
Solvent-Free Determination of Selected Polycyclic Aromatic Hydrocarbons in Plant Material Used for Food Supplements Preparation: Optimization of a Solid Phase Microextraction Method

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Optimization of the HS-SPME
2.1.1. First Experimental Design
- X1—Incubation temperature, which corresponded to the exposition temperature (TEXP);
- X2—Incubation time (tINC);
- X3—Exposition time (tEXP);
- X4—Desorption time (tDES).
2.1.2. Second Experimental Design
2.2. Method Performances
2.3. Application to Real Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrumentation and GC-MS Analysis
3.3. Experimental Designs
- X1—Incubation temperature, which corresponded to the exposition temperature (TEXP);
- X2—Incubation time (tINC);
- X3—Exposition time (tEXP);
- X4—Desorption time (tDES).
3.4. Bud Samples
3.5. Optimal HS-SPME Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stahel, W.R. The Circular Economy. Nature 2016, 531, 435–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, P.; Yang, G.; Hu, L.; Sun, J.; Shi, L.; Zhou, Y.; Wang, Z.; Yang, J. Recent Advances in the Valorization of Plant Biomass. Biotechnol. Biofuels 2021, 14, 102. [Google Scholar] [CrossRef]
- Rifna, E.J.; Misra, N.N.; Dwivedi, M. Recent Advances in Extraction Technologies for Recovery of Bioactive Compounds Derived from Fruit and Vegetable Waste Peels: A Review. Crit. Rev. Food Sci. Nutr. 2023, 63, 719–752. [Google Scholar] [CrossRef] [PubMed]
- Galal, T.M.; Al-Yasi, H.M.; Fawzy, M.A.; Abdelkader, T.G.; Hamza, R.Z.; Eid, E.M.; Ali, E.F. Evaluation of the Phytochemical and Pharmacological Potential of Taif’s Rose (Rosa damascena Mill Var. trigintipetala) for Possible Recycling of Pruning Wastes. Life 2022, 12, 273. [Google Scholar] [CrossRef]
- Grohmann, D.; Petrucci, R.; Torre, L.; Micheli, M.; Menconi, M.E. Street Trees’ Management Perspectives: Reuse of Tilia Sp.’s Pruning Waste for Insulation Purposes. Urban For. Urban Green. 2019, 38, 177–182. [Google Scholar] [CrossRef]
- Donno, D.; Turrini, F.; Boggia, R.; Guido, M.; Gamba, G.; Mellano, M.G.; Riondato, I.; Beccaro, G.L. Sustainable Extraction and Use of Natural Bioactive Compounds from the Waste Management Process of Castanea Spp. Bud-Derivatives: The FINNOVER Project. Sustainability 2020, 12, 10640. [Google Scholar] [CrossRef]
- Turrini, F.; Donno, D.; Beccaro, G.L.; Pittaluga, A.; Grilli, M.; Zunin, P.; Boggia, R. Bud-Derivatives, a Novel Source of Polyphenols and How Different Extraction Processes Affect Their Composition. Foods 2020, 9, 1343. [Google Scholar] [CrossRef]
- Viippola, V.; Rantalainen, A.-L.; Yli-Pelkonen, V.; Tervo, P.; Setälä, H. Gaseous Polycyclic Aromatic Hydrocarbon Concentrations Are Higher in Urban Forests than Adjacent Open Areas during Summer but Not in Winter—Exploratory Study. Environ. Pollut. 2016, 208, 233–240. [Google Scholar] [CrossRef]
- Orecchio, S. PAHs Associated with the Leaves of Quercus ilex L.: Extraction, GC–MS Analysis, Distribution and Sources: Assessment of Air Quality in the Palermo (Italy) Area. Atmos. Environ. 2007, 41, 8669–8680. [Google Scholar] [CrossRef]
- Howsam, M.; Jones, K.C.; Ineson, P. PAHs Associated with the Leaves of Three Deciduous Tree Species. I—Concentrations and Profiles. Environ. Pollut. 2000, 108, 413–424. [Google Scholar] [CrossRef]
- Ratola, N.; Amigo, J.M.; Alves, A. Levels and Sources of PAHs in Selected Sites from Portugal: Biomonitoring with Pinus Pinea and Pinus Pinaster Needles. Arch. Environ. Contam. Toxicol. 2010, 58, 631–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nicola, F.; Baldantoni, D.; Sessa, L.; Monaci, F.; Bargagli, R.; Alfani, A. Distribution of Heavy Metals and Polycyclic Aromatic Hydrocarbons in Holm Oak Plant–Soil System Evaluated along Urbanization Gradients. Chemosphere 2015, 134, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Penning, T.M. Processing Contaminants: Polycyclic Aromatic Hydrocarbons (PAHs). In Encyclopedia of Food Safety; Motarjemi, Y., Ed.; Academic Press: Waltham, MA, USA, 2014; pp. 416–423. ISBN 978-0-12-378613-5. [Google Scholar]
- Abbas, I.; Badran, G.; Verdin, A.; Ledoux, F.; Roumié, M.; Courcot, D.; Garçon, G. Polycyclic Aromatic Hydrocarbon Derivatives in Airborne Particulate Matter: Sources, Analysis and Toxicity. Environ. Chem. Lett. 2018, 16, 439–475. [Google Scholar] [CrossRef]
- US EPA, O. Other Carcinogenic Polycyclic Aromatic Hydrocarbons. Available online: https://www.epa.gov/risk/other-carcinogenic-polycyclic-aromatic-hydrocarbons (accessed on 14 June 2023).
