

Significance of Five-Membered Heterocycles in Human Histone Deacetylase Inhibitors

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. ZBG Scaffolds
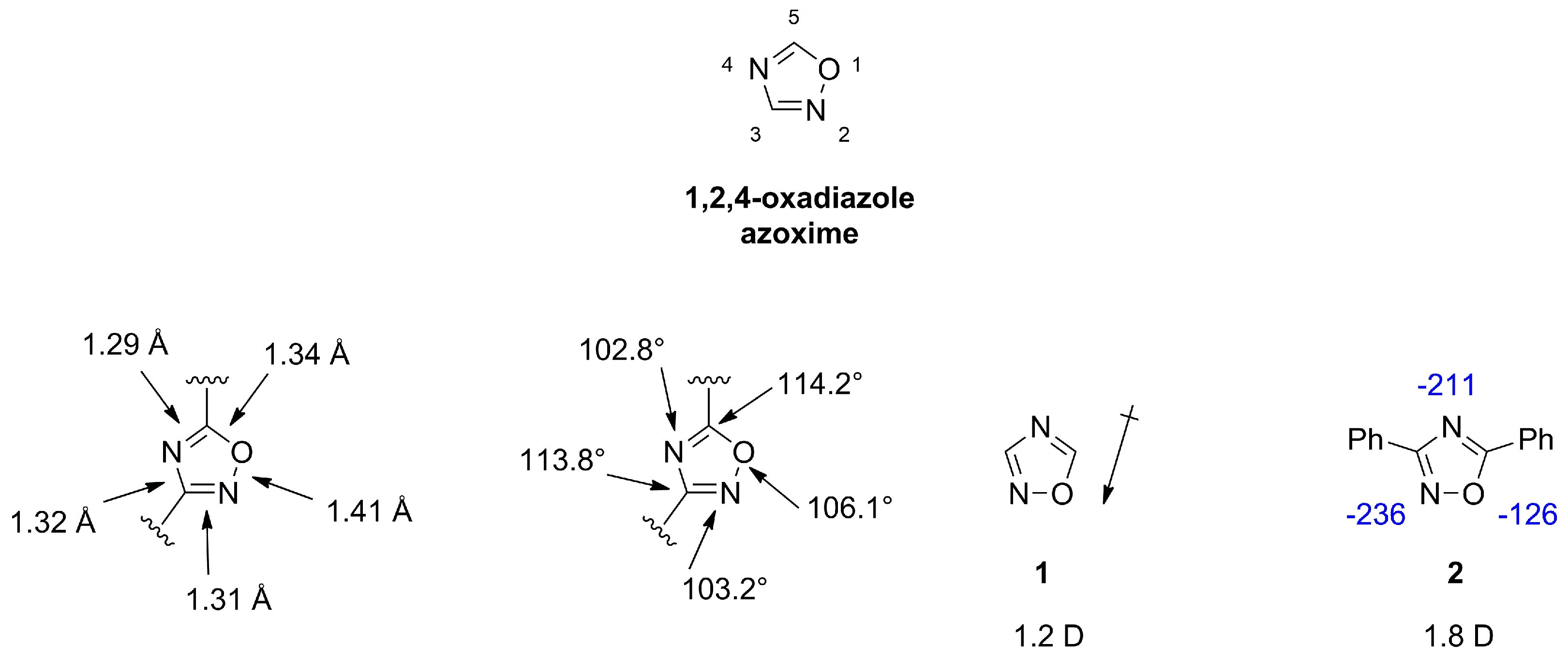
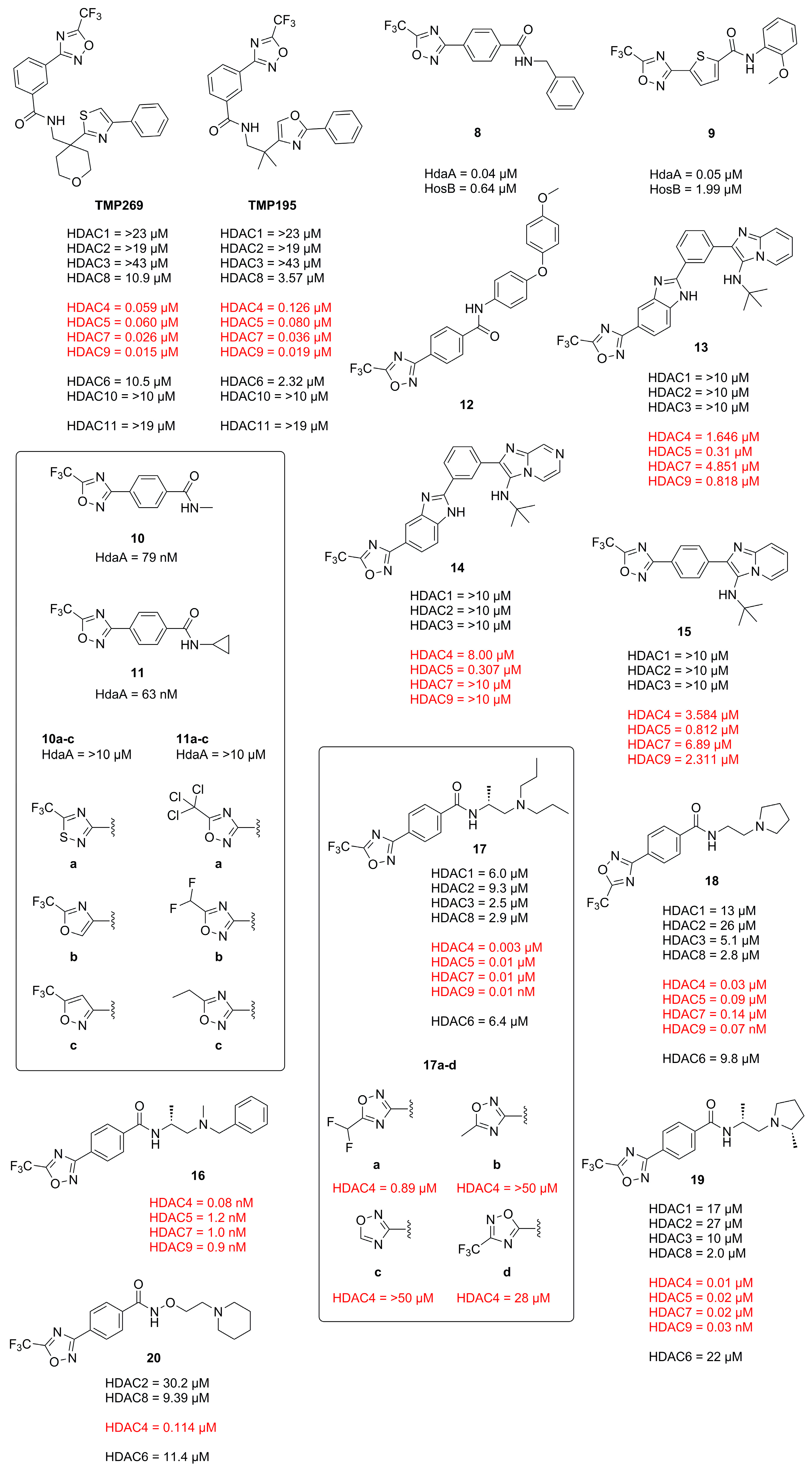
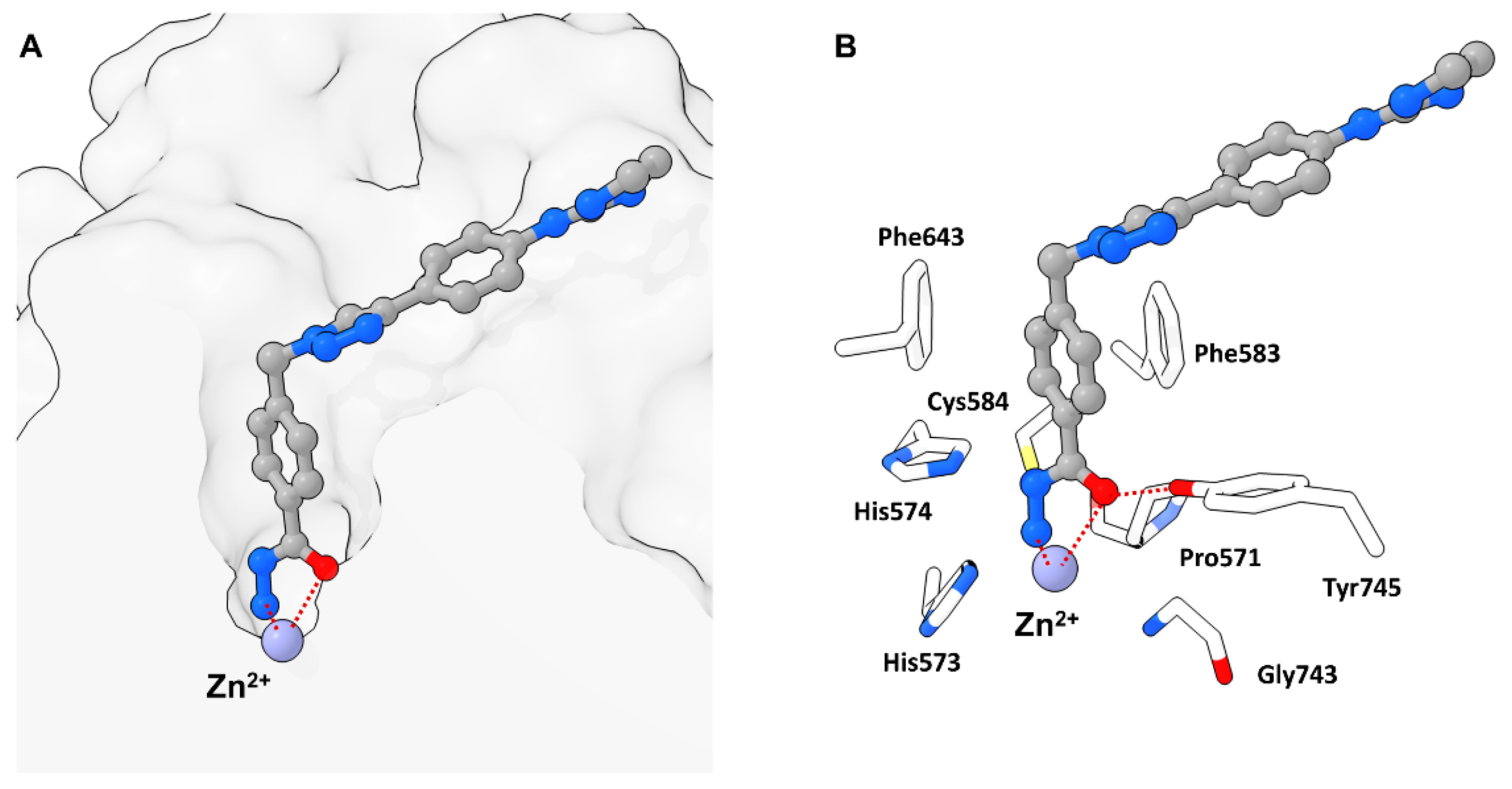
2.1. 1,2,4-Oxadiazoles
2.2. 1,3,4-Oxadiazoles
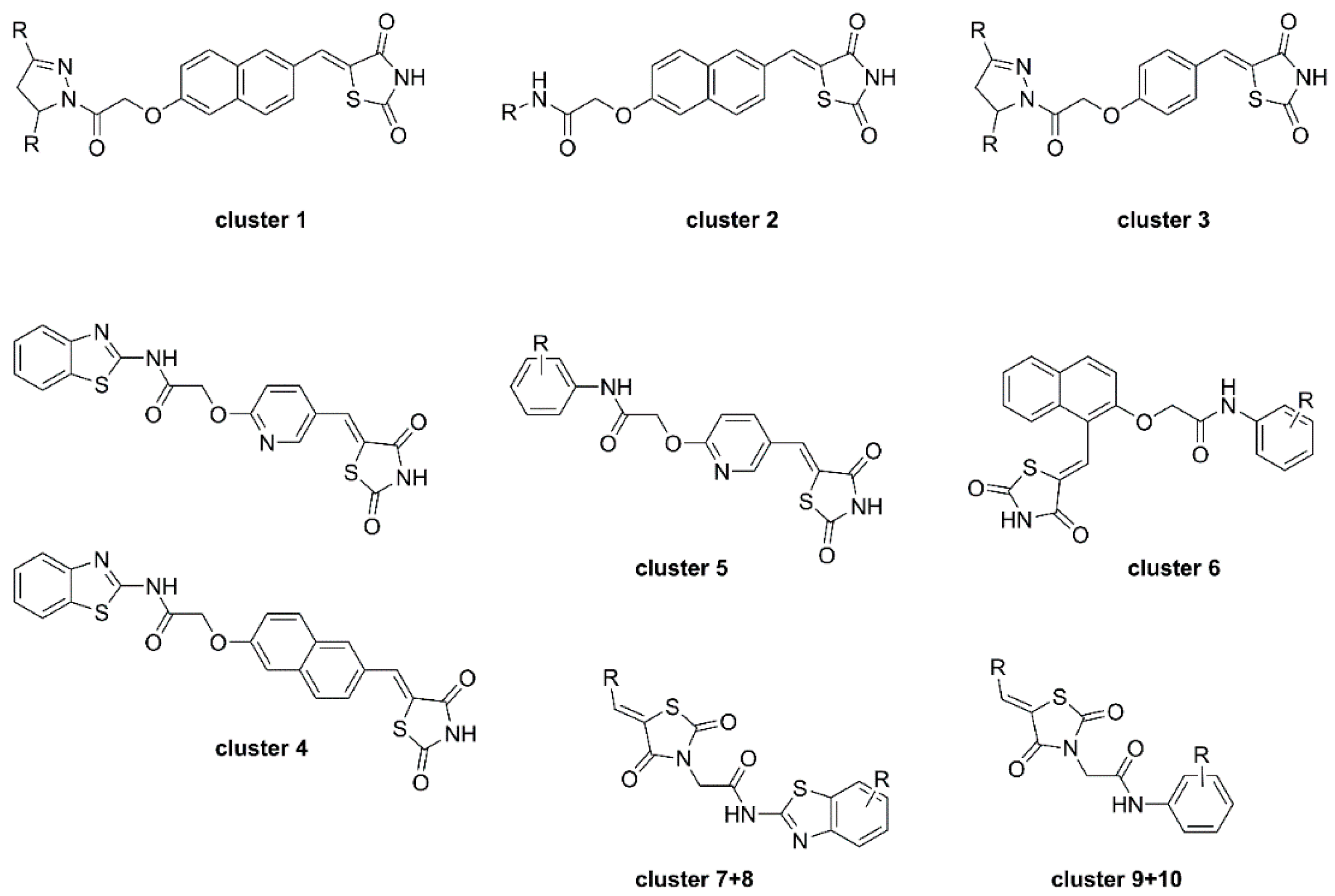
2.3. Thiazolidine-2,4-Diones
2.4. Miscellaneous Heterocycles
3. Cap Group or Linker
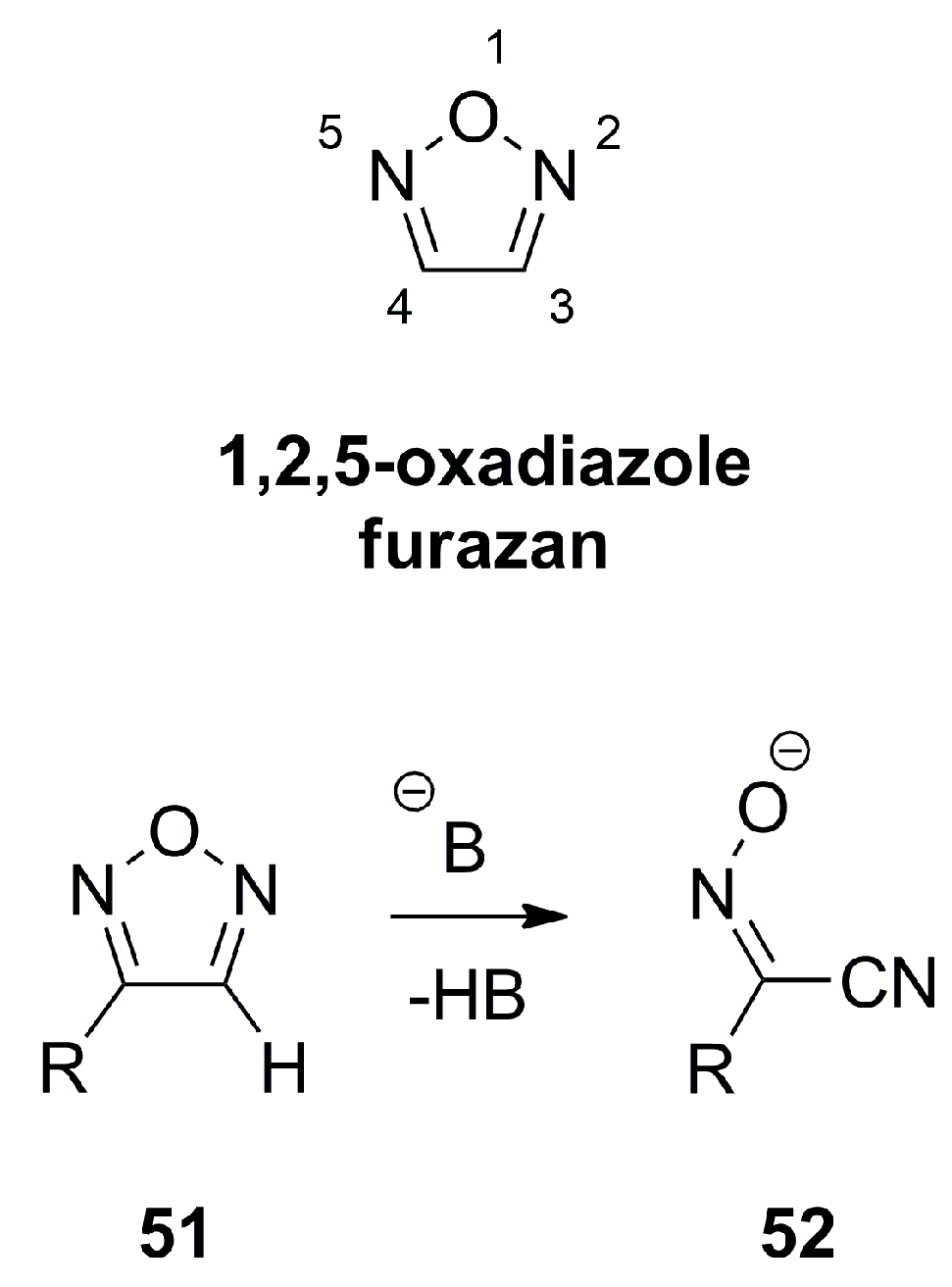
3.1. 1,2,5-Oxadiazoles
3.2. Triazoles
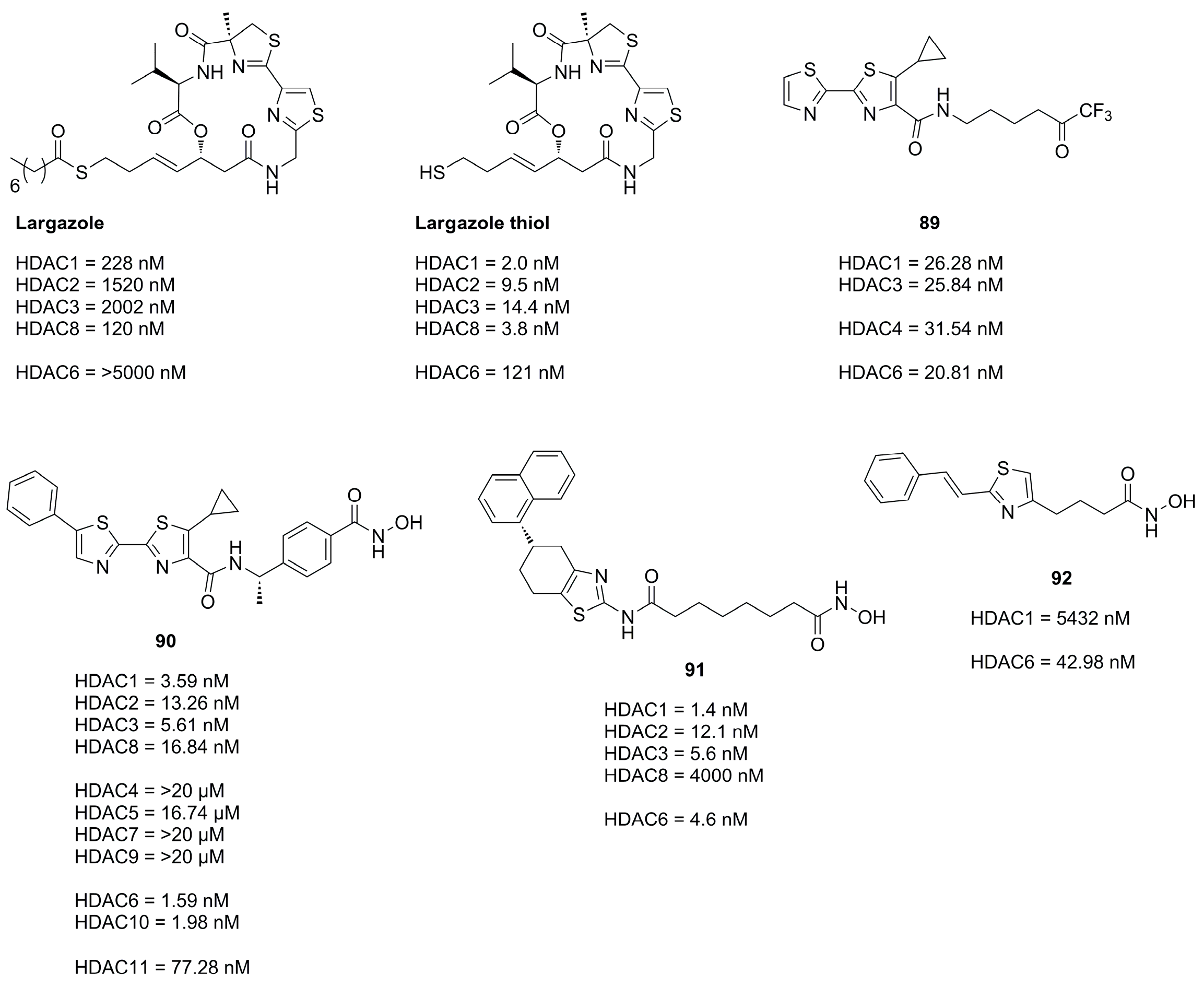
3.3. Thiazoles
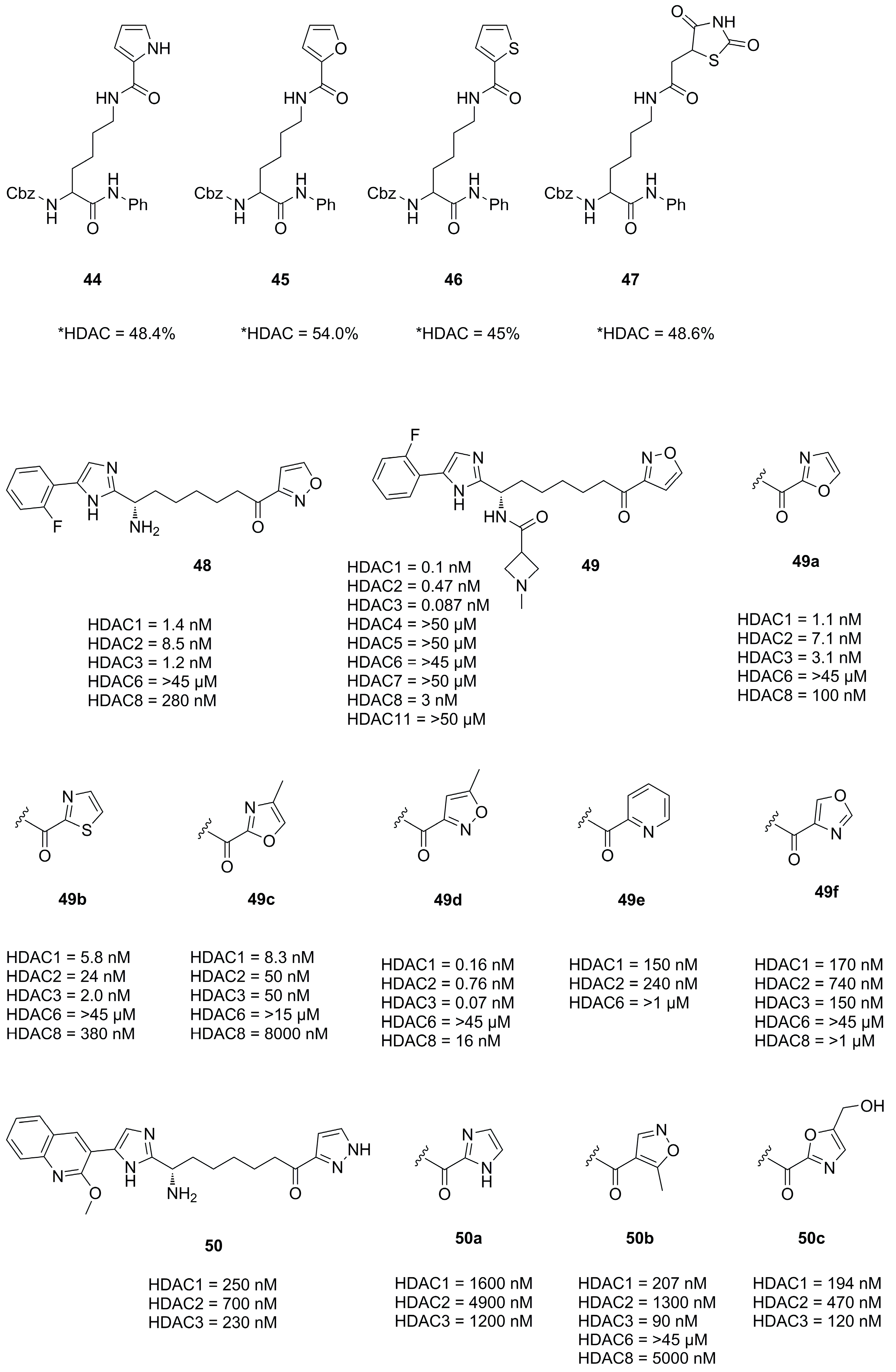
3.4. Imidazoles and Oxazoles
3.5. Pyrazoles
3.6. Thiophenes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| ADME | absorption, distribution, metabolism and excretion |
| AML | acute myeloid leukemia |
| ATP | adenosine triphosphate |
| AUC | Area under curve |
| BBB | blood brain barrier |
| BTZ | Bortezomib |
| CAM | chick chorioallantois membranes |
| CD2 | catalytic domain 2 |
| Cmax | maximum serum concentration of drug in a specified compartment of the body |
| conc. | concentrated |
| CRBN | E3 ubiquitin cereblon ligase |
| DFMO | difluoromethyl-1,3,4-oxadiazole |
| FA | formic acid |
| GI50 | concentration that causes 50% growth inhibition of cells |
| HB | hydrogen bonding |
| HD | Huntington disease |
| HDAC | Histone deacetylase |
| HDACi | Histone deacetylase inhibitor |
| HTS | hight throughput screening |
| IC50 | concentration that causes 50% enzyme inhibition |
| nM | nanomolar |
| PBMCs | peripheral blood mononuclear cells |
| PHA | phytohemagglutinin |
| PROTAC | proteolysis-targeting chimeras |
| SIRT | Sirtuins |
| smHDAC | HDAC from Schistosoma mansoni |
| t1/2 | half-life period |
| TFMO | trifluoro-1,2,4-oxadiazole |
| Tmax | time of peak plasma concentration |
| VHL | Von-Huppel–Lindau ligase |
| ZBG | Zinc binding group |
| zHDAC6 | zebrafish HDAC6 |
References
- Glomb, T.; Szymankiewicz, K.; Świątek, P. Anti-Cancer Activity of Derivatives of 1,3,4-Oxadiazole. Molecules 2018, 23, 3361. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in Medicinal Chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Carmona, A.V.; Tiwari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective Class IIa Histone Deacetylase Inhibition via a Nonchelating Zinc-Binding Group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Scarpelli, R.; Di Marco, A.; Ferrigno, F.; Laufer, R.; Marcucci, I.; Muraglia, E.; Ontoria, J.M.; Rowley, M.; Serafini, S.; Steinkühler, C.; et al. Studies of the Metabolic Stability in Cells of 5-(Trifluoroacetyl)Thiophene-2-Carboxamides and Identification of More Stable Class II Histone Deacetylase (HDAC) Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 6078–6082. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.J.; Inks, E.S.; Zhang, C.; Peterson, Y.K.; Li, J.; Chundru, K.; Lee, B.; Buchanan, A.; Miao, S.; Chou, C.J. Comparison of the Deacylase and Deacetylase Activity of Zinc-Dependent HDACs. ACS Chem. Biol. 2017, 12, 1644–1655. [Google Scholar] [CrossRef]
- Duvic, M.; Vu, J. Vorinostat in Cutaneous T-Cell Lymphoma. Drugs Today 2007, 43, 585. [Google Scholar] [CrossRef]
- Campàs-Moya, C. Romidepsin for the Treatment of Cutaneous T-Cell Lymphoma. Drugs Today 2009, 45, 787. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.-H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 4. [Google Scholar]
