Abstract
A series of cationic p-tert-butylcalix[4]arenes, with side-arms that are functionalized with imidazolium groups, have been synthesized in good yields. The parent tetrahydroxy para-t-butyl-calix[4]arene was dialkylated at the phenolic hydrogen atoms using α,ω-dibromo-alkanes to yield bis(mono-brominated) alkoxy-chains of variable length. The brominated side-arms in these compounds were then further alkylated with substituted imidazoles (N-methylimidazole, N-(2,4,6-trimethyl-phenyl)imidazole, or N-(2,6-di-isopropylphenyl)imidazole) to yield a series of dicationic calixarenes with two imidazolium groups tethered, via different numbers of methylene spacers (n = 2–4), to the calixarene moiety. Related tetracationic compounds, which contain four imidazolium units linked to the calix[4]arene backbone, were also prepared. In all of these compounds, the NMR data show that the calixarenes adopted a cone configuration. All molecules were characterized by NMR spectroscopy and by MS studies. Single crystal X-ray diffraction studies were attempted on many mono-crystals of these cations, but significant disorder problems, partly caused by occluded solvent in the lattice, and lack of crystallinity resulting from partial solvent loss, precluded the good resolution of most X-ray structures. Eventually, good structural data were obtained from an unusually disordered single crystal of 5a, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-di-[2-(N-2,6-diisopropylphenyl-imidazolium)ethoxy]calix[4]arene dibromide and its presumed structure was confirmed. The structure revealed the presence of H-bonded interactions and some evidence of π-stacking. Some of these imidazolium salts were reacted with nickelocene to form the nickel N-heterocyclic carbene (NHC) complexes 7a–7d. A bis-carbene nickel complex 8 was also isolated and its structure was established by single crystal X-ray diffraction studies. The structure was disordered and not of high quality, but the structural data corroborated the spectroscopic data.
1. Introduction
Both calixarenes and imidazolium salts have been the objects of study by a large number of different research groups in recent years [1,2]. Calixarenes, especially calix[4]arenes, which have been investigated in more detail than other calixarenes, provide a cavity that is tunable in size and in electronic properties, as both the lower and the upper rim of the calixarenes can be functionalized with a wide variety of functional groups. These molecules are under investigation as selective complexing agents and sensors for both neutral and charged ions—more commonly cations [2,3,4,5,6,7] but more recently, anions [8,9,10,11] with applications that include trapping ions, including radioactive or toxic ions [12,13]. Their size-dependent selectivity has been used in the development of catalysts [14,15,16] and has been exploited in organometallic chemistry [17], where unusual reactivity patterns have been observed [18]. Research into their chemistry has been accelerated by synthetic methods that allow the preparation of multi-gram quantities of calixarenes of various sizes in good yield, many of which are now commercially available [19,20].
Imidazolium salts, in particular, those with alkyl chains on the imidazolium nitrogen atoms, are under extensive study as low-melting ionic liquids [21,22] In addition, they are used as starting materials to access N-heterocyclic carbene complexes [23,24,25], which have become ligands of major importance in current organometallic chemistry and homogeneous catalysis [26,27].
A few years ago, we reported that para-t-butylcalix[4]arene 1 can be functionalized with alkyne groups to yield a series of molecules with one, two, or four C–C bonds. These species in turn could be reacted with [Co2(CO)8] to yield a series of cobalt carbonyl species linked to calixarene units via cobalt π-alkyne-bond interactions [28]. We also recently reported on the syntheses and anion-complexing ability of a series of calix[6]arenes that were functionalized with imidazolium ligands [29]. We are still interested in combining the relatively undeveloped organometallic chemistry of calixarenes [30] with our current research on the chemistry of N-heterocyclic carbene complexes of nickel [31,32,33,34,35,36,37,38,39]. This manuscript describes some steps in this direction via the synthesis of a series of calix[4]arene-linked imidazolium cations. Specifically, a series of new cationic para-t-butyl-calix[4]arenes, which were functionalized with imidazolium groups, were prepared. These heterocyclic groups were tethered to the calixarene skeleton via variable-length alkyl sidechains. Similar species have already been investigated by various groups in anion-binding studies [40,41,42,43]. The synthesized imidazolium salts that are reported in this manuscript have potential as anion-binding agents, and studies are underway to determine their binding ability and selectivity towards various anions. Indeed, preliminary results show that the 1H NMR spectra of the cation of 3c with PF6− as an anion showed significant 1H NMR shifts in some peaks. This indicates that the cation interactions with PF6− were different here than for those seen for the Br− anion present in 3c [44].
We also plan to see whether the calixarene-tethered nickel complexes exhibit any advantages (perhaps a cavity effect?) on Suzuki–Miyaura catalysis. We have already reported similar studies with related calix[6]arenes linked to CpNi(NHC) groups and have investigated their catalytic activity, as mentioned earlier [29,30]. Other groups have published examples of catalysis by nickel complexes attached to a supramolecular framework [45,46,47].
2. Results and Discussion
The reaction of p-tert-butylcalix[4]arene 1 with a series of α,ω-dibromoalkanes led to alkylation of two of the calixarene hydroxy groups and to the formation of a series of 1, 3-dialkylated calixarenes with –O(CH2)nBr groups (2a, n = 1; 2b, n = 3; 2c, n = 4) in moderate to excellent yields, as shown in Scheme 1.
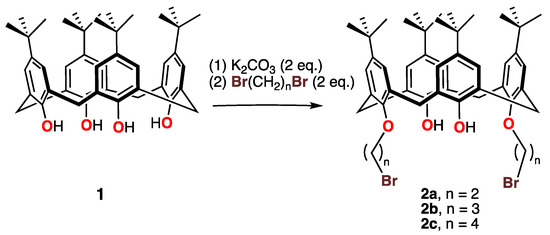
Scheme 1.
Syntheses of the dibromocalix[4]arenes 2 with various-length methylene spacers.
All reactions were carried out using an excess of α,ω-dibromoalkane. Potassium carbonate, used as a base, was added to an acetonitrile solution of 1 first and stirred for a couple of hours. Then, α,ω-dibromoalkane was introduced, and the mixture refluxed for a period of two days, as shown in the scheme.
After work-up, the series of calixarenes 2, with brominated side chains on the oxygen atoms in the 1- and 3-positions, were isolated and characterized by 1H NMR spectroscopy. The data clearly indicate that the brominated alkyl side chain was added to the calixarene moiety and that the molecule had an effective mirror plane on the 1H NMR timescale. Furthermore, the AB patterns and characteristic coupling constant (J ≈ 13 Hz) for the methylenic Ar-CH2-Ar protons and their chemical shifts (≈3.0 ± 2 ppm) indicate that, like the parent calixarene, these bromo-alkylated para-t-butylcalixarene derivatives adopted a cone conformation in solution. The presence of the phenolic OH protons was confirmed by observation via 1H NMR spectroscopy, where they appeared in the 6.6–7.4 ppm range. Detailed 1H NMR data for these compounds are listed after the synthesis of each compound.
All compounds were isolated as white or cream-colored solids. It was difficult to obtain them in an analytically pure state, as all the samples trapped solvents, presumably within or close to the calixarene cavity, and these solvents were not easily removed in vacuo. In addition, for probably the same reason, the samples did not crystallize in well-formed mono-crystals, and we were unable to obtain good mono-crystals for a quality single-crystal X-ray diffraction study.
By treating the p-tert-butylcalix[4]arene 1 with 1-4-dibromobutane and an excess of a stronger base (NaH rather than K2CO3), the tetra-substituted compound 2d was obtained in moderate (41%) yield as shown in Scheme 2.The 1H NMR spectrum of this compound showed no signals that could be attributed to phenolic protons, as expected.
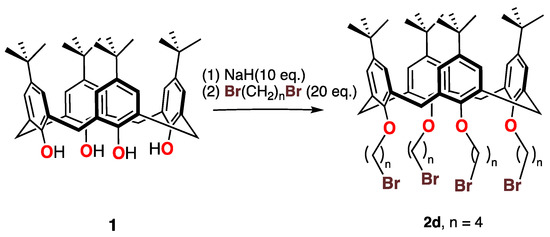
Scheme 2.