- Official Journal of the European Union, COMMISSION REGULATION (EU) 2015/1933—Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels for Polycyclic Aromatic Hydrocarbons in Cocoa Fibre, Banana Chips, Food Supplements, Dried Herbs and Dried Spices, 2015. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32015R1933 (accessed on 14 June 2023).
- Nowak, P.M.; Wietecha-Posłuszny, R.; Pawliszyn, J. White Analytical Chemistry: An Approach to Reconcile the Principles of Green Analytical Chemistry and Functionality. TrAC Trends Anal. Chem. 2021, 138, 116223. [Google Scholar] [CrossRef]
- Lancioni, C.; Castells, C.; Candal, R.; Tascon, M. Headspace Solid-Phase Microextraction: Fundamentals and Recent Advances. Adv. Sample Prep. 2022, 3, 100035. [Google Scholar] [CrossRef]
- Jalili, V.; Barkhordari, A.; Ghiasvand, A. Solid-Phase Microextraction Technique for Sampling and Preconcentration of Polycyclic Aromatic Hydrocarbons: A Review. Microchem. J. 2020, 157, 104967. [Google Scholar] [CrossRef]
- Huang, S.; He, S.; Xu, H.; Wu, P.; Jiang, R.; Zhu, F.; Luan, T.; Ouyang, G. Monitoring of Persistent Organic Pollutants in Seawater of the Pearl River Estuary with Rapid On-Site Active SPME Sampling Technique. Environ. Pollut. 2015, 200, 149–158. [Google Scholar] [CrossRef]
- Vasconcelos, P.H.M.; Camelo, A.L.M.; de Lima, A.C.A.; do Nascimento, H.O.; Vidal, C.B.; do Nascimento, R.F.; Lopes, G.S.; Longhinotti, E. Chemometric Tools Applied to Optimize a Fast Solid-Phase Microextraction Method for Analysis of Polycyclic Aromatic Hydrocarbons in Produced Water. Environ. Sci. Pollut. Res. 2021, 28, 8012–8021. [Google Scholar] [CrossRef]
- Feng, Z.; Huang, C.; Guo, Y.; Liu, W.; Zhang, L. Graphitic Carbon Nitride Derivative with Large Mesopores as Sorbent for Solid-Phase Microextraction of Polycyclic Aromatic Hydrocarbons. Talanta 2020, 209, 120541. [Google Scholar] [CrossRef]
- Arcoleo, A.; Bianchi, F.; Careri, M. Helical Multi-Walled Carbon Nanotube-Coated Fibers for Solid-Phase Microextraction Determination of Polycyclic Aromatic Hydrocarbons at Ultra-Trace Levels in Ice and Snow Samples. J. Chromatogr. A 2020, 1631, 461589. [Google Scholar] [CrossRef]
- Chen, X.-F.; Zang, H.; Wang, X.; Cheng, J.-G.; Zhao, R.-S.; Cheng, C.-G.; Lu, X.-Q. Metal–Organic Framework MIL-53(Al) as a Solid-Phase Microextraction Adsorbent for the Determination of 16 Polycyclic Aromatic Hydrocarbons in Water Samples by Gas Chromatography–Tandem Mass Spectrometry. Analyst 2012, 137, 5411–5419. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Pankajakshan, A.; Fornari, F.; Mandal, S.; Pelagatti, P.; Bacchi, A.; Mazzeo, P.P.; Careri, M. A Zinc Mixed-Ligand Microporous Metal-Organic Framework as Solid-Phase Microextraction Coating for Priority Polycyclic Aromatic Hydrocarbons from Water Samples. Microchem. J. 2020, 154, 104646. [Google Scholar] [CrossRef]