- Lu, X.; Ning, Z.; Li, Z.; Cao, H.; Wang, X. Development of Chidamide for Peripheral T-Cell Lymphoma, the First Orphan Drug Approved in China. Intractable Rare Dis. Res. 2016, 5, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.P.; Di Costanzo, L.; Gennadios, H.A.; Christianson, D.W. Evolution of the Arginase Fold and Functional Diversity. Cell. Mol. Life Sci. 2008, 65, 2039–2055. [Google Scholar] [CrossRef]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, Mechanism, and Inhibition of Histone Deacetylases and Related Metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis: UCSF ChimeraX Visualization System. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Romeo, G.; Chiacchio, U. Oxadiazoles. In Modern Heterocyclic Chemistry; Alvarez-Builla, J., Vaquero, J.J., Barluenga, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 1047–1252. ISBN 978-3-527-63773-7. [Google Scholar]
- Bird, C.W. A New Aromaticity Index and Its Application to Five-Membered Ring Heterocycles. Tetrahedron 1985, 41, 1409–1414. [Google Scholar] [CrossRef]
- Clapp, L.B. 1,2,3- and 1,2,4-Oxadiazoles. In Comprehensive Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 1984; pp. 365–391. ISBN 978-0-08-096519-2. [Google Scholar]
- Winter, C.; Fehr, M.; Craig, I.R.; Grammenos, W.; Wiebe, C.; Terteryan-Seiser, V.; Rudolf, G.; Mentzel, T.; Quintero Palomar, M.A. Trifluoromethyloxadiazoles: Inhibitors of Histone Deacetylases for Control of Asian Soybean Rust. Pest. Manag. Sci. 2020, 76, 3357–3368. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, Q.; Sun, Y.; Sun, X.; Chen, L.; Sun, L.; Gu, W. Synthesis, Antifungal Activity, DFT Study and Molecular Dynamics Simulation of Novel 4-(1,2,4-Oxadiazol-3-Yl)-N-(4-Phenoxyphenyl)Benzamide Derivatives. Chem. Biodivers. 2021, 18, e2100651. [Google Scholar] [CrossRef]
- El-Awady, R.; Saleh, E.; Hamoudi, R.; Ramadan, W.S.; Mazitschek, R.; Nael, M.A.; Elokely, K.M.; Abou-Gharbia, M.; Childers, W.E.; Srinivasulu, V.; et al. Discovery of Novel Class of Histone Deacetylase Inhibitors as Potential Anticancer Agents. Bioorg. Med. Chem. 2021, 42, 116251. [Google Scholar] [CrossRef] [PubMed]
- Turkman, N.; Liu, D.; Pirola, I. Novel Late-Stage Radiosynthesis of 5-[18F]-Trifluoromethyl-1,2,4-Oxadiazole (TFMO) Containing Molecules for PET Imaging. Sci. Rep. 2021, 11, 10668. [Google Scholar] [CrossRef] [PubMed]
- Turkman, N.; Liu, D.; Pirola, I. Design, Synthesis, Biochemical Evaluation, Radiolabeling and in Vivo Imaging with High Affinity Class-IIa Histone Deacetylase Inhibitor for Molecular Imaging and Targeted Therapy. Eur. J. Med. Chem. 2022, 228, 114011. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Landles, C.; Weiss, A.; Bradaia, A.; Seredenina, T.; Inuabasi, L.; Osborne, G.F.; Wadel, K.; Touller, C.; Butler, R.; et al. HDAC4 Reduction: A Novel Therapeutic Strategy to Target Cytoplasmic Huntingtin and Ameliorate Neurodegeneration. PLoS Biol. 2013, 11, e1001717. [Google Scholar] [CrossRef]
- Stott, A.J.; Maillard, M.C.; Beaumont, V.; Allcock, D.; Aziz, O.; Borchers, A.H.; Blackaby, W.; Breccia, P.; Creighton-Gutteridge, G.; Haughan, A.F.; et al. Evaluation of 5-(Trifluoromethyl)-1,2,4-Oxadiazole-Based Class IIa HDAC Inhibitors for Huntington’s Disease. ACS Med. Chem. Lett. 2021, 12, 380–388. [Google Scholar] [CrossRef]
- Hebach, C.; Kallen, J.; Nozulak, J.; Tintelnot-Blomley, M.; Widler, L. Novel Trifluoromethyl-Oxadiazole Derivatives and Their Use in the Treatment of. Disease. Patent WO 2013/080120 A1, 6 June 2013. [Google Scholar]