Synthesis of the tetrabromocalix[4]arene 2d.
When compounds 2a–2c were treated with N-methylimidazole, N-(2,4,6-trimethylphenyl)imidazole, or N-(2,6-di-isopropyl-phenyl)imidazole ((N-DIPP)imidazole)) in refluxing acetonitrile or in 1,4-dioxane, the unsubstituted nitrogen atom was alkylated: The products were the di-cationic imidazolium salts 3a–3c, 4a–4c, and 5a–5c, respectively, as shown in Scheme 3.
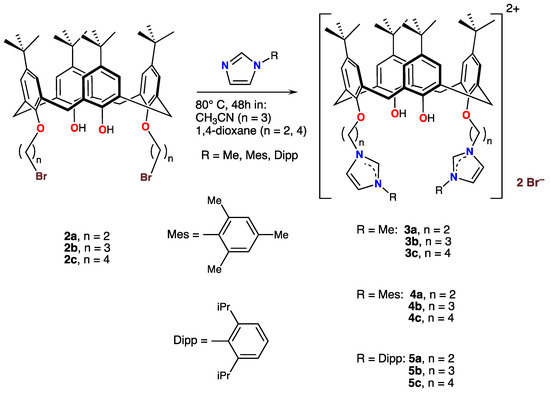
Scheme 3.
Syntheses and structures of the cationic calix[4]arene imidazolium derivatives 3, 4, and 5.
All products were obtained as colorless oils or as white or off-white hydroscopic micro-crystalline compounds, which were characterized by 1H spectroscopy. The imidazolium proton, on the carbon atom sandwiched between the two imidazolium nitrogen atoms, resonated at ≈ 8 ppm for all complexes. Again, the characteristic CH2-arene coupling constants of the calix[4]arenes clearly pointed to a cone conformation.
The reactions of the tetrasubstituted calixarene 2d with the same imidazoles used to prepare compounds 3–5 under similar conditions led to the tetraimidazolium tetracationic compounds 6a, 6b, and 6c in excellent yields, as shown in Scheme 4. All of these salts were isolated as white or off-white powders. They were soluble in polar solvents but sparingly soluble in pentane or hexane. Most salts exhibited an (M − Br)+ m/e signal in their mass spectra (MALDI-TOF, see Procedures). However, in some cases (6b, 6c), a signal for the doubly charged (M − 2Br)2+ ion was observed.
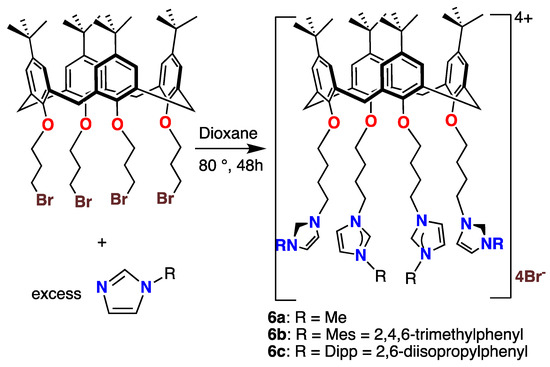
Scheme 4.
Syntheses and structures of the tetracationic calix[4]arene imidazolium species 6a–6c.
Despite repeated crystallization attempts, none of these salts produced well-formed crystals, which is, unfortunately, not an uncommon occurrence with calixarenes. Single crystals were eventually obtained from two separate samples of 5a. The data set from one was poor, though it did show the cone configuration of the calixarene.
Another data set from a crystal from the second batch exhibited the same lattice parameters as the previous structural data, but the data here were better, despite the presence of some unusual disorder. The structure was not ideal, but it clearly showed the cone conformation adopted by the di-cationic calixarene and the two substituted imidazolium groups that were linked to the calixarene via a –C2H4– side chain. One of the bromide anions in this structure was “normal,” but the other had a 50% occupancy of its crystallographic position and shared its lattice location with a methoxy anion (also at 50%). In addition, one of the four t-Bu groups of the calixarene was rotationally disordered. The crystal included H2O molecules of crystallization. The structure of the cation of 5a is shown in Figure 1.
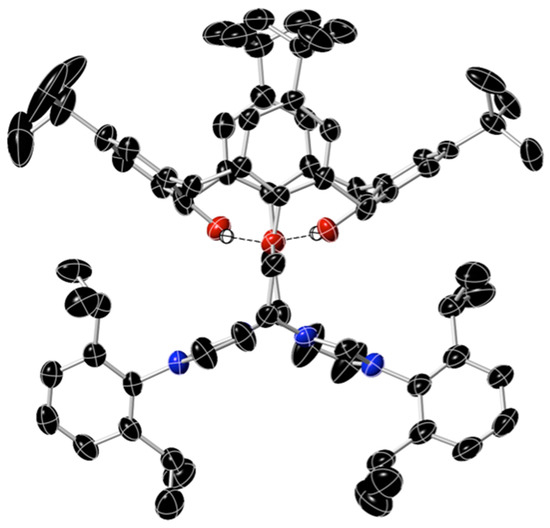
Figure 1.
The geometry of the di-cation of 5a, based on a single crystal X-ray diffraction study. Apart from the hydroxy H-atoms (shown as white spheres), only the atoms of C, O, and N are shown, respectively, as black, red, and blue ellipsoids (at a 50% probability level). One of the four t-Bu groups is rotationally disordered, so only three of the disordered Me carbons are shown around this central t-Bu carbon atom. The H-bonded O……H distances are 2.113 and 2.133 Å, whereas the O–H …… O angles are 155.3 and 167.1°.
The di-cation had approximate C2 symmetry in the solid state, as shown. 1H NMR data corroborated this cone conformation in solution. Bonds and angles for the calixarene and imidazolium groups were normal. The two calixarene phenoxy rings that contained the OH groups were significantly tilted away from each other, making an angle of 108° to each other. The other two phenoxy rings, linked to the imidazolium groups, were much closer to coplanar, deviating only 4.9° from co-planarity: This indicates the likely presence of π-π interactions between these two rings. This phenomenon was also seen in the two C6H3iPr2 aromatic rings, which exhibited significant π-stacking interactions in the solid state with rings of adjacent molecules, with an arene–arene distance of around 4 Å.
The O……O distance for the oxygen atoms of the two ether groups was 5.2 Å, whereas the HO……OH distance was 3.17 Å. The phenolic hydrogen atoms were each oriented towards an oxygen atom, and the OH……O distances of 2.113 and 2.133 Å indicate significant H-bonding interactions between these two groups in the calixarene ring.
3. Synthesis of Nickel NHC Complexes
Our research group has been interested in the chemistry and homogeneous catalysis potential of N-heterocyclic carbene complexes of nickel for the past 15 years, and we have published many papers in this area [31,32,33,34,35,36,37,38,39]. It was of interest to us to graft nickel NHC complexes onto a calixarene backbone to see whether the catalytically active organometallic fragment close to the calixarene cavity would lead to any enhanced catalytic activity. Indeed, we recently described the synthesis of nickel-NHC complexes grafted onto a calix[6]arene framework [29,30]. Thus, we decided to prepare similar complexes in which the metal carbene moiety was linked to a calix[4]arene backbone.
Imidazolium salts provide a facile entry into nickel NHC chemistry, as the reaction of nickelocene with such species leads to [Ni(NHC)(X)Cp] (X = Cl, Br, I; Cp = η5-C5H5) complexes [23,24,25]. This reaction was carried out with salts 4a–4c and 5a. When these imidazolium salts were refluxed with two molar equivalents of nickelocene for two days, the bis-carbene complexes 7a–d were obtained in satisfactory isolated yields, ranging from 64–88%, as shown in Scheme 5.
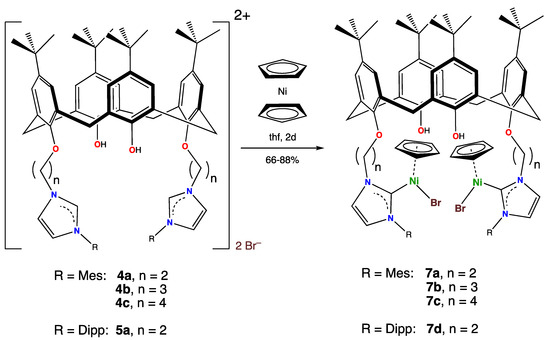
Scheme 5.
