
Treatment of Uranium-Contaminated Ground Water Using Adsorption Technology via Novel Mesoporous Silica Nanoparticles

Abstract
:1. Introduction
2. Results and Discussion
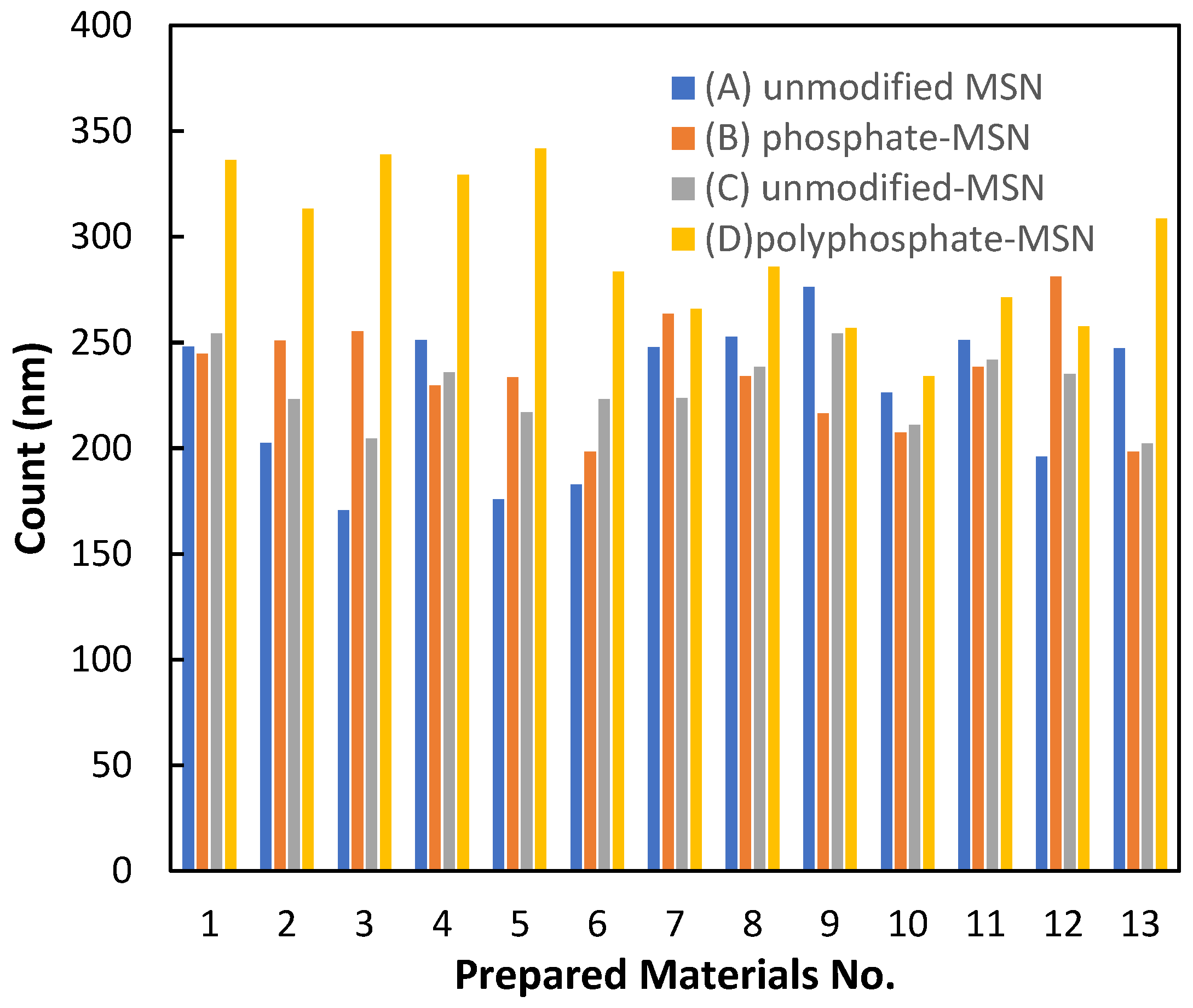
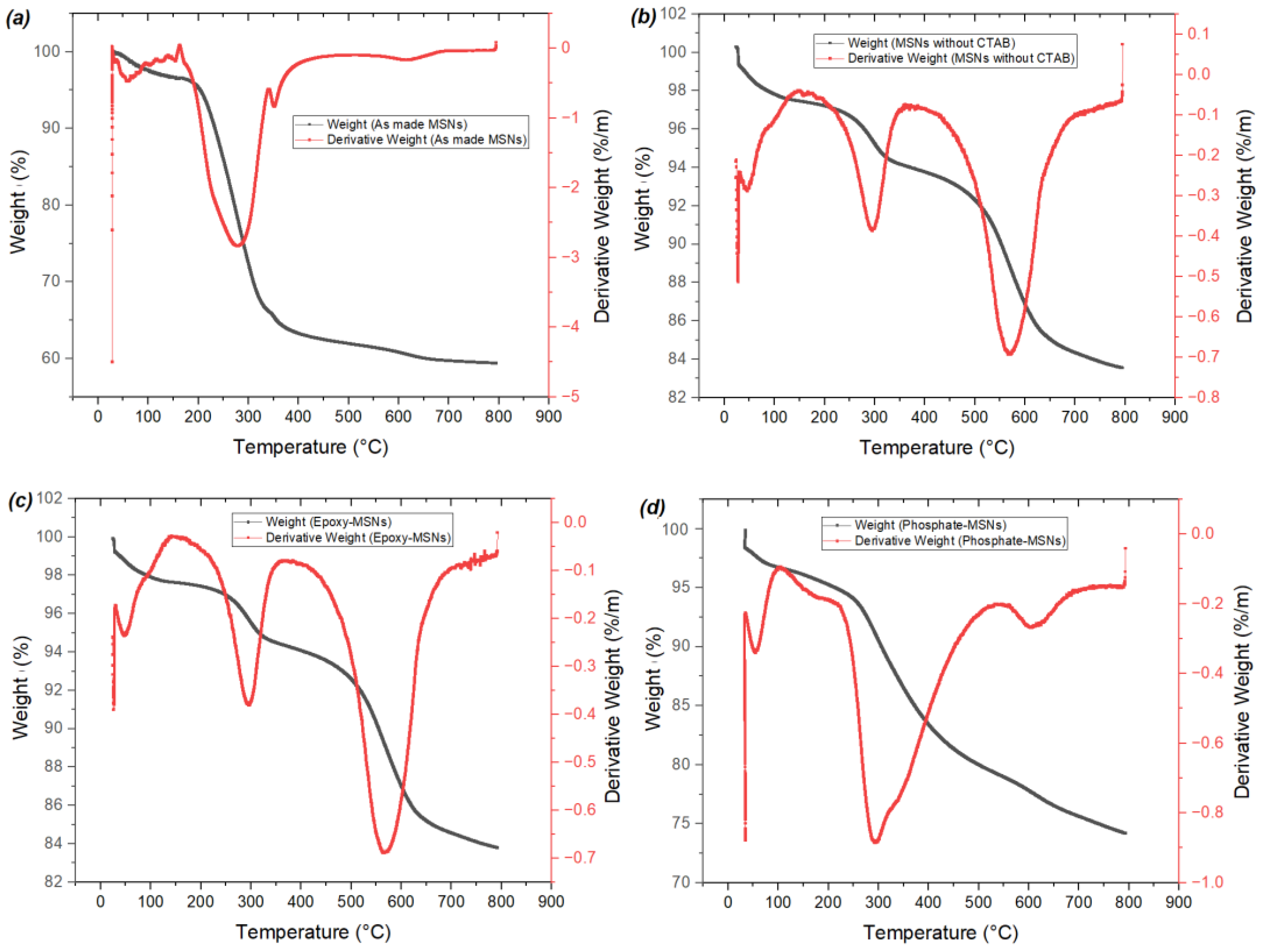
2.1. Characterization of the Materials
2.2. Mesoporous Silica Nanoparticles-Phosphate (Diphosphate)-Modified MSNs (Phosphate (Diphosphate)-MSNs)—Material (A)
2.2.1. Effect of Time on U Adsorption at Room Temperature
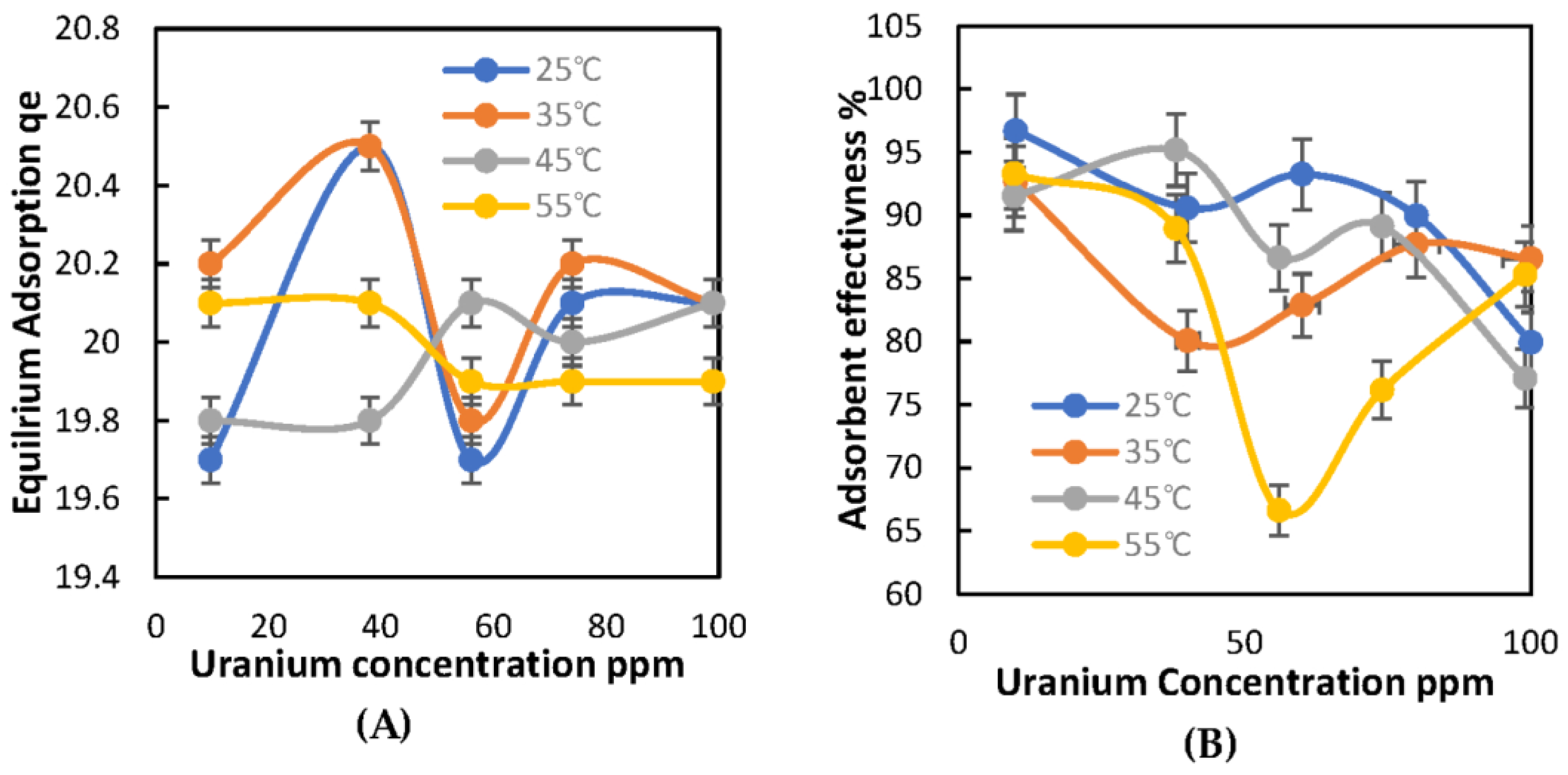
2.2.2. Effect of Temperature on U Adsorption
2.2.3. Effect of pH on Adsorption at Two U Concentrations (50 and 100 ppm) at Room Temperature
2.2.4. Effect of Material Weight at Room Temperature for 60 min
2.2.5. Effect of Mixture Solution at Room Temperature for 60 min
2.3. Mesoporous Silica Nanoparticles—Phosphate (Diphosphate)-Modified MSNs (Phosphate (Diphosphate)-MSNs)—Novel Material (B)
2.3.1. Effect of Time on U Adsorption at Room Temperature
2.3.2. Effect of Temperature on U Adsorption
2.3.3. Effect of pH on Adsorption in Two Concentrations of U (50, 100 ppm) at Room Temperature
2.3.4. Effect of Material Weight at Room Temperature for 60 min
2.3.5. Effect of Mixture Solution at Room Temperature for 60 min
3. Methodology
3.1. The Materials
3.2. The Synthesis
3.2.1. Synthesis of Mesoporous Silica Nanoparticles
3.2.2. Synthesis of 3-Glycidyloxypropyl-functionalized MSNs (Epo-MSNs)
3.2.3. Preparation of Phosphate Modified MSNs (Phosphate-MSNs)
3.2.4. Preparation of 3-Aminopropyl-functionalized MSNs (AP-MSNs)
3.2.5. Preparation of ATRP Initiator Immobilization, BiBB-MSN
3.2.6. Preparation of Glycidyl Methacrylate (GMA) Brushes Grafted GMA-MSNs
3.2.7. Preparation of Polyphosphate-MSNs
3.3. Measurement and Characterization
3.3.1. Effect of Time on U Adsorption at Room Temperature
3.3.2. Effect of Temperature on U Adsorption
3.3.3. Effect of pH on U Adsorption in Two Concentrations of U (100, 50) at Room Temperature