- Bollmann, L.M.; Skerhut, A.J.; Asfaha, Y.; Horstick, N.; Hanenberg, H.; Hamacher, A.; Kurz, T.; Kassack, M.U. The Novel Class IIa Selective Histone Deacetylase Inhibitor YAK540 Is Synergistic with Bortezomib in Leukemia Cell Lines. Int. J. Mol. Sci. 2022, 23, 13398. [Google Scholar] [CrossRef]
- Csizmar, C.M.; Kim, D.-H.; Sachs, Z. The Role of the Proteasome in AML. Blood Cancer J. 2016, 6, e503. [Google Scholar] [CrossRef]
- Lee, J.; Han, Y.; Kim, Y.; Min, J.; Bae, M.; Kim, D.; Jin, S.; Kyung, J. 1,3,4-Oxadiazole Sulfamide Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the. Same. Patent WO 2017/018805 A1, 2 February 2017. [Google Scholar]
- Kim, Y.; Lee, C.S.; Oh, J.T.; Song, H.; Choi, J.; Lee, J. Oxadiazole Amine Derivative Compounds as Histone Deacetylase 6 Inhibitor, and the Pharmaceutical Composition Comprising the Same. WO 2017/065473 A1, 20 April 2017. [Google Scholar]
- Cellupica, E.; Caprini, G.; Cordella, P.; Cukier, C.; Fossati, G.; Marchini, M.; Rocchio, I.; Sandrone, G.; Vanoni, M.A.; Vergani, B.; et al. Difluoromethyl-1,3,4-Oxadiazoles Are Slow-Binding Substrate Analog Inhibitors of Histone Deacetylase 6 with Unprecedented Isotype Selectivity. J. Biol. Chem. 2023, 299, 102800. [Google Scholar] [CrossRef]
- König, B.; Watson, P.R.; Reßing, N.; Cragin, A.D.; Schäker-Hübner, L.; Christianson, D.W.; Hansen, F.K. Difluoromethyl-1,3,4-oxadiazoles are selective, mechanism-based, and essentially irreversible inhibitors of histone deacetylase 6. ChemRxiv, 2023; preprint. [Google Scholar]
- Keuler, T.; König, B.; Bückreiß, N.; Kraft, F.B.; König, P.; Schäker-Hübner, L.; Steinebach, C.; Bendas, G.; Gütschow, M.; Hansen, F.K. Development of the First Non-Hydroxamate Selective HDAC6 Degraders. Chem. Commun. 2022, 58, 11087–11090. [Google Scholar] [CrossRef]
- Onishi, T.; Maeda, R.; Terada, M.; Sato, S.; Fujii, T.; Ito, M.; Hashikami, K.; Kawamoto, T.; Tanaka, M. A Novel Orally Active HDAC6 Inhibitor T-518 Shows a Therapeutic Potential for Alzheimer’s Disease and Tauopathy in Mice. Sci. Rep. 2021, 11, 15423. [Google Scholar] [CrossRef]
- Ptacek, J.; Snajdr, I.; Schimer, J.; Kutil, Z.; Mikesova, J.; Baranova, P.; Havlinova, B.; Tueckmantel, W.; Majer, P.; Kozikowski, A.; et al. Selectivity of Hydroxamate- and Difluoromethyloxadiazole-Based Inhibitors of Histone Deacetylase 6 In Vitro and in Cells. Int. J. Mol. Sci. 2023, 24, 4720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.-L.; Chua, K.; et al. Mice Lacking Histone Deacetylase 6 Have Hyperacetylated Tubulin but Are Viable and Develop Normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.M. Metalloenzyme Inhibitor Compounds. U.S. Patent US 10,357,493 B2, 23 July 2019. [Google Scholar]
- Form, G.R.; Raper, E.S.; Downie, T.C. The Crystal and Molecular Structure of Thiazolidine-2,4-Dione. Acta Cryst. B. Struct. Cryst. Cryst. Chem. 1975, 31, 2181–2184. [Google Scholar] [CrossRef]
- Enchev, V.; Chorbadjiev, S.; Jordanov, B. Comparative Study of the Structure of Rhodanine, Isorhodanine, Thiazolidine-2,4-Dione, and Thiorhodanine. Chem. Heterocycl. Compd. 2002, 38, 1110–1120. [Google Scholar] [CrossRef]
- Valls, N.; Segarra, V.M.; Alcalde, E.; Marin, A.; Elguero, J. Synthesis, Spectroscopy and Tautomeric Study of Thiazoles Substituted in Positions 2 and 4 by Hydroxy, Mercapto and Amino Groups. J. Prakt. Chem. 1985, 327, 251–260. [Google Scholar] [CrossRef]