Synthesis of the organometallic complexes 7a–7d from nickelocene and calix[4]arene-bound imidazolium salts.
In general, the spectroscopic signatures of the organometallic portions of complexes 7 were similar to those of many other such species that we have reported during our research over the last 15 years. The complexes were all reddish-brown to violet-red in color and exhibited 1H NMR spectra that were consistent with their structures. The imidazolium proton on the carbon atom that was sandwiched between the two nitrogen atoms was no longer visible in any of the 1H NMR spectra of complexes 7, as expected, since it was no longer there. The molecule had an effective mirror plane in solution so that only one signal was seen for the η5-C5H5 protons, which appeared (depending on the complex) in the 4.7–4.8 ppm range. The hydroxy protons were observed as singlets at 6.35–6.87 ppm. The AB signals seen for all of these species for the calixarene methylenic protons, with a coupling constant of 13.5–13.9 Hz, indicate that the cone-configuration of the calixarenes, present in the imidazolium salts, was maintained in the organometallic complexes. The MS of complexes 7 exhibited the parent peaks expected for the bis(nickel-NHC) complexes together with the expected isotopic envelope for complexes with two nickel atoms.
When the imidazolium salt 4c was reacted with Ni(η-C5H5)2 under more forcing conditions (refluxing 1,4-dioxane for 4 days), a different product, 8, was obtained (Scheme 6) in moderate 45% yield. Its structure was determined via a single crystal X-ray diffraction study, but despite many attempts, only small crystals could be obtained. The resolved structure did not appear to be significantly disordered, apart from commonly observed rotational disorder for some t-Bu groups, but the data were not good due to many weak or absent reflections, and so the esds were high and the structure was not fully refined. Nevertheless, we believe the structure is useful, as it nevertheless established the structure of the compound and yielded further geometric information.
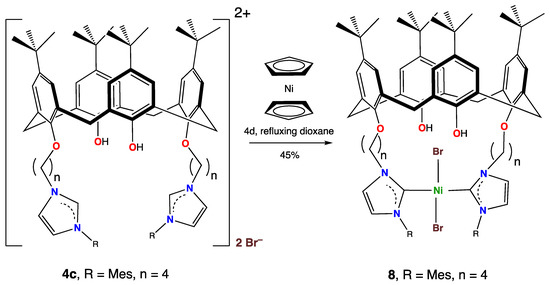
Scheme 6.
Synthesis of 8 from the reaction of Ni(η-C5H5)2 and 4c in refluxing dioxane.
It is quite unusual for an 18-electron organometallic complex to lose a η5-C5H5 ligand, as these ligands are typically inert and not substitutionally labile. A well-known textbook states, “The Cp group […] (is) the most firmly bound … polyenyl and the most inert to nucleophilies or electrophiles” [48]. Although a few examples have been reported [49], they remain unusual.
The X-ray data show (Figure 2) that 8 is a bis-Ni(NHC) complex with no cyclopentadienyl ligands bound to the nickel atom. The nickel was in a trans-square-planar geometry with two substituted NHC ligands and two bromides forming the coordination sphere of the metal. The geometry of the complex is analogous to many other complexes of the type trans-[NiX2(NHC)2] (X = Cl, Br, I) that have been reported and have been known for the last 25 years [50,51,52,53]. The coordination of the ligand around the nickel in 8 is broadly similar to what has been observed in such complexes. The calixarene part of this molecule adopted a cone configuration.
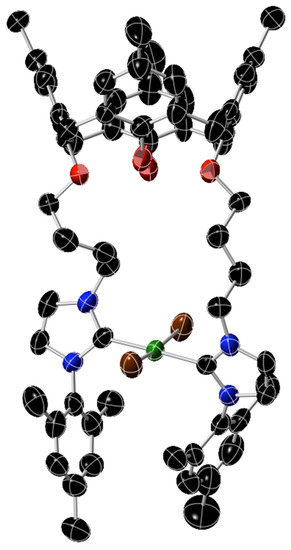
Figure 2.
Structure of complex 8. The atoms are color coded: C, black; O, red; N, blue; Ni, green; Br, brown. The 50% ellipsoids are shown. The structure remains preliminary and is not completely refined as the crystal is small and reflections are weak. Thus, distances and angles are not provided.
The cleavage of Ni–Cp bonds in the formation of 8 is unusual, but other reports of Ni–Cp bond cleavage have been seen for Ni–NHC complexes [54]. Ni–(NHC) bonds decompose in the presence of OH groups and water [55], but this does not appear to have taken place significantly, since it was the Ni–Cp bonds and not the Ni–NHC bonds that were broken in forming complex 8.
4. Conclusions
A series of bis-(bromoalkoxy)calix[4]arene derivatives 2a–2c were synthesized by treating the parent p-t-butylcalix[4]arene with two equivalents of α,ω-dibromoalkanes and a base (potassium carbonate). A related tetrabromoalkoxycalix[4]arene (2d) was also prepared. All of these bromocalix[4]arenes were alkylated further with imidazoles to afford the dicationic imidazolium species 3–5 and the related tetracationic imidazolium salts 6. In all of these species, the imidazolium unit was tethered to the calixarene skeleton by a variable-length methylene spacer (–(CH2)n, n = 2, 4, 6). These salts were fully characterized by 1H NMR spectroscopy and by MS, and a single crystal X-ray structural determination was carried out for compound 5a. Four of these bis-imidazolium cations were treated with nickelocene to generate bis-(NHC NiCpBr) species (7a–d) in which the NHC ligands were each linked to the calix[4]arene via (CH2)n (n = 2–4) spacer groups. Prolonged reaction of 4c with nickelocene led to unusual loss of the nickel-bound C5H5 ligand and formed complex 8 with two NHC ligands bound trans-in a square planar geometry to a NiBr2 unit. The preliminary structure of 8 was determined by an X-ray diffraction study, and it corroborated the spectroscopic data.
Further studies are planned along two research directions. We plan to investigate the Ni–NHC complexes in catalysis with a view to determining whether the calixarene-bound organometallic complexes show any enhanced catalytic effect resulting from the calixarene. Furthermore, as many of the synthesized molecules are di- or tetra-cations, we intend to probe their anion recognition properties and indeed do have some preliminary unpublished results [44].
5. Procedures
General. Spectroscopic data are listed for each compound below, but synthetic experimental data are given here only for 2a, 2d, 3a, and 7a (as representative syntheses) since the procedures for the synthesis of the calixarenes with brominated side chains 2a–2c, the imidazolium cations 3–6, and the organometallic nickel NHC complexes 7b–7d, respectively, are similar to each other. Full synthetic data may be found in the Supplementary Material Tables.
All reactions were carried out under an Ar atmosphere. SiO2 (Geduran 1.11567) was used for column chromatography. For the calixarene synthesis, reagents (puriss, p. a., grade) were commercial and used without further purification. Solvents were distilled under Ar over sodium/benzophenone (diethyl ether, pentane) or CaH2 (dichloromethane) prior to use. DMF (N-N-dimethyl-formamide) was purified by standing over molecular sieves and then was distilled at reduced pressure, and p-t-butylcalix[4]arene was prepared following published procedures [19,20]. All 1H NMR data were obtained on FT-Bruker Ultra Shield 300 or Spectrospin 400 spectrometers operating at 300.13 or 400.14 MHz, respectively, at room temperature; chemical shifts (∂) are in ppm, relative to residual deuterated solvent peaks in CDCl3 (unless otherwise stated). Values of the coupling constant (J) are in Hz; peaks are singlets unless otherwise stated; d = doublet, t = triplet, q = quartet, m = multiplet, qn, Jap = apparent quintet and coupling constant. Mass spectra were recorded on a Bruker micrOTOF-Q mass spectrometer by the Mass Spectroscopy Service (UMR CNRS 7177) at the University of Strasbourg.
