
Spontaneous Water-Promoted Self-Aggregation of a Hydrophilic Gold(I) Complex Due to Ligand Sphere Rearrangement


Abstract
:
1. Introduction
2. Results and Discussion
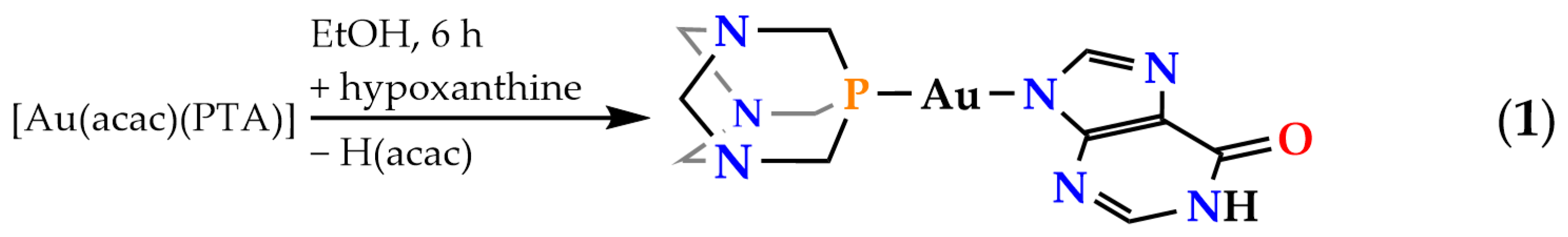
2.1. Synthesis and Characterization of Complex [Au(N9-Hypoxanthinate)(PTA)] (1)
2.2. Synthesis and Characterization of Complexes [Au(PTA)2](ClO4) (2) and (NBu4)[Au(N9-Hypoxanthinate)2] (3)
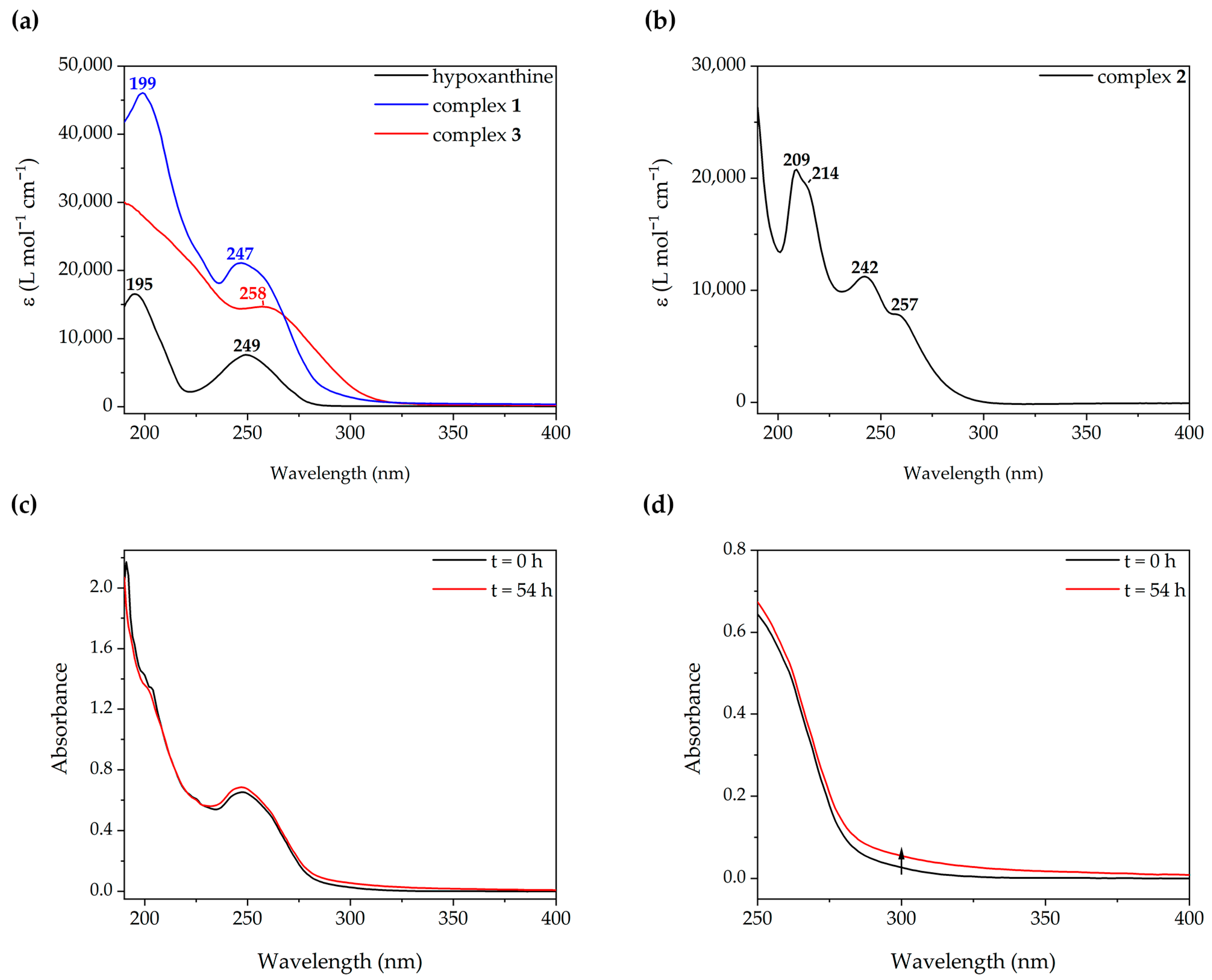
2.3. Photophysical Studies
2.4. Pulsed-Field Gradient Spin Echo NMR Studies
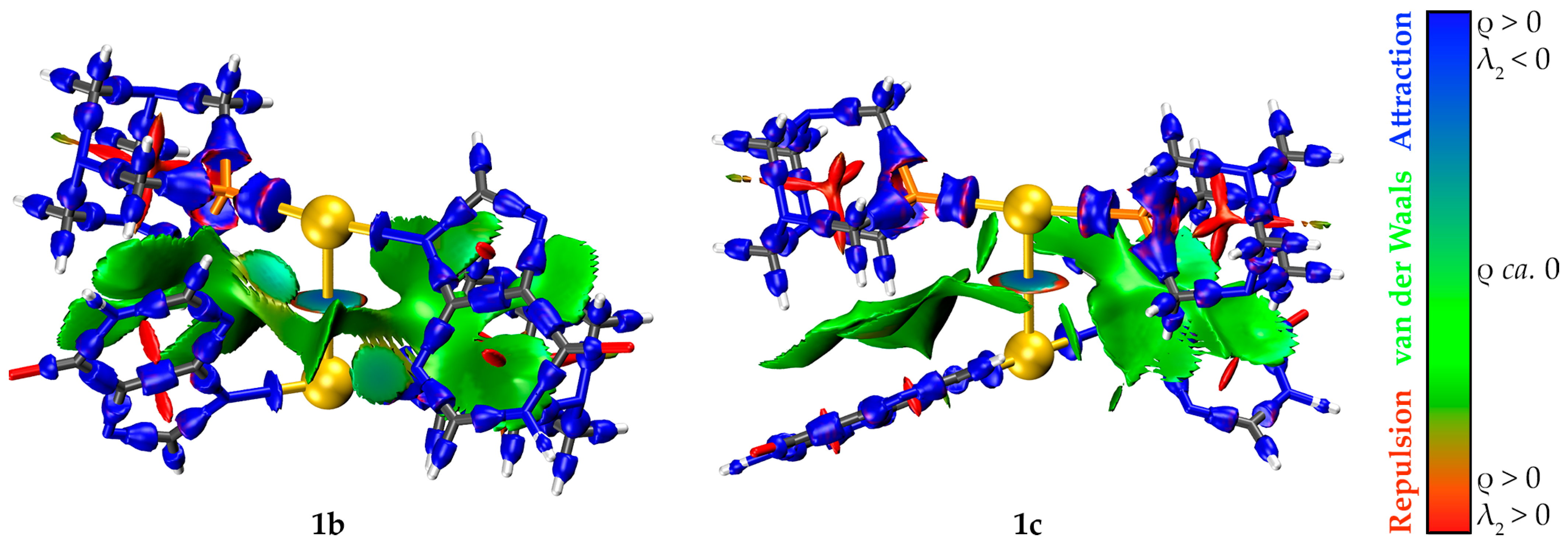
2.5. Computational Studies
3. Materials and Methods
3.1. General Procedures
3.2. Instrumentation
3.3. Computational Details
3.4. Synthesis of Complex [Au(N9-Hypoxanthinate)(PTA)] (1)
3.5. Synthesis of Complex [Au(PTA)2](ClO4) (2)
3.6. Synthesis of Complex (NBu4)[Au(N9-Hypoxanthinate)2] (3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hristova, Y.R.; Kemper, B.; Besenius, P. Water-Soluble Au(I) Complexes, Their Synthesis and Applications. Tetrahedron 2013, 69, 10525–10533. [Google Scholar] [CrossRef]
- Wei, C.; Li, C.-J. A Highly Efficient Three-Component Coupling of Aldehyde, Alkyne, and Amines via C−H Activation Catalyzed by Gold in Water. J. Am. Chem. Soc. 2003, 125, 9584–9585. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Li, C.-J. Water-Triggered and Gold(I)-Catalyzed Cascade Addition/Cyclization of Terminal Alkynes with Ortho-Alkynylaryl Aldehyde. Org. Lett. 2006, 8, 1953–1955. [Google Scholar] [CrossRef] [PubMed]
- Sanz, S.; Jones, L.A.; Mohr, F.; Laguna, M. Homogenous Catalysis with Gold: Efficient Hydration of Phenylacetylene in Aqueous Media. Organometallics 2007, 26, 952–957. [Google Scholar] [CrossRef]
- Elie, B.T.; Levine, C.; Ubarretxena-Belandia, I.; Varela-Ramírez, A.; Aguilera, R.J.; Ovalle, R.; Contel, M. Water-Soluble (Phosphane)Gold(I) Complexes—Applications as Recyclable Catalysts in a Three-Component Coupling Reaction and as Antimicrobial and Anticancer Agents. Eur. J. Inorg. Chem. 2009, 2009, 3421–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, D.; Zhang, X.; Zhou, Y.; Zhang, D.; Zhang, L.; Wang, H.; Jiang, H.; Liu, H. Gold- and Silver-Catalyzed Intramolecular Hydroamination of Terminal Alkynes: Water-Triggered Chemo- and Regioselective Synthesis of Fused Tricyclic Xanthines. Adv. Synth. Catal. 2009, 351, 2770–2778. [Google Scholar] [CrossRef]