- Sohda, T.; Mizuno, K.; Kawamatsu, Y. Studies on Antidiabetic Agents. VI. Asymmetric Transformation of (.+-.)-5-(4-(1-Methylcyclohexylmethoxy)Benzyl)-2,4-Thiazolidinedione(Ciglitazone) with Optically Active 1-Phenylethylamines. Chem. Pharm. Bull. 1984, 32, 4460–4465. [Google Scholar] [CrossRef]
- Bharatam, P.V.; Khanna, S. Rapid Racemization in Thiazolidinediones: A Quantum Chemical Study. J. Phys. Chem. A 2004, 108, 3784–3788. [Google Scholar] [CrossRef]
- Ibrahim, A.M.; Shoman, M.E.; Mohamed, M.F.A.; Hayallah, A.M.; Abuo-Rahma, G.E.-D.A. Chemistry and Applications of Functionalized 2,4-Thiazolidinediones. Eur. J. Org. Chem. 2023, 26, e202300184. [Google Scholar] [CrossRef]
- Chrysanthopoulos, P.K.; Mujumdar, P.; Woods, L.A.; Dolezal, O.; Ren, B.; Peat, T.S.; Poulsen, S.-A. Identification of a New Zinc Binding Chemotype by Fragment Screening. J. Med. Chem. 2017, 60, 7333–7349. [Google Scholar] [CrossRef]
- Tilekar, K.; Upadhyay, N.; Jänsch, N.; Schweipert, M.; Mrowka, P.; Meyer-Almes, F.J.; Ramaa, C.S. Discovery of 5-Naphthylidene-2,4-Thiazolidinedione Derivatives as Selective HDAC8 Inhibitors and Evaluation of Their Cytotoxic Effects in Leukemic Cell Lines. Bioorg. Chem. 2020, 95, 103522. [Google Scholar] [CrossRef]
- Upadhyay, N.; Tilekar, K.; Jänsch, N.; Schweipert, M.; Hess, J.D.; Henze Macias, L.; Mrowka, P.; Aguilera, R.J.; Choe, J.; Meyer-Almes, F.-J.; et al. Discovery of Novel N-Substituted Thiazolidinediones (TZDs) as HDAC8 Inhibitors: In-Silico Studies, Synthesis, and Biological Evaluation. Bioorg. Chem. 2020, 100, 103934. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Sharma, A.K.; Gupta, S.; Ramaa, C.S. Design, Synthesis, and Biological Evaluation of Novel 2,4-Thiazolidinedione Derivatives as Histone Deacetylase Inhibitors Targeting Liver Cancer Cell Line. Med. Chem. Res. 2012, 21, 1156–1165. [Google Scholar] [CrossRef]
- Hou, X.; Du, J.; Liu, R.; Zhou, Y.; Li, M.; Xu, W.; Fang, H. Enhancing the Sensitivity of Pharmacophore-Based Virtual Screening by Incorporating Customized ZBG Features: A Case Study Using Histone Deacetylase 8. J. Chem. Inf. Model. 2015, 55, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Tilekar, K.; Hess, J.D.; Upadhyay, N.; Schweipert, M.; Flath, F.; Gutierrez, D.A.; Loiodice, F.; Lavecchia, A.; Meyer-Almes, F.; Aguilera, R.J.; et al. HDAC4 Inhibitors with Cyclic Linker and Non-hydroxamate Zinc Binding Group: Design, Synthesis, HDAC Screening and in Vitro Cytotoxicity Evaluation. ChemistrySelect 2021, 6, 6748–6763. [Google Scholar] [CrossRef]
- Tilekar, K.; Hess, J.D.; Upadhyay, N.; Bianco, A.L.; Schweipert, M.; Laghezza, A.; Loiodice, F.; Meyer-Almes, F.-J.; Aguilera, R.J.; Lavecchia, A.; et al. Thiazolidinedione “Magic Bullets” Simultaneously Targeting PPARγ and HDACs: Design, Synthesis, and Investigations of Their In Vitro and In Vivo Antitumor Effects. J. Med. Chem. 2021, 64, 6949–6971. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, N.; Tilekar, K.; Safuan, S.; Kumar, A.P.; Schweipert, M.; Meyer-Almes, J.; Ramaa, C.S. Multi-Target Weapons: Diaryl-Pyrazoline Thiazolidinediones Simultaneously Targeting VEGFR-2 and HDAC Cancer Hallmarks. RSC Med. Chem. 2021, 12, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, N.; Tilekar, K.; Safuan, S.; Kumar, A.P.; Schweipert, M.; Meyer-Almes, F.-J.; Ramaa, C.S. Double-Edged Swords: Diaryl Pyrazoline Thiazolidinediones Synchronously Targeting Cancer Epigenetics and Angiogenesis. Bioorg. Chem. 2021, 116, 105350. [Google Scholar] [CrossRef]
