4-1-4) → CPC Hexane-EtOAc-MeOH-H2O (0.5-4-1-4) | CC GPC LH-20 (EtOH) → CC RP C18 (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O w/FA) | [47] | |
| Spiro-flavostilbenoids | 38–43 | Acetone (r.t.) | CC RP DMS (gr. MeOH-H2O-Acetone) | CC GPC LH-20 (MeOH) | HPLC RP C18 (MeOH-H2O) → HPLC RP C18 (CH3CN-H2O) | [11] |
| 38, 39 | MeOH | LLE H2O-EtOAc-n-BuOH | CC NP Silica gel (gr. n-Hexane-EtOAc-MeOH) | CC NP Silica gel (n-Hexane-EtOAc) → CC GPC LH-20 (MeOH) | [19] | |
| 36–40 | MeOH (r.t.) | CC RP C18 (gr. MeOH-H2O) | CC RP C18 (gr. CH3CN-H2O H3PO4) or CC RP C18 (gr. CH3CN-H2O) | [30,33] | ||
| 41, 44, 46–48 | MeOH (r.t.) | LLE H2O-n-Hexane-EtOAc | CC GPC LH-20 (MeOH) | CC NP Silica gel (gr. CHCl3-Acetone-AcOH) → HPLC RP C18 (CH3CN/MeOH-H2O) | [31] | |
| 38–40, 44–48 | MeOH-H2O (80%, v/v) | LLE H2O-EtOAc | CC GPC LH-20 (MeOH) | HPLC RP C18 (gr CH3CN-H2O w/TFA) | [28,29] | |
| Scillascillin-type homoisoflavonoids | 49, 52, 56 | DCM (agitation or shaker, r.t.) | CC NP Silica gel (unknown solvent or DCM) | [16,23] | ||
| 49, 50 | DCM → EtOAc → MeOH (shaker, successively) | CC NP Silica gel (n-Hexane-DCM-MeOH) | CC NP Silica gel (DCM-Et2O) | [22] | ||
| 49 | DCM → MeOH (agitation, r.t.) | CC NP Silica gel (DCM) | [18] | |||
| 63, 64 | DCM → MeOH (shaker, successively) | CC NP Silica gel (gr. n-Hexane-DCM-MeOH) | TLC NP Silica gel (EtOAc-DCM) | [22] | ||
| 50, 56, 60–62, 64 | DCM-MeOH (1:1, v/v) → MeOH (maceration, r.t., successively) | LLE MeOH-CHCl3-H2O (lower phase) | CC NP Silica gel (gr. DCM-MeOH) | CC NP Silica gel (gr. CHCl3-Light Petroleum) → recrystallization (MeOH) for 50 or CC GPC LH-20 (unknown solvent) → CC NP Silica gel (gr. CHCl3-MeOH) → TLC (unknown solvent) for 60 or CC GPC LH-20 (unknown solvent) → CC NP Silica gel (gr. CHCl3-Light Petroleum) → TLC (CHCl3-Light Petroleum-MeOH) for 56, 61, 62, 64 | [15] | |
| 53 | DCM-MeOH (1:1, v/v) → MeOH (maceration, r.t., successively) | LLE (MeOH-H2O, 70%, v/v)-Light Petroleum-CHCl3-EtOAc-n-BuOH (lower phase) | CC NP Silica gel (gr. Light Petroleum-EtOAc) | CC NP Silica gel (gr. Light Petroleum-CHCl3-MeOH) | [15] | |
| 58, 61 | DCM-MeOH (1:1, v/v) → MeOH (maceration, r.t., successively) | CC NP Silica gel (gr. Petrol-EtOAc-MeOH) | TLC NP Silica gel (CHCl3-MeOH) | TLC NP Silica gel (CHCl3) | [20] | |
| 52, 54, 56 | Light petrol → Et2O → MeOH (Soxhlet, successively) | CC NP Silica gel (gr. n-Hexane-Et2O-MeOH) | CC NP Silica gel (gr. n-Hexane-Acetone) | HPLC NP Silica gel (CHCl3-MeOH) → PLC NP Silica gel (CHCl3-Acetone) | [24] | |
| 53–56 | Light petrol → Et2O → MeOH (Soxhlet, successively) | CC NP Silica gel (CHCl3-EtOAc) | PLC NP Silica gel (Benzene-EtOAc) → PLC NP Silica gel (CHCl3-Acetone) or PLC NP Silica gel (Benzene-EtOAc) → crystallization (MeOH) | [26] | ||
| 57 | MeOH (reflux) | CC RP Diaion HP20 (gr. MeOH-H2O → MeOH → EtOH → EtOAc) | CC NP Silica gel (gr CHCl3-MeOH-H2O) | CC NP Silica gel (CHCl3-MeOH-H2O) → CC RP C18 (MeOH-H2O) | [13] | |
| 49, 51, 52, 59 | MeOH (r.t.) | LLE H2O-EtOAc | CC NP Silica gel (gr. n-Hexane-Acetone) | CC RP C18 (gr. MeOH-H2O) → HPLC RP C18 (MeOH-H2O) → crystallization (n-Hexane-Acetone) or CC RP C18 (gr. MeOH-H2O) → HPLC RP C18 (MeOH-H2O) → HPLC NP Silica gel (CHCl3-MeOH) | [27] | |
| 53 | Petrol → Et2O (Soxhlet, successively) | CC NP Silica gel (gr. n-Hexane-Et2O) | CC NP Silica gel (gr. CHCl3-EtOAc) | CC NP Silica gel (Benzene-EtOAc) → TLC NP Silica gel (n-Hexane-Et2O-dioxane) | [25] | |
| 49–52 | Petrol → Et2O → MeOH (Soxhlet, successively) | CC NP Silica gel (gr. CHCl3-MeOH) | TLC NP Silica gel (Benzene-EtOAc) | PLC NP Silica gel (Benzene-EtOAc) | [14] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecio, Ł.; Pecio, S.; Mroczek, T.; Oleszek, W. Spiro-Flavonoids in Nature: A Critical Review of Structural Diversity and Bioactivity. Molecules 2023, 28, 5420. https://doi.org/10.3390/molecules28145420
Pecio Ł, Pecio S, Mroczek T, Oleszek W. Spiro-Flavonoids in Nature: A Critical Review of Structural Diversity and Bioactivity. Molecules. 2023; 28(14):5420. https://doi.org/10.3390/molecules28145420
Chicago/Turabian StylePecio, Łukasz, Solomiia Pecio, Tomasz Mroczek, and Wiesław Oleszek. 2023. "Spiro-Flavonoids in Nature: A Critical Review of Structural Diversity and Bioactivity" Molecules 28, no. 14: 5420. https://doi.org/10.3390/molecules28145420
APA StylePecio, Ł., Pecio, S., Mroczek, T., & Oleszek, W. (2023). Spiro-Flavonoids in Nature: A Critical Review of Structural Diversity and Bioactivity. Molecules, 28(14), 5420. https://doi.org/10.3390/molecules28145420