- Almássy, A.; Nagy, C.E.; Bényei, A.C.; Joó, F. Novel Sulfonated N-Heterocyclic Carbene Gold(I) Complexes: Homogeneous Gold Catalysis for the Hydration of Terminal Alkynes in Aqueous Media. Organometallics 2010, 29, 2484–2490. [Google Scholar] [CrossRef]
- Czégéni, C.E.; Papp, G.; Kathó, Á.; Joó, F. Water-Soluble Gold(I)–NHC Complexes of Sulfonated IMes and SIMes and Their Catalytic Activity in Hydration of Alkynes. J. Mol. Catal. A Chem. 2011, 340, 1–8. [Google Scholar] [CrossRef]
- Yeo, C.I.; Ooi, K.K.; Tiekink, E.R.T. Gold-Based Medicine: A Paradigm Shift in Anti-Cancer Therapy? Molecules 2018, 23, 1410. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Ma, X.; Chang, X.; Liang, Z.; Lv, L.; Shan, M.; Lu, Q.; Wen, Z.; Gust, R.; Liu, W. Recent Development of Gold(I) and Gold(III) Complexes as Therapeutic Agents for Cancer Diseases. Chem. Soc. Rev. 2022, 51, 5518–5556. [Google Scholar] [CrossRef]
- Moreno-Alcántar, G.; Picchetti, P.; Casini, A. Gold Complexes in Anticancer Therapy: From New Design Principles to Particle-Based Delivery Systems. Angew. Chem. Int. Ed. 2023, 62, e202218000. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Schier, A. A Briefing on Aurophilicity. Chem. Soc. Rev. 2008, 37, 1931–1951. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. Aurophilic Interactions as a Subject of Current Research: An up-Date. Chem. Soc. Rev. 2012, 41, 370–412. [Google Scholar] [CrossRef]
- Mirzadeh, N.; Privér, S.H.; Blake, A.J.; Schmidbaur, H.; Bhargava, S.K. Innovative Molecular Design Strategies in Materials Science Following the Aurophilicity Concept. Chem. Rev. 2020, 120, 7551–7591. [Google Scholar] [CrossRef] [PubMed]
- Romo-Islas, G.; Gavara, R. Recent Progress on Supramolecular Luminescent Assemblies Based on Aurophilic Interactions in Solution. Inorganics 2021, 9, 32. [Google Scholar] [CrossRef]