- Schweipert, M.; Jänsch, N.; Upadhyay, N.; Tilekar, K.; Wozny, E.; Basheer, S.; Wurster, E.; Müller, M.; CS, R.; Meyer-Almes, F.-J. Mechanistic Insights into Binding of Ligands with Thiazolidinedione Warhead to Human Histone Deacetylase 4. Pharmaceuticals 2021, 14, 1032. [Google Scholar] [CrossRef]
- Wang, F.; Wang, C.; Wang, J.; Zou, Y.; Chen, X.; Liu, T.; Li, Y.; Zhao, Y.; Li, Y.; He, B. Nɛ-Acetyl Lysine Derivatives with Zinc Binding Groups as Novel HDAC Inhibitors. R. Soc. Open. Sci. 2019, 6, 190338. [Google Scholar] [CrossRef]
- Liu, J.; Kelly, J.; Yu, W.; Clausen, D.; Yu, Y.; Kim, H.; Duffy, J.L.; Chung, C.C.; Myers, R.W.; Carroll, S.; et al. Selective Class I HDAC Inhibitors Based on Aryl Ketone Zinc Binding Induce HIV-1 Protein for Clearance. ACS Med. Chem. Lett. 2020, 11, 1476–1483. [Google Scholar] [CrossRef]
- Tu, S.; Yuan, H.; Hu, J.; Zhao, C.; Chai, R.; Cao, H. Design, Synthesis and Biological Evaluation of Nitro Oxide Donating N-Hydroxycinnamamide Derivatives as Histone Deacetylase Inhibitors. Chem. Pharm. Bull. 2014, 62, 1185–1191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duan, W.; Hou, J.; Chu, X.; Li, X.; Zhang, J.; Li, J.; Xu, W.; Zhang, Y. Synthesis and Biological Evaluation of Novel Histone Deacetylases Inhibitors with Nitric Oxide Releasing Activity. Bioorg. Med. Chem. 2015, 23, 4481–4488. [Google Scholar] [CrossRef] [PubMed]
- Illi, B.; Colussi, C.; Grasselli, A.; Farsetti, A.; Capogrossi, M.C.; Gaetano, C. NO Sparks off Chromatin: Tales of a Multifaceted Epigenetic Regulator. Pharmacol. Ther. 2009, 123, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Borretto, E.; Lazzarato, L.; Spallotta, F.; Cencioni, C.; D’Alessandra, Y.; Gaetano, C.; Fruttero, R.; Gasco, A. Synthesis and Biological Evaluation of the First Example of NO-Donor Histone Deacetylase Inhibitor. ACS Med. Chem. Lett. 2013, 4, 994–999. [Google Scholar] [CrossRef][Green Version]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 Blockade by Nitric Oxide and Histone Deacetylase Inhibitors Reveals a Common Target in Duchenne Muscular Dystrophy Treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef]
- Cai, J.; Wei, H.; Hong, K.H.; Wu, X.; Zong, X.; Cao, M.; Wang, P.; Li, L.; Sun, C.; Chen, B.; et al. Discovery, Bioactivity and Docking Simulation of Vorinostat Analogues Containing 1,2,4-Oxadiazole Moiety as Potent Histone Deacetylase Inhibitors and Antitumor Agents. Bioorg. Med. Chem. 2015, 23, 3457–3471. [Google Scholar] [CrossRef]
- Cai, J.; Wei, H.; Hong, K.H.; Wu, X.; Cao, M.; Zong, X.; Li, L.; Sun, C.; Chen, J.; Ji, M. Discovery and Preliminary Evaluation of 2-Aminobenzamide and Hydroxamate Derivatives Containing 1,2,4-Oxadiazole Moiety as Potent Histone Deacetylase Inhibitors. Eur. J. Med. Chem. 2015, 96, 1–13. [Google Scholar] [CrossRef]
- Guan, P.; Wang, L.; Hou, X.; Wan, Y.; Xu, W.; Tang, W.; Fang, H. Improved Antiproliferative Activity of 1,3,4-Thiadiazole-Containing Histone Deacetylase (HDAC) Inhibitors by Introduction of the Heteroaromatic Surface Recognition Motif. Bioorg. Med. Chem. 2014, 22, 5766–5775. [Google Scholar] [CrossRef]
- Yang, F.; Shan, P.; Zhao, N.; Ge, D.; Zhu, K.; Jiang, C.-S.; Li, P.; Zhang, H. Development of Hydroxamate-Based Histone Deacetylase Inhibitors Containing 1,2,4-Oxadiazole Moiety Core with Antitumor Activities. Bioorg. Med. Chem. Lett. 2019, 29, 15–21. [Google Scholar] [CrossRef]
- Pidugu, V.R.; Yarla, N.S.; Pedada, S.R.; Kalle, A.M.; Satya, A.K. Design and Synthesis of Novel HDAC8 Inhibitory 2,5-Disubstituted-1,3,4-Oxadiazoles Containing Glycine and Alanine Hybrids with Anti Cancer Activity. Bioorg. Med. Chem. 2016, 24, 5611–5617. [Google Scholar] [CrossRef]