X-ray data were collected on a single crystal of 5a grown from a dichloromethane solution layered with diethylether. Diffraction data were collected at 173(2) K on a Bruker APEX II DUO Kappa-CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device using Mo-Kα radiation (λ = 0.71073 Å). The crystal-detector distance was 38 mm. The cell parameters were determined from reflections taken from 3 sets of 12 frames, each at 10 s exposure. The structure was solved using the program SHELXT-2014. The refinement and all further calculations were carried out using SHELXL-2014 [56]. Hydrogen atoms were included in calculated positions and treated as riding atoms using SHELXL default parameters. The non-hydrogen atoms were refined anisotropically using weighted full-matrix least-squares on F2. The SQUEEZE instruction in PLATON was applied. The residual electron density was assigned to three molecules of the methanol solvent.
2a, (1,3)-cone-5,11,17,23-tetra-t-butyl-25,27-dihydroxy-bis(2-bromo-ethoxy)calix[4]-arene.
A mixture of p-t-butylcalix[4]arene (3.244 g 5.00 mmol), K2CO3 (1.383 g, 10.00 mmol), and acetone (50 mL) was stirred for 2 h. Subsequently, a solution of 1,3-dibromopropane (2.834 g, 15.00 mmol) in acetone (50 mL) was added and the mixture was refluxed for 4 d. Methanol (5 mL) was then added, and all solvents were evaporated under vacuum. The residue was dissolved in dichloromethane (100 mL) and extracted with water (150 mL). The organic phase was dried with anhydrous Na2SO4 and then evaporated to dryness. Washing of the residue with petroleum ether and its subsequent removal afforded 2a, C48H62O4Br2 (3.354 g, 3.887 mmol, 77%), as a fine off-white powder. C48H62O4Br2 862.812 g mol−1.
1H NMR: 7.062 (4H, Ar–H), 6.785 (4H, Ar’–H), 6.625 (2H, Ar–OH), 4.313 and 3.330 (8H, AB-type doublet, Ar–CH2–Ar’, JAB = 13), 4.310 (t, 4H, CH2CH2O, JHH = 6.6), 3.844 (t, 4H, CH2CH2O, JHH = 6.6), 1.294 (18H, t-Bu), 0.950 (18H, t-Bu’).
2b, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-dihydroxy-26,28-bis-(3-bromopropoxy)-calix[4]arene.
1H NMR: 7.664 (2H, Ar–OH), 7.049 (4H, Ar–H), 6.876 (4H, Ar’–H), 4.260 and 3.353 (8H, AB-type doublet, Ar–CH2–Ar’, JAB = 13), 4.110 (t, J = 5.5, 4H, CH2O,), 4.010 (t, J = 6.4, 4H, CH2Br,), 2.524 (qn, Japp = 6.1, 4H, CH2CH2CH2), 1.273 (18H, t-Bu), 1.017 (18H, t-Bu’).
2c, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-bis-(4-bromobutoxy)calix[4]arene dibromide.
1H NMR: 7.435 (2H, Ar–OH), 7.059 (4H, Ar–H), 6.805 (4H, Ar’–H), 4.237 and 3.316 (8H, AB doublet, JAB = 13, Ar–CH2–Ar’), 4.006 (t, 4H, CH2O, J = 5.8), 3.643 (t, J = 6.6, 4H, CH2Br), 2.335 (qn Jap = 4.3, 4H, CH2CH2O), 2.162 (qn, Japp = 4.2, CH2CH2Br), 1.292 (18H, t-Bu), 0.967 (18H, t-Bu’).
2d, (1,3)-Cone-5,11,17,23-tetra-t-butyl-tetrakis(4-bromobutoxy)calix[4]arene.
A mixture of p-t-butylcalix[4]arene (7.788g, 12 mmol) and NaH (12 g, 0.5 mmol) was placed in a 500 mL round-bottomed flask under an argon atmosphere, and oxygen and water-free DMF (240 mL) were added. The mixture was stirred for 2 h, and then a solution of dried and freeze-thaw degassed 1,4-dibromobutane (29 mL, 0.240 mol) in DMF (120 mL) was added and the mixture heated under reflux for 4 d. The solvent was then removed under vacuum, and the residue was dissolved in CH2Cl2 (300 mL) and water 400 mL) was added. The mixture was stirred vigorously and then placed in a large separating funnel, and the organic layer was removed and placed in anhydrous Na2SO4 to remove any residual water. The dichloroethane solution was then concentrated: The addition of methanol led to the precipitation of 2d as an off-white powder (C60H84Br4O4, 1188,9 g mol−1, 5.85 g, 4.92 mmol, 41%).
1H NMR: 6.78 (8H, Ar–H), 4.35 and 3.13 (AB doublet, JAB = 12.4, 8H, Ar–CH2-Ar), 3.91 (t, 8H, CH2O), 3.51 (t, 8H, CH2Br), 2.15 (qn, Japp = 7.5, 8H, CH2CH2O), 2.01 (qn, Japp = 7.5, 8H, CH2CH2Br), 0.95 (36H, tBu).
3a, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-bis-(2-(N-methylimida-zolium)ethoxy)calix[4]arene dibromide.
MS (MALDI TOF): m/z 947.48 (M − Br)+.
1H NMR: 10.29 (2H, NCHN), 8.34 (2H, NCH), 7.08 (2H, NCH’), 7.26 (4H, Ar-H), 6.67 (4H, Ar-H), 6.23 (4H, Ar’-H), 6.15 (2H, OH), 5.20 (t, J = 5.0, 4H, CH2CH2O), 4.38 (t, J = 5.2, 4H, CH2CH2N), 4.00 (6H, NMe), 3.72 and 3.32 (AB doublet, JAB = 13, 8H, Ar–CH2–Ar’), 1.33 (18H, t-Bu), 0.87 (18H, t-Bu’).
3b, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-bis-(4-(N-methylimida-zolium)propoxy)calix[4]arene dibromide.
MS MALDI TOF, m/z 975.51 (M − Br)+.
1H NMR: 10.27 (2H, N-CH-N), 7.85 (2H, NCH-), 7.54 (2H, NCH’-), 7.40 (2H, OH), 7.07 (4H, Ar-H), 6.82 (4H, Ar’-H), 4.84 (t, J = 7.5, 4H, CH2-CH2-O), 4.12 and 3.36 (AB doublet, JAB = 13.4, 8H, Ar–CH2–Ar’), 4.08 (6H, N-CH3), 4.07 (t, J = 6.3, 4H, CH2-CH2-N), 2.68 (qn, Japp = 6.6, 4H, CH2-CH2-CH2), 1.28 (18H, t-Bu), 0.97 (18H t-Bu’).
3c, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-bis-(4-(N-methylimida-zolium)butoxy)calix[4]arene dibromide.
MS (MALDI TOF): m/z 1003.6 (M − Br)+.
1H NMR: 9.75 (2H, N-CH-N), 7.82 (2H, NCH-), 7.51 (2H, NCH’-), 7.08 (4H, Ar-H), 7.00 (2H, OH), 6.71 (4H, Ar-H), 4.65 (t, J = 7.5, 4H, CH2-CH2-O), 4.15 and 3.34 (AB doublet, JAB = 13.6, 8H, Ar–CH2–Ar’), 4.07 (6, N-CH3), 3.96 (t, J = 5.8, 4H, CH2-CH2-N), 2.24 (qn, Japp = 6.8 Hz, 4H, CH2-CH2-O), 1.87 (qn, Japp = 6.3, 4H, CH2-CH2-N), 1.30 (18H, t-Bu), 0.88 (18H t-Bu’).
4a, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-bis-[2-(N-2,4,6-trimethyl-phenylimidazolium)ethoxy] calix[4]arene dibromide.
MS (MALDI TOF): m/z 1155.61 (M − Br)+.
1H NMR: 10.48 (2H, N-CH-N), 8.36 (2H, NCH-), 7.32 (2H, NCH’-), 7.02 (4H, Ar-H), 6.94 (4H, Ar’-H), 6.57 (4H, Ar’-H), 5.86 (2H, OH), 5.35 (t, J = 4.5, 4H, CH2-CH2-O), 4.37 (t, J = 4.35, 4H, CH2-CH2-N), 3.87 and 3.20 (AB doublet, JAB = 13.6, 8H, Ar–CH2–Ar’), 2.34 (6H, Ar-CH3), 2.00 (12H, Ar-CH3), 1.30 (18H, t-Bu), 0.81 (18H t-Bu’).