3.3.4. Effect of Material Weight at Room Temperature for 60 min
3.3.5. Effect of Mixture Solution at Room Temperature for 60 min
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Guidelines for Drinking-Water Quality: Fourth Edition Incorporating First Addendum, 4th ed + 1st Add; World Health Organization: Geneva, Switzerland, 2017; Available online: https://apps.who.int/iris/handle/10665/254637 (accessed on 25 December 2022).
- U.S. Environmental Protection Agency. 2012 Edition of the Drinking Water Standards and Health Advisories; EPA 822-S-12-001; U.S. Environmental Protection Agency: Washington, DC, USA, 2012; 20p.
- Shabana, E.; Kinsara, A. Radioactivity in the groundwater of a high background radiation area. J. Environ. Radioact. 2014, 137, 181–189. [Google Scholar] [CrossRef]
- Shabana, E.-S.I.; Abulfaraj, W.H.; Kinsara, A.A.; Abu Rizaiza, O.S. Natural radioactivity in the groundwater of Wadi Nu’man, Mecca Province, Saudi Arabia. Radiochim. Acta 2013, 101, 461–470. [Google Scholar] [CrossRef]
- Giannakoudakis, D.A.; Ioannou, E.; Gournis, D.; Deligiannakis, Y. Supreme and selective capture of one of the most dangerous metal, URANIUM, by phospho-nate-functionalized ordered mesoporous silica: Surface chemistry matters the most. In EGU General Assembly Conference Abstracts 2021, Proceedings of the 23rd EGU General Assembly, Online, 19–30 April 2021; Copernicus GmbH: Göttingen, Germany, 2021; p. EGU21-16251. [Google Scholar] [CrossRef]
- Michailidou, G.; Koumentakou, I.; Liakos, E.V.; Lazaridou, M.; Lambropoulou, D.A.; Bikiaris, D.N.; Kyzas, G.Z. Adsorption of Uranium, Mercury, and Rare Earth Elements from Aqueous Solutions onto Magnetic Chitosan Adsorbents: A Review. Polymers 2021, 13, 3137. [Google Scholar] [CrossRef] [PubMed]
- da Silva Alves, D.C.; Healy, B.; Pinto, L.A.d.A.; Cadaval, T.R.S., Jr.; Breslin, C.B. Recent Developments in Chitosan-Based Adsorbents for the Removal of Pollutants from Aqueous Environments. Molecules 2021, 26, 594. [Google Scholar] [CrossRef] [PubMed]