- Pidugu, V.R.; Yarla, N.S.; Bishayee, A.; Kalle, A.M.; Satya, A.K. Novel Histone Deacetylase 8-Selective Inhibitor 1,3,4-Oxadiazole-Alanine Hybrid Induces Apoptosis in Breast Cancer Cells. Apoptosis 2017, 22, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shen, M.; Tang, M.; Zhang, W.; Cui, X.; Zhang, Z.; Pei, H.; Li, Y.; Hu, M.; Bai, P.; et al. Discovery of 1,2,4-Oxadiazole-Containing Hydroxamic Acid Derivatives as Histone Deacetylase Inhibitors Potential Application in Cancer Therapy. Eur. J. Med. Chem. 2019, 178, 116–130. [Google Scholar] [CrossRef]
- Suzuki, T.; Ota, Y.; Kasuya, Y.; Mutsuga, M.; Kawamura, Y.; Tsumoto, H.; Nakagawa, H.; Finn, M.G.; Miyata, N. An Unexpected Example of Protein-Templated Click Chemistry. Angew. Chem. 2010, 122, 6969–6972. [Google Scholar] [CrossRef]
- Suzuki, T.; Muto, N.; Bando, M.; Itoh, Y.; Masaki, A.; Ri, M.; Ota, Y.; Nakagawa, H.; Iida, S.; Shirahige, K.; et al. Design, Synthesis, and Biological Activity of NCC149 Derivatives as Histone Deacetylase 8-Selective Inhibitors. ChemMedChem 2014, 9, 657–664. [Google Scholar] [CrossRef]
- Suzuki, T.; Kasuya, Y.; Itoh, Y.; Ota, Y.; Zhan, P.; Asamitsu, K.; Nakagawa, H.; Okamoto, T.; Miyata, N. Identification of Highly Selective and Potent Histone Deacetylase 3 Inhibitors Using Click Chemistry-Based Combinatorial Fragment Assembly. PLoS ONE 2013, 8, e68669. [Google Scholar] [CrossRef] [PubMed]
- Ingham, O.J.; Paranal, R.M.; Smith, W.B.; Escobar, R.A.; Yueh, H.; Snyder, T.; Porco, J.A.; Bradner, J.E.; Beeler, A.B. Development of a Potent and Selective HDAC8 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Huong, T.-T.-L.; Dung, D.-T.-M.; Huan, N.-V.; Cuong, L.-V.; Hai, P.-T.; Huong, L.-T.-T.; Kim, J.; Kim, Y.-G.; Han, S.-B.; Nam, N.-H. Novel N -Hydroxybenzamides Incorporating 2-Oxoindoline with Unexpected Potent Histone Deacetylase Inhibitory Effects and Antitumor Cytotoxicity. Bioorg. Chem. 2017, 71, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Dung, D.T.M.; Hai, P.-T.; Anh, D.T.; Huong, L.-T.-T.; Yen, N.T.K.; Han, B.W.; Park, E.J.; Choi, Y.J.; Kang, J.S.; Hue, V.-T.-M.; et al. Novel Hydroxamic Acids Incorporating 1-((1H-1,2,3-Triazol-4-Yl)Methyl)-3-Hydroxyimino-Indolin-2-Ones: Synthesis, Biological Evaluation, and SAR Analysis. J. Chem. Sci. 2018, 130, 63. [Google Scholar] [CrossRef]
- Dung, D.T.M.; Huan, N.V.; Cam, D.M.; Hieu, D.C.; Hai, P.-T.; Huong, L.-T.-T.; Kim, J.; Choi, J.E.; Kang, J.S.; Han, S.-B.; et al. Novel Hydroxamic Acids Incorporating 1-((1H-1,2,3-Triazol-4-Yl)Methyl)- 3-Substituted-2-Oxoindolines: Synthesis, Biological Evaluation and SAR Analysis. Med. Chem. 2018, 14, 831–850. [Google Scholar] [CrossRef]
- Sun, N.; Yang, K.; Yan, W.; Yao, M.; Yu, C.; Duan, W.; Gu, X.; Guo, D.; Jiang, H.; Xie, C.; et al. Design and Synthesis of Triazole-Containing HDAC Inhibitors That Induce Antitumor Effects and Immune Response. J. Med. Chem. 2023, 66, 4802–4826. [Google Scholar] [CrossRef]
- Aboeldahab, A.M.A.; Beshr, E.A.M.; Shoman, M.E.; Rabea, S.M.; Aly, O.M. Spirohydantoins and 1,2,4-Triazole-3-Carboxamide Derivatives as Inhibitors of Histone Deacetylase: Design, Synthesis, and Biological Evaluation. Eur. J. Med. Chem. 2018, 146, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Ramos-Morales, E.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural Basis of the Antiproliferative Activity of Largazole, a Depsipeptide Inhibitor of the Histone Deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar] [CrossRef]