- Lima, J.; Rodríguez, L. Supramolecular Gold Metallogelators: The Key Role of Metallophilic Interactions. Inorganics 2014, 3, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Blasco, D.; López-de-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Rodríguez-Castillo, M.; Amaveda, H.; Mora, M.; García Sakai, V.; Martínez-González, J.A. Multidisciplinary Study on the Hydrogelation of the Digold(I) Complex [Au(9N-Adeninate)2(μ-dmpe)]: Optical, Rheological, and Quasi-Elastic Neutron Scattering Perspectives. Inorg. Chem. Front. 2021, 8, 3707–3715. [Google Scholar] [CrossRef]
- Niermeier, P.; Wickemeyer, L.; Neumann, B.; Stammler, H.-G.; Goett-Zink, L.; Kottke, T.; Mitzel, N.W. Aurophilicity in Action: Stepwise Formation of Dinuclear Au(I) Macrocycles with Rigid 1,8-Dialkynylanthracenes. Dalton Trans. 2019, 48, 4109–4113. [Google Scholar] [CrossRef] [PubMed]
- Blasco, D.; Rodríguez-Castillo, M.; Olmos, M.E.; Monge, M.; López-de-Luzuriaga, J.M. Spontaneous in Situ Generation of Photoemissive Aurophilic Oligomers in Water Solution Based on the 2-Thiocytosine Ligand. RSC Adv. 2022, 12, 8466–8473. [Google Scholar] [CrossRef]
- Daigle, D.J.; Pepperman, A.B., Jr.; Vail, S.L. Synthesis of a Monophosphorus Analog of Hexamethylenetetramine. J. Heterocycl. Chem. 1974, 11, 407–408. [Google Scholar] [CrossRef]
- Phillips, A.D.; Gonsalvi, L.; Romerosa, A.; Vizza, F.; Peruzzini, M. Coordination Chemistry of 1,3,5-Triaza-7-Phosphaadamantane (PTA): Transition Metal Complexes and Related Catalytic, Medicinal and Photoluminescent Applications. Coord. Chem. Rev. 2004, 248, 955–993. [Google Scholar] [CrossRef]
- Bravo, J.; Bolaño, S.; Gonsalvi, L.; Peruzzini, M. Coordination Chemistry of 1,3,5-Triaza-7-Phosphaadamantane (PTA) and Derivatives. Part II. The Quest for Tailored Ligands, Complexes and Related Applications. Coord. Chem. Rev. 2010, 254, 555–607. [Google Scholar] [CrossRef]
- Guerriero, A.; Peruzzini, M.; Gonsalvi, L. Coordination Chemistry of 1,3,5-Triaza-7-Phosphatricyclo[3.3.1.1]Decane (PTA) and Derivatives. Part III. Variations on a Theme: Novel Architectures, Materials and Applications. Coord. Chem. Rev. 2018, 355, 328–361. [Google Scholar] [CrossRef]