- Elwakeel, K.Z.; Hamza, M.F.; Guibal, E. Effect of agitation mode (mechanical, ultrasound and microwave) on uranium sorption using amine- and dithizone-functionalized magnetic chitosan hybrid materials. Chem. Eng. J. 2021, 411, 128553. [Google Scholar] [CrossRef]
- Zhao, F.; Sillanpää, M. Cross-linked chitosan and β-cyclodextrin as functional adsorbents in water treatment. In Advanced Water Treatment; Elsevier: Amsterdam, The Netherlands, 2020; pp. 161–264. [Google Scholar] [CrossRef]
- Barczak, M. Functionalization of mesoporous silica surface with carboxylic groups by Meldrum’s acid and its application for sorption of proteins. J. Porous Mater. 2018, 26, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Barczak, M.; Dobrowolski, R.; Borowski, P.; Giannakoudakis, D.A. Pyridine-, thiol- and amine-functionalized mesoporous silicas for adsorptive removal of pharmaceuticals. Microporous Mesoporous Mater. 2020, 299, 110132. [Google Scholar] [CrossRef]
- Olchowski, R.; Zięba, E.; Giannakoudakis, D.A.; Anastopoulos, I.; Dobrowolski, R.; Barczak, M. Tailoring Surface Chemistry of Sugar-Derived Ordered Mesoporous Carbons towards Efficient Removal of Diclofenac from Aquatic Environments. Materials 2020, 13, 1625. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Nazir, M.A.; Jamshaid, M.; Kumar, O.P.; Najam, T.; Shah, S.S.A.; Rehman, A.U. Synthesis of nanoadsorbent entailed mesoporous organosilica for decontamination of methylene blue and methyl orange from water. Int. J. Environ. Anal. Chem. 2021, 1–14. [Google Scholar] [CrossRef]
- Shen, J.; Schäfer, A. Removal of fluoride and uranium by nanofiltration and reverse osmosis: A review. Chemosphere 2014, 117, 679–691. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, H.; Zhang, Y.; Wu, J.; Zhang, H.; Wang, G.; Li, J. Pseudocapacitive deionization of uranium(VI) with WO3/C electrode. Chem. Eng. J. 2020, 398, 125460. [Google Scholar] [CrossRef]