- Almaliti, J.; Al-Hamashi, A.A.; Negmeldin, A.T.; Hanigan, C.L.; Perera, L.; Pflum, M.K.H.; Casero, R.A.; Tillekeratne, L.M.V. Largazole Analogues Embodying Radical Changes in the Depsipeptide Ring: Development of a More Selective and Highly Potent Analogue. J. Med. Chem. 2016, 59, 10642–10660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shan, G.; Zheng, Y.; Yu, X.; Ruan, Z.-W.; Li, Y.; Lei, X. Synthesis and Preliminary Biological Evaluation of Two Fluoroolefin Analogs of Largazole Inspired by the Structural Similarity of the Side Chain Unit in Psammaplin A. Mar. Drugs 2019, 17, 333. [Google Scholar] [CrossRef]
- Gong, C.-J.; Gao, A.-H.; Zhang, Y.-M.; Su, M.-B.; Chen, F.; Sheng, L.; Zhou, Y.-B.; Li, J.-Y.; Li, J.; Nan, F.-J. Design, Synthesis and Biological Evaluation of Bisthiazole-Based Trifluoromethyl Ketone Derivatives as Potent HDAC Inhibitors with Improved Cellular Efficacy. Eur. J. Med. Chem. 2016, 112, 81–90. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Gong, C.-J.; Su, M.-B.; Chen, F.; He, T.; Zhang, Y.-M.; Shen, Q.-Q.; Su, Y.; Ding, J.; Li, J.; et al. Synthesis and in Vitro and in Vivo Biological Evaluation of Tissue-Specific Bisthiazole Histone Deacetylase (HDAC) Inhibitors. J. Med. Chem. 2020, 63, 804–815. [Google Scholar] [CrossRef]
- Nam, G.; Jung, J.M.; Park, H.-J.; Baek, S.Y.; Baek, K.S.; Mok, H.Y.; Kim, D.E.; Jung, Y.H. Structure-Activity Relationship Study of Thiazolyl-Hydroxamate Derivatives as Selective Histone Deacetylase 6 Inhibitors. Bioorg. Med. Chem. 2019, 27, 3408–3420. [Google Scholar] [CrossRef]
- Bresciani, A.; Ontoria, J.M.; Biancofiore, I.; Cellucci, A.; Ciammaichella, A.; Di Marco, A.; Ferrigno, F.; Francone, A.; Malancona, S.; Monteagudo, E.; et al. Improved Selective Class I HDAC and Novel Selective HDAC3 Inhibitors: Beyond Hydroxamic Acids and Benzamides. ACS Med. Chem. Lett. 2019, 10, 481–486. [Google Scholar] [CrossRef]
- Clausen, D.J.; Liu, J.; Yu, W.; Duffy, J.L.; Chung, C.C.; Myers, R.W.; Klein, D.J.; Fells, J.; Holloway, K.; Wu, J.; et al. Development of a Selective HDAC Inhibitor Aimed at Reactivating the HIV Latent Reservoir. Bioorg. Med. Chem. Lett. 2020, 30, 127367. [Google Scholar] [CrossRef]
- Yu, W.; Liu, J.; Yu, Y.; Zhang, V.; Clausen, D.; Kelly, J.; Wolkenberg, S.; Beshore, D.; Duffy, J.L.; Chung, C.C.; et al. Discovery of Ethyl Ketone-Based HDACs 1, 2, and 3 Selective Inhibitors for HIV Latency Reactivation. Bioorg. Med. Chem. Lett. 2020, 30, 127197. [Google Scholar] [CrossRef] [PubMed]
- Bürli, R.W.; Luckhurst, C.A.; Aziz, O.; Matthews, K.L.; Yates, D.; Lyons, K.A.; Beconi, M.; McAllister, G.; Breccia, P.; Stott, A.J.; et al. Design, Synthesis, and Biological Evaluation of Potent and Selective Class IIa Histone Deacetylase (HDAC) Inhibitors as a Potential Therapy for Huntington’s Disease. J. Med. Chem. 2013, 56, 9934–9954. [Google Scholar] [CrossRef] [PubMed]