- Siele, V.I. Some Reactions of 1,3,5-Triaza-7-Phosphaadamantane and Its 7-Oxide. J. Heterocycl. Chem. 1977, 14, 337–339. [Google Scholar] [CrossRef]
- Mahmoud, A.G.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. 3,7-Diacetyl-1,3,7-Triaza-5-Phosphabicyclo[3.3.1]Nonane (DAPTA) and Derivatives: Coordination Chemistry and Applications. Coord. Chem. Rev. 2021, 429, 213614. [Google Scholar] [CrossRef]
- Gavara, R.; Llorca, J.; Lima, J.C.; Rodríguez, L. A Luminescent Hydrogel Based on a New Au(I) Complex. Chem. Commun. 2013, 49, 72–74. [Google Scholar] [CrossRef]
- Aguiló, E.; Gavara, R.; Lima, J.C.; Llorca, J.; Rodríguez, L. From Au(I) Organometallic Hydrogels to Well-Defined Au(0) Nanoparticles. J. Mater. Chem. C 2013, 1, 5538–5547. [Google Scholar] [CrossRef]
- Svahn, N.; Sanz, I.; Rissanen, K.; Rodríguez, L. Supramolecular Assemblies and Photophysical Properties of Ionic Homo- and Heteronuclear Metallophilic Complexes. J. Organomet. Chem. 2019, 897, 170–177. [Google Scholar] [CrossRef]
- Blasco, D.; López-de-Luzuriaga, J.M.; Monge, M.; Elena Olmos, M.; Pascual, D.; Rodríguez-Castillo, M. Time-Dependent Molecular Rearrangement of [Au(N9-Adeninate)(PTA)] in Aqueous Solution and Aggregation-Induced Emission in a Hydrogel Matrix. Inorg. Chem. 2021, 60, 3667–3676. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.K.; Choquesillo-Lazarte, D.; Domínguez-Martín, A.; Brandi-Blanco, M.P.; María González-Pérez, J.M.; Castiñeiras, A.; Niclós-Gutiérrez, J. Chelating Ligand Conformation Driving the Hypoxanthine Metal Binding Patterns. Inorg. Chem. 2011, 50, 10549–10551. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.K.; Domínguez-Martín, A.; Brandi-Blanco, M.P.; Choquesillo-Lazarte, D.; Nurchi, V.M.; Niclós-Gutiérrez, J. Metal Ion Binding Modes of Hypoxanthine and Xanthine versus the Versatile Behaviour of Adenine. Coord. Chem. Rev. 2012, 256, 193–211. [Google Scholar] [CrossRef]