4b, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-bis-[3-(N-2,4,6-trimethyl-phenyl)imidazolium)propoxy]calix[4]arene dibromide.
MS (MALDI TOF): m/z 1183.65 (M − Br)+.
1H NMR: 10.21 (2H, N-CH-N), 8.42 (2H, NCH-), 7.72 (2H, NCH’-), 7.04 (4H, Ar-H), 6.97 (4H, Ar-H), 6.96 (2H, OH), 6.86 (4H, Ar-H), 5.29 (t, J = 7.5, 4H, CH2-CH2-O), 4.12 (t, J = 7.5, 4H, CH2-CH2-N), 4.13 and 3.34 (AB doublet, JAB = 13.2, Ar–CH2–Ar’), 2.79 (qn, Japp = 6.2, 4H, CH2CH2O), 2.32 (6H, Ar-CH3), 2.08 (12H, Ar-CH3), 1.26 (18H, t-Bu), 1.01 (18H t-Bu’).
4c, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-di-[4-(N-2,4,6-trimethylphenyl)imidazolium)butoxy]calix[4]arene dibromide.
MS (MALDI TOF): m/z 1211.58 (M − Br)+. M = 1291.42 g.mol−1.
1H NMR: 1H NMR: 10.25 (2H, N-CH-N), 8.61 (2H, NCH-) 7.16 (4H, NCH’- and OH), 6.95 (4H, Ar-H), 6.72 (4H, Ar-H), 5.12 (t, 4H, J = 7.5, CH2-CH2-O), 4.12 and 3.34 (AB doublet, JAB = 13.6, 8H, Ar–CH2–Ar’), 4.07 (t, 4H, J = 7.5, CH2-CH2-N), 2.48 (qn Jap 7.0, 4H CH2CH2O), 2.32 (6H, Ar-CH3), 2.05 (12H, Ar-CH3), 1.98 (qn Jap = 6.4, 4H, CH2CH2N), 1.30 (18H, t-Bu), 0.90 (18H t-Bu’).
5a, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-di-[2-(N-2,6-diisopropyl-phenylimidazolium)ethoxy] calix[4]arene dibromide.
MS (MALDI TOF):, m/z 1239.7 (M − Br).
1H NMR: 10.63 (2H, NCHN), 8.31 (2H, NCH), 7.51 (t, J = 7.5, 2H, Ar-H4), 7.46 (2H, NCH’), 7.28 (d, 4H J = 7.5, Ar-H(3), Ar-H(5)), 7.06 (4H, Ar-H), 6.62 (4H, Ar’-H), 6.01 (2H, OH), 5.46 (t, J = 4.4, 4H, CH2CH2O), 4.53 (t, J = 4.8, 4H, CH2CH2N), 3.96 and 3.27 (AB doublet, JAB = 13, 8H, Ar–CH2–Ar’), 2.26 (m, 4H, CHMe2), 1.31 (18H, t-Bu), 1.19 (d, J = 6.9, 6H, CHMe2), 1.02 (d, J = 6.7, 6H, CHMe2), 0.83 (18H, t-Bu’).
5b, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-di-hydroxy-26,28-bis-[3-(N-2,6-diisopropyl-phenyl)imidazolium)propoxy]calix[4]arene dibromide.
MS (MALDI TOF): m/z = 1267.73 (M − Br)+.
1H NMR: 10.35 (2H, NCHN), 8.52 (2H, NCH), 7.91 (2H, NCH’), 7.49 (t, J = 4.9, 2H, Ar-H4), 7.28 (d, J = 4.9, 4H, Ar-H(3), Ar-H(5)), 7.23 (2H, OH), 7.02 (4H, Ar-H), 6.89 (4H, Ar’-H), 5.44 (t, J = 6.5, 4H, CH2CH2O), 4.14 (t, 4H, CH2CH2N), 4.14 and 3.34(AB doublet, JAB = 12.6, 8H, Ar–CH2–Ar’), 2.78 (qn, Japp = 6.7, 4H, CH2CH2N), (2.29, m, 4H, CHMe2), 1.25 (18H, t-Bu), 1.18 (d, J = 6.7, 6H CHMe2), 1.15 (d, J = 6.7, 6H, CHMe2), 1.05 (18H, t-Bu’).
5c, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,27-dihydroxy-26,28-di-[4-(N-2,6-diisopropyl-phenyl)imidazolium)butoxy]calix[4]arene dibromide.
MS (MALDI TOF): 1295.77 (M − Br)+, 100%.
1H NMR: 10.31 (2H, NCHN), 8.67 (2H, NCH), 7.51 (t, 2H, J = 8, Ar-H4), 7.28(d, 4H, J = 8, Ar-H(3), Ar-H(5)), 7.23 (2H, OH), 7.15 (2H, NCH’), 7.07(4H, Ar-H), 6.76 (4H, Ar’-H), 4.19 and 3.37 (AB-type doublet, JAB = 13.6, 8H, Ar–CH2–Ar’), 4.12 (t, 4H, J = 5.3 CH2CH2N), 4.10 (t, 4H, J = 5.3, CH2CH2O), 2.52 (qn, J = 7.2, 4H, CH2-CH2-O), 2.30 (m, J = 6.6, 4H, CHMe2), 2.03 (qn Jap = 5.8, 4H, CH2CH2N), 1.29 (18H, t-Bu), 1.20 (d, J = 6.6, 6H, CHMe2), 1.13 (d, J = 6.6, 6H, CHMe2), 0.92 (18H, t-Bu).
6a, (1,3)-Cone-5,11,17,23-tetra-t-butyl-25,26,27,28-tetra-[4-(1-N-methylimidazolium)] butoxy-calix[4]arene tetrabromide.
MS (MALDI TOF): m/z 1437.59 (M − Br)+.
1H NMR: 10.36 (4H, NCHN), 8.27 (4H, NCH), 7.16 (2H, NCH’), 6.77 (4H, Ar-H), 4.72 (t, J = 6.9, 8H, CH2CH2O), 4.31 and 3.14 (AB doublet, JAB = 14.7, 8H, Ar–CH2–Ar), 4.00 (12H, NMe), 3.94 (t, J = 6.5, 8H, CH2CH2N), 2.15 (qnapp, Japp = 7, 8H, CH2CH2O), 1.84 (qnapp, Japp = 7, 8H, CH2CH2N), 1.07 (36H, t-Bu).
6b, (1,3)-Cone-5,11,17,23-tetra(t-butyl)-25,26,27,28-tetra(4-N-(2,4,6-trimethylphenyl-imidazolium)butoxy- calix[4]arene tetrabromide.
MS (MALDI TOF): m/z 886.96 (M − 2Br)+/2.
1H-NMR: 10.34 (4H, N–CH–N), 9.02 (4H, N–CH), 7.02 (4H N–CH’), 6.95 (8H, Ar–H), 6.75 (8H, Ar–H’), 5.08 (t, J = 7.4 Hz, 8H, CH2-CH2-O), 4.35 and 3.13 (AB doublet, JAB = 12.4, 8H, Ar-CH2–Ar), 3.98 (t, 8H, J = 7.4 Hz, CH2-CH2-N), 2.31 (12H, Ar-pMe), 2.22 (qn, Jap = 7.4, CH2-CH2-O), 2.06 (qn, Jap = 7.4, 8H, CH2-CH2-N), 2.00 (24H, Ar–oMe), 1.06 (36H, t-Bu).
6c, (1,3)-Cone-5,11,17,23-tetra(t-butyl)-25,26,27,28-tetra-4-N-(2,6-diisopropylphenyl-imidazolium)butoxy- calix[4]arene tetrabromide.
MS (MALDI TOF): 971.06 (M − 2Br)+/2.