- Yang, C.; Wang, J.; Li, D. Microextraction Techniques for the Determination of Volatile and Semivolatile Organic Compounds from Plants: A Review. Anal. Chim. Acta 2013, 799, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Sarafraz-Yazdi, A.; Ghaemi, F.; Amiri, A. Comparative Study of the Sol–Gel Based Solid Phase Microextraction Fibers in Extraction of Naphthalene, Fluorene, Anthracene and Phenanthrene from Saffron Samples Extractants. Microchim. Acta 2012, 176, 317–325. [Google Scholar] [CrossRef]
- Limmer, M.A.; Balouet, J.-C.; Karg, F.; Vroblesky, D.A.; Burken, J.G. Phytoscreening for Chlorinated Solvents Using Rapid in Vitro SPME Sampling: Application to Urban Plume in Verl, Germany. Environ. Sci. Technol. 2011, 45, 8276–8282. [Google Scholar] [CrossRef] [PubMed]
- Popek, R.; Łukowski, A.; Bates, C.; Oleksyn, J. Accumulation of Particulate Matter, Heavy Metals, and Polycyclic Aromatic Hydrocarbons on the Leaves of Tilia cordata Mill. in Five Polish Cities with Different Levels of Air Pollution. Int. J. Phytoremediat. 2017, 19, 1134–1141. [Google Scholar] [CrossRef]
- De Nicola, F.; Alfani, A.; Maisto, G. Polycyclic Aromatic Hydrocarbon Contamination in an Urban Area Assessed by Quercus Ilex Leaves and Soil. Environ. Sci. Pollut. Res. 2014, 21, 7616–7623. [Google Scholar] [CrossRef] [Green Version]
- Klingberg, J.; Strandberg, B.; Sjöman, H.; Taube, M.; Wallin, G.; Pleijel, H. Polycyclic Aromatic Hydrocarbon (PAH) Accumulation in Quercus Palustris and Pinus Nigra in the Urban Landscape of Gothenburg, Sweden. Sci. Total Environ. 2022, 805, 150163. [Google Scholar] [CrossRef]
- Liang, J.; Fang, H.; Zhang, T.; Wang, X. Polycyclic Aromatic Hydrocarbons in the Leaves of Twelve Plant Species along an Urbanization Gradient in Shanghai, China. Environ. Sci. Pollut. Res. 2017, 24, 9361–9369. [Google Scholar] [CrossRef]
- Jabali, Y.; Millet, M.; El-Hoz, M. Determination of 16 Polycyclic Aromatic Hydrocarbons (PAHs) in Surface and Groundwater in North Lebanon by Using SPME Followed by GC–ITMS/MS. Euro-Mediterr. J. Environ. Integr. 2018, 3, 24. [Google Scholar] [CrossRef]
- Beiranvand, M.; Ghiasvand, A. Design and Optimization of the VA-TV-SPME Method for Ultrasensitive Determination of the PAHs in Polluted Water. Talanta 2020, 212, 120809. [Google Scholar] [CrossRef]