- Rosopulos, Y.; Nagel, U.; Beck, W. Metallkomplexe Mit Biologisch Wichtigen Liganden, XXXIV. Allyl-Palladium(II)- Und Triphenylphosphan-Gold(I)-Komplexe Mit Nucleobasen Und Nucleosiden. Chem. Ber. 1985, 118, 931–942. [Google Scholar] [CrossRef]
- Tiekink, E.R.T.; Kurucsev, T.; Hoskins, B.F. X-Ray Structure and UV Spectroscopic Studies of (Adeninato-N9)Triethylphosphinegold(I). J. Crystallogr. Spectrosc. Res. 1989, 19, 823–839. [Google Scholar] [CrossRef]
- Blasco, D.; López-De-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Pascual, D.; Rodríguez-Castillo, M. Cooperative Au(I)⋯Au(I) Interactions and Hydrogen Bonding as Origin of a Luminescent Adeninate Hydrogel Formed by Ultrathin Molecular Nanowires. Inorg. Chem. 2018, 57, 3805–3817. [Google Scholar] [CrossRef]
- Forward, J.M.; Bohmann, D.; Fackler, J.P., Jr.; Staples, R.J. Luminescence Studies of Gold(I) Thiolate Complexes. Inorg. Chem. 2002, 34, 6330–6336. [Google Scholar] [CrossRef]
- Zuccaccia, D.; Macchioni, A. An Accurate Methodology to Identify the Level of Aggregation in Solution by PGSE NMR Measurements: The Case of Half-Sandwich Diamino Ruthenium(II) Salts. Organometallics 2005, 24, 3476–3486. [Google Scholar] [CrossRef]
- Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Determining Accurate Molecular Sizes in Solution through NMR Diffusion Spectroscopy. Chem. Soc. Rev. 2008, 37, 479–489. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; Laguna, M.; Jiménez, J.; Gómez, M.P.; Sainz, A.; Jones, P.G. Gold Complexes with Heterocyclic Thiones as Ligands. X-Ray Structure Determination of [Au(C5H5NS)2]ClO4. J. Chem. Soc. Dalton Trans. 1990, 11, 3457–3463. [Google Scholar] [CrossRef]
- Vicente, J.; Chicote, M.-T.; Saura-Llamas, I.; Lagunas, M.-C. A Facile, Novel Way to Prepare Anionic, Neutral and Cationic Gold(I) Complexes. J. Chem. Soc., Chem. Commun. 1992, 12, 915–916. [Google Scholar] [CrossRef]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular Program Suite for Ab Initio Quantum-Chemical and Condensed-Matter Simulations. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef]