- Reinoso-Maset, E.; Ly, J. Study of uranium(VI) and radium(II) sorption at trace level on kaolinite using a multisite ion exchange model. J. Environ. Radioact. 2016, 157, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Bhalara, P.D.; Punetha, D.; Balasubramanian, K. A review of potential remediation techniques for uranium(VI) ion retrieval from contaminated aqueous environment. J. Environ. Chem. Eng. 2014, 2, 1621–1634. [Google Scholar] [CrossRef]
- Dressler, A.; Leydier, A.; Grandjean, A. Effects of Impregnated Amidophosphonate Ligand Concentration on the Uranium Extraction Behavior of Mesoporous Silica. Molecules 2022, 27, 4342. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Mei, P.; Xiao, J.; Huang, X.; Ishag, A.; Sun, Y. Carbon materials for extraction of uranium from seawater. Chemosphere 2021, 278, 130411. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, P.; Dong, F.; Nie, X.; Ding, C.; Wang, S.; Zhang, Y.; Liu, H.; Zhou, S. Polyamine and amidoxime groups modified bifunctional polyacrylonitrile-based ion exchange fibers for highly efficient extraction of U(VI) from real uranium mine water. Chem. Eng. J. 2019, 367, 198–207. [Google Scholar] [CrossRef]
- Sarafraz, H.; Alahyarizadeh, G.; Minuchehr, A.; Modaberi, H.; Naserbegi, A. Economic and Efficient phosphonic functional groups mesoporous silica for uranium selective adsorption from aqueous solutions. Sci. Rep. 2019, 9, 9686. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Li, Y.; Wang, X.; Hu, S.; Wen, J.; Xiong, J.; Asiri, A.M.; Marwani, H.M. Phosphate-Functionalized Polyethylene with High Adsorption of Uranium(VI). ACS Omega 2017, 2, 3267–3275. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Sun, Q.; Zhang, Y.; Abney, C.W.; Aguila, B.; Lin, W.; Ma, S. Functionalized Porous Aromatic Framework for Efficient Uranium Adsorption from Aqueous Solutions. ACS Appl. Mater. Interfaces 2017, 9, 12511–12517. [Google Scholar] [CrossRef]