1H NMR: 10.49 (4H, NCHN), 9.23 (4H, CHN), 7.46 (t, 4H, ArH-4), 7.23 (d, 8H, ArH-3, ArH-5), 7.03 (4H, CHN), 6.75 (8H, Ar-H), 5.16 (t, J = 7.0, 8H, CH2CH2O), 4.38 and 3.13, (AB doublet, J = 12.8, 8H, Ar-CH2-Ar), 4.02 (t, J = 7.0, 8H, CH2CH2N), 2.37 (qnapp, J = 7.0, 8H, CH2CH2-O), 2.24 (m, 8, CHMe2), 2.24 (qnapp, 8H, CH2CH2N), 1.16 (d, J = 6.4, 12H, CHMe), 1.10 (d, J = 7.1, 12H, CHMe), 1.06 (36H, tBu).
5.1. Synthesis of 7a
A mixture of nickelocene (44 mg, 0.232 mmol) and 4a (143 mg, 0.116 mmol) was placed anaerobically in a Schlenk tube equipped with a reflux condenser, and thf (10 mL) was added. The dark green solution was refluxed for 2 d, and it slowly turned a deep red-violet color during this period. The mixture was cooled and subsequently filtered through a Celite pad, and the solvent was then removed under vacuum. The solid residue was washed with pentane (3 × 10 mL) and again dried under vacuum to afford the NHC complex 7a, C82H98N4O4Br2Ni2, as a red-violet powder (144 mg, 0.0972 mmol, 84%, 1480.9 g mol−1).
MS (MALDI TOF): 1401.0 (M − Br)+.
1H NMR (400 MHz): 8.02 (2H, CH-N), 7.08 (4H, Ar-H), 7.04 (4H, Ar-H), 6.88 (2H, CH-N), 6.70 (4H, Ar-H), 6.48 (2H, OH), 5.61 (t, J = 4.5, CH2-CH2-N), 4.77 (10H, C5H5), 4.55 (t, J = 4.7, 4H, CH2-CH2-O), 4.11 and 3.32 (AB doublets, J = 13.5, Ar-CH2-Ar), 2.40 (6H, Ar-CH3), 1.33 (30H, 12 H Ar-CH3 and 18H, t-Bu), 0.87 (18H, t-Bu).
7b, (C84H102N4O4Br2Ni2, M = 1508.9 g mol−1,110 mg, 73 mmol, 64%).
MS (MALDI TOF) 1429.58 (M − Br)+.
1H-NMR: 7.66 (2H, CH-N), 7.62 (2H, CH-N), 7.09 (4H, Ar-H), 7.06 (4H, Ar-H), 6.89 (2H, OH), 6.86 (8H, Ar-H), 5.34 (t, 4H, J = 6.6, CH2-CH2-N), 4.74 (10H, Cp), 4.12 and 3.38 (AB doublets, J = 13.2, Ar-CH2-Ar), 4.12 (t, 4H, J = 5.9, CH2-CH2-O), 2.99 (qu, 4H, J = 5.8, CH2-CH2-O), 2.41 (6H, Ar-para-CH3), 2.10 (qn, 4H, J = 6.3, CH2-CH2-O), 1.30 (12H, br., Ar-ortho-CH3), 1.30 (18H, t-Bu), 0.99 (18H, t-Bu).
7c, (C86H106N4O4Br2Ni2, M = 1536.98 g mol−1).
MS (MALDI TOF) 1457.6 (M − Br)+, 100%.
1H-NMR: 7.52 (2H, CH-N), 7.47 (2H, CH-N), 7.09 (4H, Ar-H), 7.06 4(4H, Ar-H), 6.87 (2H, OH), 6.81 (4H, Ar-H), 5.18 (t, 4H, J = 7, CH2-CH2-N), 4.78 (10H, Cp), 4.32 and 3.37, (8H, AB doublets, J = 13.5, Ar-CH2-Ar), 4.12 (t, 4H, J = 6.3, CH2-CH2-O), 2.52 (qn, 4H, J = 7.0, CH2-CH2-N), 2.41 (6H, Ar-para-CH3), 2.10 (qn, 4H, J = 6.3, CH2-CH2-O), 1.55 (12H, br., Ar-ortho-CH3), 1.30 (18H, t-Bu), 0.96 (18H, t-Bu).
7d, C88H110N4O4Br2Ni2, M = 1565 g mol−1, 98 mg, 63 mmol, 70%.
MS (MALDI TOF): m/z = 1485.63 (M − Br)+, 100%.
1H-NMR: 7.90 (2H, CH-N), 7.53 (t, 2H, J = 7.5, Ar-H), 7.35 (d, 4H, Ar-H), 7.10 (4H, Ar-H), 7.01 (2H, CH-N), 6.70 (4H, Ar-H), 6.35 (2H, OH), 4.73 (10H, Cp), 4.64 (t, 4H, J = 4.8, CH2-CH2-O), 4.09 and 3.31 (8H, AB doublets, J = 13.5 Hz, Ar-CH2-Ar), 1.86 (m, 4H, J = 3.2, CHMe2), 1.19 (18H, t-Bu), 1.19 (d, 6H, J = 6.9 Hz, CHMe2), 0.88 (d, 6H, CHMe2), 0.88 (18H, t-Bu).
5.2. Synthesis of Complex 8
Nickelocene (104 mg, 0.554 mmol), the imidazolium salt 4c (357 mg, 0.277 mmol), and 1,4-dioxane (10 mL) were introduced into a 50 mL round-bottomed flask equipped with a reflux condenser under Ar, and the mixture was heated, refluxed for 4 d and then allowed to cool. The solution was filtered over Celite, and the solvent was removed under vacuum. The resulting solid was washed with pentane, dried, and then redissolved in toluene. Layering with pentane yielded small crystals of 8, C76H96Br2N4O4Ni (1348.10 g mol−1, 45%, 170 mg, 0.126 mmol).
MS (MALDI TOF): m/z = 1268.20 (M − Br)+ 100%.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28155697/s1. Synthetic experimental details, 1H NMR data, and the Checkcif file for 5a are included in the supplementary information tables. Crystal data for complex 5a has been deposited in the CCDC database as deposition number 2258058 and can be accessed from there. The cif file for 8 is available as supplementary information on request.
Author Contributions
Conceptualization, Resources, Writing—review and editing, M.J.C.; Methodology, Software, Formal Analysis, Validation, Writing—original draft preparation, Visualization, and funding acquisition, M.J.C. and H.N.; Validation, data curation, supervision and project administration, M.J.C. and A.H.; Investigation, L.K. and M.J.C. All authors have read and agreed to the published version of the manuscript.