- Treutler, O.; Ahlrichs, R. Efficient Molecular Numerical Integration Schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for Mixing Exact Exchange with Density Functional Approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjusted Ab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary Basis Sets for Main Row Atoms and Transition Metals and Their Use to Approximate Coulomb Potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem.Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Häser, M.; Treutler, O.; Ahlrichs, R. Calculation of Excitation Energies within Time-Dependent Density Functional Theory Using Auxiliary Basis Set Expansions. Chem. Phys. Lett. 1997, 264, 573–578. [Google Scholar] [CrossRef]
- Furche, F.; Rappoport, D. Density Functional Methods for Excited States: Equilibrium Structure and Electronic Spectra. In Computational Photochemistry; Olivucci, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Casida, M.E.; Huix-Rotllant, M. Progress in Time-Dependent Density-Functional Theory. Annu. Rev. Phys. Chem. 2012, 63, 287–323. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Interaction Region Indicator: A Simple Real Space Function Clearly Revealing Both Chemical Bonds and Weak Interactions. Chem.-Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 27–28, 33–38. [Google Scholar] [CrossRef]


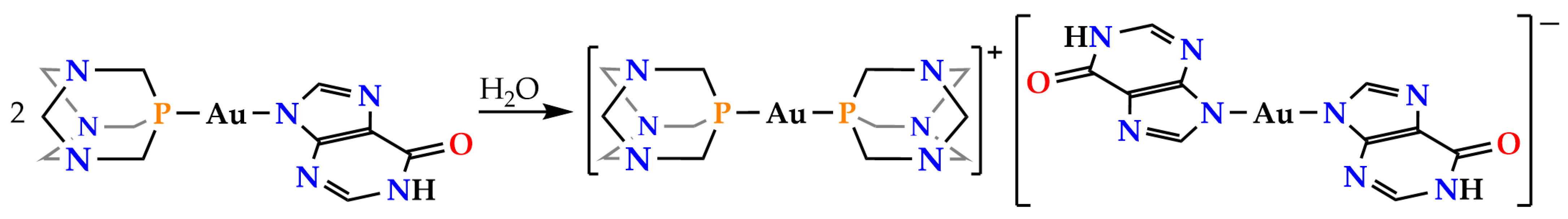








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (h) | PTA Signal (4.32 ppm) | Hypoxanthinate Signal (8.01 ppm) | ||
|---|---|---|---|---|
| Dt·10−10 (m s−1) | rH (Å) | Dt·10−10 (m s−1) | rH (Å) | |
| 0 | 4.16 | 4.93 | 5.80 | 3.54 |
| 48 | 4.13 | 4.96 | 5.51 | 3.72 |
| Excitation | Energy (nm) | Oscillator Strength | Orbital Contributions (%) | |||
|---|---|---|---|---|---|---|
| Model 1a | ||||||
| S1 ← S0 | 253 | 0.00 | HOMO-1 (85) | → | LUMO (87) | (51.8) |
| HOMO (86) | → | LUMO+1 (88) | (32.7) | |||
| S5 ← S0 | 247 | 0.20 | HOMO-1 (85) | → | LUMO+1 (88) | (68.6) |
| HOMO (86) | → | LUMO (87) | (20.5) | |||
| S20 ← S0 | 205 | 0.41 | HOMO-4 (82) | → | LUMO (87) | (74.7) |
| HOMO-1 (85) | → | LUMO+4 (91) | (13.2) | |||
| T1 ← S0 | 336 | - a | HOMO-2 (84) | → | LUMO+2 (89) | (57.2) |