- Sihn, Y.H.; Byun, J.; Patel, H.A.; Lee, W.; Yavuz, C.T. Rapid extraction of uranium ions from seawater using novel porous polymeric adsorbents. RSC Adv. 2016, 6, 45968–45976. [Google Scholar] [CrossRef]
- Yu, B.; Bai, Y.; Ming, Z.; Yang, H.; Chen, L.; Hu, X.; Feng, S.; Yang, S.-T. Adsorption behaviors of tetracycline on magnetic graphene oxide sponge. Mater. Chem. Phys. 2017, 198, 283–290. [Google Scholar] [CrossRef]
- Bratskaya, S.; Schwarz, S.; Chervonetsky, D. Comparative study of humic acids flocculation with chitosan hydrochloride and chitosan glutamate. Water Res. 2004, 38, 2955–2961. [Google Scholar] [CrossRef] [PubMed]
- Alswieleh, A.M.; Alshahrani, M.M.; Alzahrani, K.E.; Alghamdi, H.S.; Niazy, A.; Alsilme, A.S.; Beagan, A.M.; Alsheheri, B.M.; Alghamdi, A.A.; Almeataq, M.S. Surface modification of pH-responsive poly(2-(tert-butylamino)ethyl methacrylate) brushes grafted on mesoporous silica nanoparticles. Des. Monomers Polym. 2019, 22, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Magied, M.O.A.; Dhmees, A.S.; Abd El-Hamid, A.A.M.; Eldesouky, E.M. Uranium extraction by sulfonated mesoporous silica derived from blast furnace slag. J. Nucl. Mater. 2018, 509, 295–304. [Google Scholar] [CrossRef]
- Alswieleh, A.M.; Albahar, H.Y.; Alfawaz, A.M.; Alsilme, A.S.; Beagan, A.M.; Alsalme, A.M.; Almeataq, M.S.; Alshahrani, A.; Alotaibi, K.M. Evaluation of the Adsorption Efficiency of Glycine-, Iminodiacetic Acid-, and Amino Propyl-Functionalized Silica Nanoparticles for the Removal of Potentially Toxic Elements from Contaminated Water Solution. J. Nanomater. 2021, 2021, 6664252. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | BET Surface-Area (m2.g−1) | Pore-Volume (cm3.g−1) |
|---|---|---|
| MSNs | 1048 | 1.51 |
| phosphate-MSNs | 570 | 0.95 |
| Polyphosphate-MSNs | 10.02 | 0.0166 |
| A. Effect of Time on U Adsorption at Room Temperature | ||||
| U++ Concentration (ppm) | t (Minute) | Ce (mg/L) | % | Qt (mg/g) |