Funding
We are grateful to the University of Strasbourg, (université de Strasbourg), the French National Center for Scientific Research (CNRS, Centre National de la Recherche Scientifque) and the Tunisian government (for a Ph.D. scholarship for H.N.) for their financial help.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Corinne Bailly from the University of Strasbourg X-ray service for the structural determination of compound 8. We also acknowledge the CNRS and the University of Strasbourg for supporting and partially funding this work. The authors also thank E. Wasielewski and M. Chessé, who are responsible for the research platforms and facilities (Strasbourg NMR platform and Strasbourg chromatography facilities) of LIMA (UMR7042 CNRS-Unistra-UHA) and contributed, by their valuable technical and scientific support, to the achievement of this research project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gutsch, C.D. (Ed.) Calixarenes: An Introduction, 2nd ed.; Royal Society of Chemistry Publishing: London, UK, 2008; 276p. [Google Scholar]
- Mandolini, L.; Ungaro, R. (Eds.) Calixarenes in Action; Imperial College Press: London, UK, 2000; 271p. [Google Scholar]
- Casnati, A. Calixarenes and cations: A time-lapse photography of the big-bang. Chem. Commun. 2013, 49, 6827. [Google Scholar] [CrossRef] [PubMed]
- Morohashi, N.; Iijima, S.; Akasaka, K.; Hattori, T. Selective extraction of Pd(II) by p-tert-butylcalix[4]arenedicarboxylic acid. N. J. Chem. 2017, 41, 2231. [Google Scholar] [CrossRef]
- He, Q.; Zhang, Z.; Brewster, J.T.; Lynch, V.M.; Kim, S.K.; Sessler, J.L. Hemispherand-Strapped Calix[4]pyrrole: An Ion-pair Receptor for the Recognition and Extraction of Lithium Nitrite. J. Am. Chem. Soc. 2016, 138, 9779–9782. [Google Scholar] [CrossRef] [PubMed]
- Yeon, Y.; Leem, S.; Wagen, C.; Lynch, V.M.; Kim, S.K.; Sessler, J.L. 3-(Dicyanomethylidene)indan-1-one-Functionalized Calix[4]arene-Calix[4]pyrrole Hybrid: An Ion-Pair Sensor for Cesium Salts. Org. Lett. 2016, 18, 4396. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.I.; Barata, P.D.; Fialho, C.B.; Prata, J.V. Highly sensitive and selective fluorescent probes for Cu (II) detection based on calix[4]arene-oxacyclophane architectures. Molecules 2020, 25, 2456. [Google Scholar] [CrossRef]
- Canard, G.; Edzang, J.A.; Chen, Z.; Chesse, M.; Elhabiri, M.; Giorgi, M.; Siri, O. 1,3-Alternate Tetraamido-Azacalix[4]arenes as Selective Anion Receptors. Chem. Eur. J. 2016, 22, 5756. [Google Scholar] [CrossRef] [PubMed]
- Marcos, P.M.; Teixeira, F.A.; Segurado, M.A.P.; Ascenso, J.R.; Bernardino, R.J.; Michel, S.; Hubscher-Bruder, V.J. Bidentate Urea Derivatives of p-tert-Butyldihomooxacalix[4]arene: Neutral Receptors for Anion Complexation. Org. Chem. 2014, 79, 742. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Hay, B.P.; Lynch, V.M.; Li, H.; Sessler, J.L.; Kim, S.K. Calix[4]pyrrole-Based Molecular Capsule: Dihydrogen Phosphate-Promoted 1:2 Fluoride Anion Complexation. J. Am. Chem. Soc. 2022, 144, 16996. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Nanda Kishore, M.V.; Zhou, W.; He, Q. Recent advances in selective recognition of fluoride with macrocyclic receptors. Coord. Chem. Rev. 2022, 461, 214480. [Google Scholar] [CrossRef]
- Duncan, N.C.; Roach, B.D.; Williams, N.J.; Bonnesen, P.V.; Rajbanshi, A.; Moyer, B.A. N, N’-Dicyclohexyl-N’’-Isotridecylguanidine as Suppressor for the Next Generation Caustic Side Solvent Extraction (NG-CSSX) Process. Sep. Sci. Technol. 2012, 47, 2074. [Google Scholar]
- Casnati, A.; Della, C.N.; Fontanella, M.; Sansone, F.; Ugozzoli, F.; Ungaro, R.; Liger, K.; Dozol, J.-F. Calixarene-based picolinamide extractants for selective An/Ln separation from radioactive waste. Eur. J. Org. Chem. 2005, 11, 2338. [Google Scholar] [CrossRef]
- Jeunesse, C.; Armspach, D.; Matt, D. Playing with podands based on cone-shaped cavities. How can a cavity influence the properties of an appended metal center? Chem. Commun. 2005, 45, 5603–5614. [Google Scholar] [CrossRef] [PubMed]
- Peramo, A.; Abdellah, I.; Pecnard, S.; Mougin, J.; Martini, C.; Couvreur, P.; Huc, V.; Desmaeele, D. A Self-Assembling NHC-Pd-Loaded Calixarene as a Potent Catalyst for the Suzuki-Miyaura Cross-Coupling Reaction in Water. Molecules 2020, 25, 1459. [Google Scholar] [CrossRef]
- Zhang, X.; Tong, S.; Zhu, J.; Wang, M.-X. Inherently chiral calixarenes by a catalytic enantioselective desymmetrizing crossdehydrogenative coupling. Chem. Sci. 2023, 14, 827. [Google Scholar] [CrossRef] [PubMed]
- Notestein, J.M.; Iglesia, E.; Katz, A.J. Grafted Metallocalixarenes as Single-Site Surface Organometallic Catalysts. Am. Chem. Soc. 2004, 126, 6478. [Google Scholar] [CrossRef]
- Gramage-Doria, R.; Armspach, D.; Matt, D. Metalated cavitands (calixarenes, resorcinarenes, cyclodextrins) with internal coordination sites. Coord. Chem. Rev. 2013, 257, 776. [Google Scholar] [CrossRef]
- Gutsche, C.D.; Iqbal, M. p-tert-butylcalix[4]arene. Org. Synth. 1990, 68, 234. [Google Scholar]
- Gutsche, C.D.; Muthukrishnan, R. Calixarenes. 1. Analysis of the product mixtures produced by the base-catalyzed condensation of formaldehyde with para-substituted phenols. J. Org. Chem. 1978, 43, 4905. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X. Imidazolium Ionic Liquids, Imidazolylidene Heterocyclic Carbenes, and Zeolitic Imidazolate Frameworks for CO2 Capture and Photochemical Reduction. Angew. Chem. Int. Ed. 2016, 55, 2308. [Google Scholar] [CrossRef]
- Stassen, H.K.; Ludwig, R.; Wulf, A. Dupont, Imidazolium Salt Ion Pairs in Solution. J. Chem. Eur. J. 2015, 21, 8324. [Google Scholar] [CrossRef]
- Ritleng, V.; Barth, C.; Brenner, E.; Milosevic, S.; Chetcuti, M.J. Synthesis, structure, and solution dynamics of pentamethylcyclopentadienyl nickel complexes bearing N-heterocyclic carbene ligands. Organometallics 2008, 27, 4223. [Google Scholar] [CrossRef]
- Abernethy, C.D.; Cowley, A.H.; Jones, R.A.J. Reaction of nickelocene with 1,3-dimesitylimidazolium chloride. Organomet. Chem. 2000, 596, 3. [Google Scholar] [CrossRef]
- Kelly, R.A.; Scott, N.M.; Díez-González, S.; Stevens, E.D.; Nolan, S.P. Simple Synthesis of CpNi(NHC)Cl Complexes (Cp = Cyclopentadienyl; NHC = N-Heterocyclic Carbene). Organometallics 2005, 24, 3442. [Google Scholar] [CrossRef]
- Huynh, H.V. (Ed.) The Organometallic Chemistry of N-Heterocyclic Carbenes; Wiley: Hoboken, NJ, USA; West Sussex, UK, 2017. [Google Scholar]
- Nolan, S.; Cazin, C. Science of Synthesis: N-Heterocyclic Carbenes in Catalytic Organic Synthesis 1; Georg Thieme Verlag: Stuttgart, NY, USA, 2017. [Google Scholar]
- Chetcuti, M.J.; Devoille, A.M.J.; Ben Othman, A.; Souane, R.; Thiéury, P.; Vicens, J. Synthesis of mono-, di- and tetra-alkyne functionalized calix[4]arenes: Reactions of these multipodal ligands with dicobalt octacarbonyl to give complexes which contain up to eight cobalt atoms. Dalton Trans. 2009, 16, 2999. [Google Scholar] [CrossRef] [PubMed]