| HOMO-2 (84) | → | LUMO (87) | (34.5) | |||
| Model 1b | ||||||
| S1 ← S0 | 272 | 0.05 | HOMO (172) | → | LUMO (173) | (77.7) |
| HOMO-1 (171) | → | LUMO (173) | (18.1) | |||
| S3 ← S0 | 271 | 0.08 | HOMO-1 (171) | → | LUMO (173) | (63.8) |
| HOMO-2 (170) | → | LUMO (173) | (15.1) | |||
| HOMO (172) | → | LUMO (173) | (14.7) | |||
| S12 ← S0 | 247 | 0.12 | HOMO (172) | → | LUMO+1 (174) | (46.5) |
| HOMO-4 (168) | → | LUMO+1 (174) | (23.6) | |||
| S25 ← S0 | 232 | 0.31 | HOMO-5 (167) | → | LUMO+3 (176) | (43.4) |
| HOMO-4 (168) | → | LUMO+4 (177) | (34.1) | |||
| T1 ← S0 | 337 | - a | HOMO-4 (168) | → | LUMO+1 (174) | (32.9) |
| HOMO-5 (167) | → | LUMO+2 (175) | (31.8) | |||
| Model 1c | ||||||
| S1 ← S0 | 288 | 0.00 | HOMO (172) | → | LUMO+1 (174) | (96.1) |
| S4 ← S0 | 281 | 0.13 | HOMO-2 (170) | → | LUMO+1 (174) | (60.4) |
| HOMO (172) | → | LUMO (173) | (19.8) | |||
| HOMO-1 (171) | → | LUMO+1 (174) | (16.8) | |||
| S5 ← S0 | 281 | 0.13 | HOMO (172) | → | LUMO (173) | (44.4) |
| HOMO-2 (170) | → | LUMO+1 (174) | (34.9) | |||
| HOMO-1 (171) | → | LUMO+1 (174) | (17.5) | |||
| S14 ← S0 | 253 | 0.36 | HOMO-6 (166) | → | LUMO (173) | (90.5) |
| T1 ← S0 | 336 | - a | HOMO-4 (168) | → | LUMO+2 (175) | (42.5) |
| HOMO-5 (167) | → | LUMO+3 (176) | (40.8) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Gobernado, A.; Blasco, D.; Monge, M.; López-de-Luzuriaga, J.M. Spontaneous Water-Promoted Self-Aggregation of a Hydrophilic Gold(I) Complex Due to Ligand Sphere Rearrangement. Molecules 2023, 28, 5680. https://doi.org/10.3390/molecules28155680
Rodríguez-Gobernado A, Blasco D, Monge M, López-de-Luzuriaga JM. Spontaneous Water-Promoted Self-Aggregation of a Hydrophilic Gold(I) Complex Due to Ligand Sphere Rearrangement. Molecules. 2023; 28(15):5680. https://doi.org/10.3390/molecules28155680
Chicago/Turabian StyleRodríguez-Gobernado, Ainhoa, Daniel Blasco, Miguel Monge, and José M. López-de-Luzuriaga. 2023. "Spontaneous Water-Promoted Self-Aggregation of a Hydrophilic Gold(I) Complex Due to Ligand Sphere Rearrangement" Molecules 28, no. 15: 5680. https://doi.org/10.3390/molecules28155680