| 100 | 5 | 29.78 | 70.22 | 20.1 |
| 10 | 23.17 | 76.83 | 20.1 | |
| 15 | 28.79 | 71.21 | 20.2 | |
| 30 | 22.04 | 77.74 | 19.9 | |
| 60 | 22.61 | 77.16 | 20.1 | |
| 120 | 14.5 | 85.35 | 20.2 | |
| 180 | 16.53 | 83.3 | 20 | |
| 240 | 14.27 | 85.59 | 20.1 | |
| B. Effect of temperature on U adsorption (COF) at 25 °C. | ||||
| U++ concentration (ppm) | Ce (mg/L) | % | qe (mg/g) | Ce/qe |
| 10 | 0.32 | 96.7 | 19.7 | 0.02 |
| 40 | 3.58 | 90.58 | 20.5 | 0.17 |
| 60 | 3.77 | 93.27 | 19.7 | 0.19 |
| 80 | 7.43 | 89.96 | 20.1 | 0.37 |
| 100 | 19.87 | 79.93 | 20.1 | 0.99 |
| C. Effect of temperature on U adsorption (COF) at 35 °C. | ||||
| U++ concentration (ppm) | Ce (mg/L) | % | qe (mg/g) | Ce/qe |
| 10 | 0.71 | 92.68 | 20.2 | 0.04 |
| 40 | 7.57 | 80.08 | 20.5 | 0.37 |
| 60 | 9.58 | 82.89 | 19.8 | 0.48 |
| 80 | 9.09 | 87.72 | 20.2 | 0.45 |
| 100 | 13.3 | 86.57 | 20.1 | 0.66 |
| D. Effect of temperature on U adsorption (COF) at 45 °C. | ||||
| U++ concentration (ppm) | Ce (mg/L) | % | qe (mg/g) | Ce/qe |
| 10 | 0.82 | 91.55 | 19.8 | 20 |
| 40 | 1.83 | 95.18 | 19.8 | 20 |
| 60 | 7.47 | 86.66 | 20.1 | 20 |
| 80 | 8.05 | 89.12 | 20 | 20 |
| 100 | 22.69 | 77.08 | 20.1 | 20 |
| E. Effect of temperature on U adsorption (COF) at 55°C. | ||||
| U++ concentration (ppm) | Ce (mg/L) | % | qe (mg/g) | Ce/qe |
| 10 | 0.65 | 93.3 | 20.1 | 0.03 |
| 40 | 4.19 | 88.97 | 20.1 | 0.21 |
| 60 | 18.69 | 66.63 | 19.9 | 0.94 |
| 80 | 17.64 | 76.16 | 19.9 | 0.89 |
| 100 | 14.53 | 85.32 | 19.9 | 0.73 |
| A. The Effect of pH at Concentration of U (100 ppm) at Room Temperature. | ||||
| pH | Ci (mg/L) | Ce (mg/L) | % | qe (mg/g) |
| 2 | 100 | 87.3 | 12.7 | 19.9 |
| 4 | 56.11 | 43.89 | 20 | |
| 6 | 27.15 | 72.85 | 20.1 | |
| 8 | 44.06 | 55.94 | 19.9 | |
| B. The effect of pH at concentration of U (50 ppm) and room temperature. | ||||
| pH | Ci (mg/L) | Ce (mg/L) | % | qe (mg/g) |
| 2 | 50 | 44.37 | 11.26 | 20.1 |
| 4 | 24.38 | 51.24 | 20.1 | |