- Aloui, L.; Elhabiri, M.; Platas-Iglesias, C.; Esteban-Gomez, D.; Abidi, R.; Chetcuti, M.J. Synthesis and Characterization of Positively Charged tris-Imidazolium Calix[6]arene Hosts for Anion Recognition. ChemistrySelect 2019, 4, 321. [Google Scholar] [CrossRef]
- Aloui, L.; Abidi, R.; Chetcuti, M.J. We recently reported the syntheses and catalytic properties of calix[6]arenes tethered via N-heterocyclic carbene ligands, to (cyclopentadienyl)nickel complexes. Syntheses and characterization of nickel-N-heterocyclic carbenes linked to a calix[6]arene platform and their applications in Suzuki-Miyaura cross-coupling catalysis. Chim. Acta. 2020, 505, 119494. [Google Scholar]
- Ritleng, V.; Henrion, M.; Chetcuti, M.J. Nickel N-Heterocyclic Carbene-Catalyzed C-Heteroatom Bond Formation, Reduction, and Oxidation: Reactions and Mechanistic Aspects. ACS Catal. 2016, 6, 890. [Google Scholar] [CrossRef]
- Henrion, M.; Ritleng, V.; Chetcuti, M.J. Nickel N-Heterocyclic Carbene-Catalyzed C-C Bond Formation: Reactions and Mechanistic Aspects. ACS Catal. 2015, 5, 1283. [Google Scholar] [CrossRef]
- Rocquin, M.; Ritleng, V.; Barroso, S.; Martins, A.M.; Chetcuti, M.J. Synthesis of inexpensive chiral half-sandwich nickel N-heterocyclic carbene complexes: X-ray diffraction study of the D-menthyl-functionalized complex [Ni(iPr2Ph-NHC-CH2OMent)ClCp]. J. Organomet. Chem. 2016, 808, 57. [Google Scholar] [CrossRef]
- Henrion, M.; Chetcuti, M.J.; Ritleng, V. From acetone metalation to the catalytic α-arylation of acyclic ketones with NHC-nickel(II) complexes. Chem. Commun. 2014, 50, 4624. [Google Scholar] [CrossRef] [PubMed]
- Shahane, S.d.P.; Cardoso, B.; Chetcuti, M.J.; Ritleng, V. Benzothiazole nickelation: An obstacle to the catalytic arylation of azoles by cyclopentadienyl nickel N-heterocyclic carbene complexes. Catalysts 2019, 9, 76. [Google Scholar] [CrossRef]
- Ulm, F.; Poblador-Bahamonde, A.I.; Choppin, S.; Bellemin-Laponnaz, S.; Chetcuti, M.J.; Achard, T.; Ritleng, V. Synthesis, characterization, and catalytic application in aldehyde hydrosilylation of half-sandwich nickel complexes bearing (κ1-C)- and hemilabile (κ2-C,S)-thioether-functionalized NHC ligands. Dalton Trans. 2018, 47, 17134–17145. [Google Scholar] [CrossRef]
- Cardoso, B.d.P.; Bernard-Schaaf, J.-M.; Shahane, S.; Veiros, L.F.; Chetcuti, M.J.; Ritleng, V. Displacement of η5-cyclopentadienyl ligands from half-sandwich C,C-(NHC-cyanoalkyl)-nickel(II) metallacycles: Further insight into the structure of the resulting Cp-free nickelacycles and a catalytic activity study. Dalton Trans. 2018, 47, 1535. [Google Scholar] [CrossRef] [PubMed]
- Ulm, F.; Djukic, J.-P.; Chetcuti, M.J.; Ritleng, V. Hydroboration of Alkenes Catalysed by a Nickel N-Heterocyclic Carbene Complex: Reaction and Mechanistic Aspects. Chem. Eur. J. 2020, 26, 8916. [Google Scholar] [CrossRef] [PubMed]
- Ulm, F.; Shahane, S.; Truong-Phuoc, L.; Romero, T.; Papaefthimiou, V.; Chessé, M.; Chetcuti, M.J.; Pham-Huu, C.; Michon, C.; Ritleng, V. Half-Sandwich Nickel(II) NHC-Picolyl Complexes as Catalysts for the Hydrosilylation of Carbonyl Compounds: Evidence for NHC-Nickel Nanoparticles under Harsh Reaction Conditions. Eur. J. Inorg. Chem. 2021, 2021, 3074. [Google Scholar] [CrossRef]
- Bullough, E.K.; Kilner, C.A.; Little, M.A.; Willans, C.E. Tetrakis(methylimidazole) and tetrakis(methylimidazolium) calix[4]arenes: Competitive anion binding and deprotonation. Org. Biomolec. Chem. 2012, 10, 2824. [Google Scholar] [CrossRef] [PubMed]
- Botha, F.; Budka, J.; Eigner, V.; Hudeček, O.; Vrzal, L.; Císařová, I.; Lhoták, P. Recognition of chiral anions using calix[4]arene-based ureido receptor in the 1,3-alternate conformation. Tetrahedron 2014, 70, 477. [Google Scholar] [CrossRef]
- Tabakci, M.; Tabakci, B.; Beduk, A.D. Synthesis and application of an efficient calix[4]arene-based anion receptor bearing imidazole groups for Cr(VI) anionic species. Tetrahedron 2012, 68, 4182. [Google Scholar] [CrossRef]
- Sabater, P.; Zapata, F.; Caballero, A.; De La Visitación, N.; Alkorta, I.; Elguero, J.; Molina, P.J. Comparative Study of Charge-Assisted Hydrogen- and Halogen-Bonding Capabilities in Solution of Two-Armed Imidazolium Receptors toward Oxoanions. Org. Chem. 2016, 81, 7448. [Google Scholar] [CrossRef] [PubMed]
- Naghmouchi, H.; Chetcuti, M.J. Unpublished Results; University of Strasbourg: Strasbourg, France, 2016. [Google Scholar]
- Natarajan, N.; Chavagnan, T.; Semeril, D.; Brenner, E.; Matt, D.; Ramesh, R.; Toupet, L. Cavitand Chemistry: Nickel Half-Sandwich Complexes with Imidazolylidene Ligands Bearing One or Two Resorcinarenyl Substituents. Eur. J. Inorg. Chem. 2018, 7, 890. [Google Scholar] [CrossRef]
- Monnereau, L.; Semeril, D.; Matt, D.; Gourlaouen, C. Catalytic Behaviour of Calixarenylphosphanes in Nickel-Catalysed Suzuki-Miyaura Cross-Coupling. Eur. J. Inorg. Chem. 2017, 3, 581. [Google Scholar] [CrossRef]
- Sahin, N.; Semeril, D.; Brenner, E.; Matt, D.; Oezdemir, I.; Kaya, C.; Toupet, L. Catalytic Behaviour of Calixarenylphosphanes in Nickel-Catalysed Suzuki-Miyaura Cross-Coupling. Eur. J. Org. Chem. 2013, 20, 4443. [Google Scholar]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 6th ed.; Wiley Interscience: Hoboken, NJ, USA, 2014; p. 147. [Google Scholar]
- Henrion, M.; Oertel, A.M.; Ritleng, V.; Chetcuti, M.J. Facile displacement of η5-cyclopentadienyl ligands from half-sandwich alkyl,NHC-nickel complexes: An original route to robust cis-C,C-nickel square planar complexes. Chem. Commun. 2013, 49, 6424. [Google Scholar] [CrossRef]
- McGuinness, D.S.; Cavell, K.J.; Skelton, B.W.; White, A.H. Zerovalent Palladium and Nickel Complexes of Heterocyclic Carbenes: Oxidative Addition of Organic Halides, Carbon-Carbon Coupling Processes, and the Heck Reaction. Organometallics 1999, 18, 1596. [Google Scholar] [CrossRef]
- Huynh, H.V.; Holtgrewe, C.; Pape, T.; Koh, L.L.; Hahn, E. Synthesis and Structural Characterization of the First Bis(benzimidazolin-2-ylidene) Complexes of Nickel(II). Organometallics 2006, 25, 245. [Google Scholar] [CrossRef]
- Matsubara, K.; Ueno, K.; Shibata, Y. Synthesis and Structures of Nickel Halide Complexes Bearing Mono- and Bis-coordinated N-Heterocyclic Carbene Ligands, Catalyzing Grignard Cross-Coupling Reactions. Organometallics 2006, 25, 3422. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Xu, Y.-C.; Xie, L.-Z.; Sun, H.-M.; Shen, Q.; Zhang, Y. Controlled synthesis of nickel(ii) dihalides bearing two different or identical N-heterocyclic carbene ligands and the influence of carbene ligands on their structures and catalysis. Dalton Trans. 2011, 40, 4697. [Google Scholar] [CrossRef]
- Milosevic, S.; Brenner, E.; Ritleng, V.; Chetcuti, M.J. Unsaturated dinickel-molybdenum clusters with N-heterocyclic carbene ligands. Dalton Trans. 2008, 15, 1973. [Google Scholar] [CrossRef] [PubMed]
- Astakhov, A.V.; Khazipov, O.V.; Degtyareva, E.S.; Khrustalev, V.N.; Chernyshev, V.M.; Ananikov, V.P. Facile Hydrolysis of Nickel(II) Complexes with N-Heterocyclic Carbene Ligands. Organometallics 2015, 34, 5759. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3. [Google Scholar]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).