- Senger, J.; Melesina, J.; Marek, M.; Romier, C.; Oehme, I.; Witt, O.; Sippl, W.; Jung, M. Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors. J. Med. Chem. 2016, 59, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Anh, D.T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E.J.; Jun, H.W.; Kang, J.S.; Kwon, J.-H.; Dung, D.T.M.; Anh, V.T.; Hue, V.T.M.; et al. Exploration of Certain 1,3-Oxazole- and 1,3-Thiazole-Based Hydroxamic Acids as Histone Deacetylase Inhibitors and Antitumor Agents. Bioorg. Chem. 2020, 101, 103988. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Liao, C.; Li, Z.; Wang, Z.; Sun, Q.; Liu, C.; Yang, Y.; Tu, Z.; Jiang, S. Design, Synthesis, and Biological Evaluation of 1, 3-Disubstituted-Pyrazole Derivatives as New Class I and IIb Histone Deacetylase Inhibitors. Eur. J. Med. Chem. 2014, 86, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Niu, Q.; Liu, J.; Bao, Y.; Yang, J.; Luan, S.; Fan, Y.; Liu, D.; Zhao, L. Novel Thiol-Based Histone Deacetylase Inhibitors Bearing 3-Phenyl-1 H -Pyrazole-5-Carboxamide Scaffold as Surface Recognition Motif: Design, Synthesis and SAR Study. Bioorg. Med. Chem. Lett. 2016, 26, 375–379. [Google Scholar] [CrossRef]
- Wen, J.; Bao, Y.; Niu, Q.; Yang, J.; Fan, Y.; Li, J.; Jing, Y.; Zhao, L.; Liu, D. Identification of N-(6-Mercaptohexyl)-3-(4-Pyridyl)-1H-Pyrazole-5-Carboxamide and Its Disulfide Prodrug as Potent Histone Deacetylase Inhibitors with in Vitro and in Vivo Anti-Tumor Efficacy. Eur. J. Med. Chem. 2016, 109, 350–359. [Google Scholar] [CrossRef]
- Xu, Q.; Yu, S.; Cai, Y.; Yang, J.; Zhao, L.; Liu, D. Design, Synthesis and Biological Evaluation of Novel Selective Thiol-Based Histone Deacetylase(HDAC) VI Inhibitors Bearing Indeno[1,2-c]Pyrazole or Benzoindazole Scaffold. Chem. Res. Chin. Univ. 2018, 34, 75–83. [Google Scholar] [CrossRef]
- Zagni, C.; Citarella, A.; Oussama, M.; Rescifina, A.; Maugeri, A.; Navarra, M.; Scala, A.; Piperno, A.; Micale, N. Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker. Int. J. Mol. Sci. 2019, 20, 945. [Google Scholar] [CrossRef]
- Gediya, P.; Vyas, V.K.; Carafa, V.; Sitwala, N.; Della Torre, L.; Poziello, A.; Kurohara, T.; Suzuki, T.; Sanna, V.; Raguraman, V.; et al. Discovery of Novel Tetrahydrobenzo[b]Thiophene-3-Carbonitriles as Histone Deacetylase Inhibitors. Bioorg. Chem. 2021, 110, 104801. [Google Scholar] [CrossRef]
- Yang, F.; Han, L.; Zhao, N.; Yang, Y.; Ge, D.; Zhang, H.; Chen, Y. Synthesis and Biological Evaluation of Thiophene-Based Hydroxamate Derivatives as HDACis with Antitumor Activities. Future Med. Chem. 2020, 12, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Bottomley, M.J.; Lo Surdo, P.; Di Giovine, P.; Cirillo, A.; Scarpelli, R.; Ferrigno, F.; Jones, P.; Neddermann, P.; De Francesco, R.; Steinkühler, C.; et al. Structural and Functional Analysis of the Human HDAC4 Catalytic Domain Reveals a Regulatory Structural Zinc-Binding Domain. J. Biol. Chem. 2008, 283, 26694–26704. [Google Scholar] [CrossRef] [PubMed]


























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frühauf, A.; Behringer, M.; Meyer-Almes, F.-J. Significance of Five-Membered Heterocycles in Human Histone Deacetylase Inhibitors. Molecules 2023, 28, 5686. https://doi.org/10.3390/molecules28155686
Frühauf A, Behringer M, Meyer-Almes F-J. Significance of Five-Membered Heterocycles in Human Histone Deacetylase Inhibitors. Molecules. 2023; 28(15):5686. https://doi.org/10.3390/molecules28155686
Chicago/Turabian StyleFrühauf, Anton, Martin Behringer, and Franz-Josef Meyer-Almes. 2023. "Significance of Five-Membered Heterocycles in Human Histone Deacetylase Inhibitors" Molecules 28, no. 15: 5686. https://doi.org/10.3390/molecules28155686
APA StyleFrühauf, A., Behringer, M., & Meyer-Almes, F.-J. (2023). Significance of Five-Membered Heterocycles in Human Histone Deacetylase Inhibitors. Molecules, 28(15), 5686. https://doi.org/10.3390/molecules28155686