| 6 | 4.057 | 91.89 | 19.9 | |
| 8 | 11.95 | 76.1 | 20.1 | |
| A. 60 min at U Concentration 100 ppm. | |||
| Weight (mg) | Ce (mg/L) | % | qe (mg/g) |
| 10 | 46.37 | 53.63 | 10.1 |
| 15 | 36.7 | 63.3 | 15 |
| 20 | 34.94 | 65.06 | 20.1 |
| 25 | 10.7 | 89.3 | 25 |
| 30 | 16.42 | 83.58 | 29.9 |
| 50 | 5.43 | 94.57 | 49.9 |
| B. 60 min at U concentration 50 ppm. | |||
| Weight (mg) | Ce (mg/L) | % | qe (mg/g) |
| 10 | 27 | 46 | 10 |
| 15 | 17.63 | 64.74 | 15 |
| 20 | 18.86 | 62.28 | 19.9 |
| 25 | 2.42 | 95.16 | 25.1 |
| 30 | 12.44 | 96.12 | 29.9 |
| 50 | 0.63 | 98.74 | 49.9 |
| A. The Effect of Adsorption with Pb. | ||||
| Element | Ci (mg/L) | Ce (mg/L) | % | qe (mg/g) |
| Cr | 100 | 77 | 23 | 49.9 |
| Ni | 100 | 60.19 | 39.81 | |
| Cu | 100 | 88.69 | 11.31 | |
| Zn | 100 | 93 | 7 | |
| Cd | 100 | 99 | 1 | |
| Pb | 100 | 76 | 24 | |
| U | 100 | 41.73 | 58.27 | |
| B. The effect of adsorption in the absence of Pb. | ||||
| Element | Ci (mg/L) | Ce (mg/L) | % | qe (mg/g) |
| Cr | 100 | 41 | 59 | 50.1 |
| Ni | 100 | 30.18 | 69.82 | |
| Cu | 100 | 44.52 | 55.48 | |
| Zn | 100 | 46.53 | 53.47 | |
| Cd | 100 | 50 | 50 | |
| U | 100 | 2.41 | 97.59 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshammari, A.S.; Almeataq, M.S.; Basfar, A.A. Treatment of Uranium-Contaminated Ground Water Using Adsorption Technology via Novel Mesoporous Silica Nanoparticles. Molecules 2023, 28, 5642. https://doi.org/10.3390/molecules28155642
Alshammari AS, Almeataq MS, Basfar AA. Treatment of Uranium-Contaminated Ground Water Using Adsorption Technology via Novel Mesoporous Silica Nanoparticles. Molecules. 2023; 28(15):5642. https://doi.org/10.3390/molecules28155642
Chicago/Turabian StyleAlshammari, Abdulmalik S., Mohammed S. Almeataq, and Ahmed A. Basfar. 2023. "Treatment of Uranium-Contaminated Ground Water Using Adsorption Technology via Novel Mesoporous Silica Nanoparticles" Molecules 28, no. 15: 5642. https://doi.org/10.3390/molecules28155642