- Sulej-Suchomska, A.M.; Polkowska, Ż.; Chmiel, T.; Dymerski, T.M.; Kokot, Z.J.; Namieśnik, J. Solid Phase Microextraction–Comprehensive Two-Dimensional Gas Chromatography–Time-of-Flight Mass Spectrometry: A New Tool for Determining PAHs in Airport Runoff Water Samples. Anal. Methods 2016, 8, 4509–4520. [Google Scholar] [CrossRef] [Green Version]
- Vera Candioti, L.; De Zan, M.M.; Cámara, M.S.; Goicoechea, H.C. Experimental Design and Multiple Response Optimization. Using the Desirability Function in Analytical Methods Development. Talanta 2014, 124, 123–138. [Google Scholar] [CrossRef]
- Price, P.S.; Jayjock, M.A. Available Data on Naphthalene Exposures: Strengths and Limitations. Regul. Toxicol. Pharmacol. 2008, 51, 15–21. [Google Scholar] [CrossRef]
- Araujo, F.M.; Azevedo, G.C.; Nogueira, F.d.S.; Matos, R.C.; Matos, M.A.C. Eco-Friendly Method for the Determination of Polycyclic Aromatic Hydrocarbons in Sediments by HS-SPME-GC/MS. Chromatographia 2020, 83, 261–271. [Google Scholar] [CrossRef]
- Benedetti, B.; Di Carro, M.; Magi, E. Multivariate Optimization of an Extraction Procedure Based on Magnetic Molecular Imprinted Polymer for the Determination of Polycyclic Aromatic Hydrocarbons in Sea Water. Microchem. J. 2019, 145, 1199–1206. [Google Scholar] [CrossRef]
- Maghsoudi, S.; Noroozian, E. HP-SPME of Volatile Polycyclic Aromatic Hydrocarbons from Water Using Multiwalled Carbon Nanotubes Coated on a Steel Fiber through Electrophoretic Deposition. Chromatographia 2012, 75, 913–921. [Google Scholar] [CrossRef]
- Doong, R.; Chang, S.; Sun, Y. Solid-Phase Microextraction for Determining the Distribution of Sixteen US Environmental Protection Agency Polycyclic Aromatic Hydrocarbons in Water Samples. J. Chromatogr. A 2000, 879, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Ratola, N.; Lacorte, S.; Alves, A.; Barceló, D. Analysis of Polycyclic Aromatic Hydrocarbons in Pine Needles by Gas Chromatography–Mass Spectrometry: Comparison of Different Extraction and Clean-up Procedures. J. Chromatogr. A 2006, 1114, 198–204. [Google Scholar] [CrossRef]
- De Nicola, F.; Concha Graña, E.; Aboal, J.R.; Carballeira, A.; Fernández, J.Á.; López Mahía, P.; Prada Rodríguez, D.; Muniategui Lorenzo, S. PAH Detection in Quercus Robur Leaves and Pinus Pinaster Needles: A Fast Method for Biomonitoring Purpose. Talanta 2016, 153, 130–137. [Google Scholar] [CrossRef]
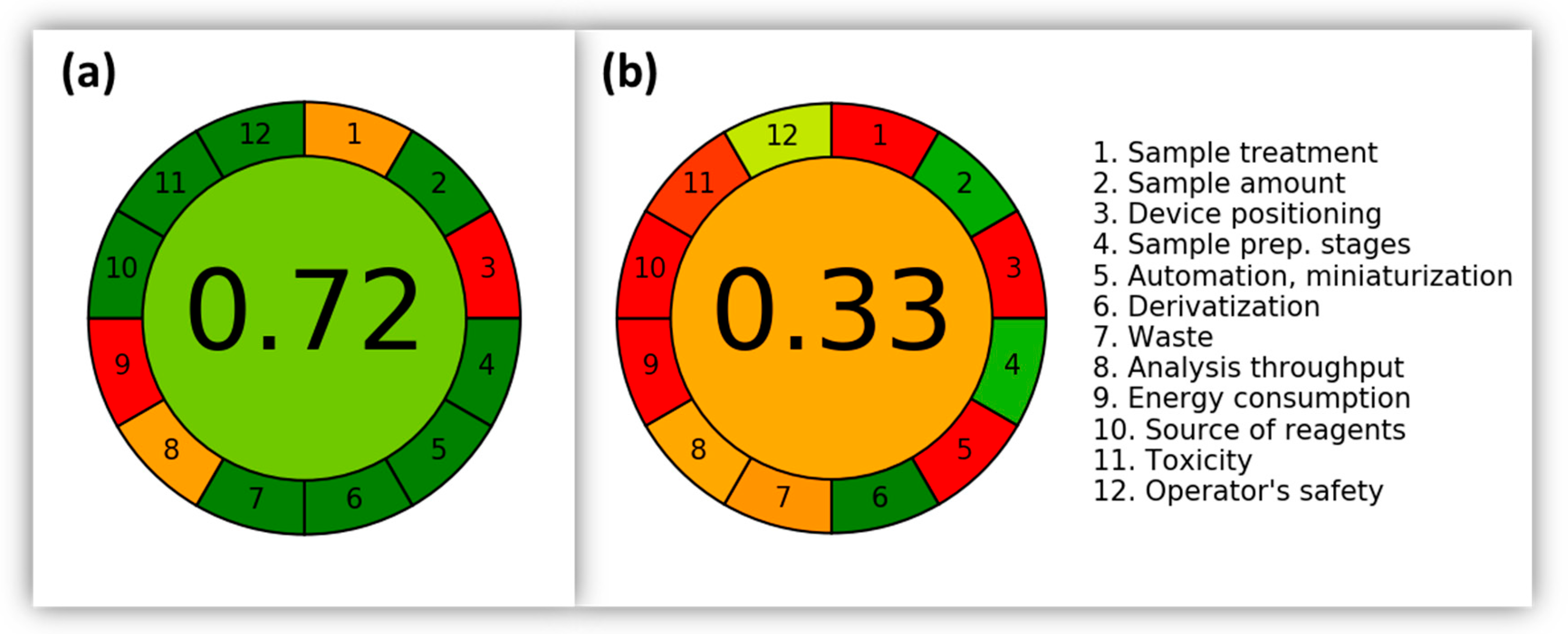
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Mao, Y.; Ke, Y.; Liu, W.; Cheng, C.; Shi, M.; Zhang, Z.; Zhang, J.; Qi, S.; Xing, X. Spatial and Seasonal Variations of PAHs in Soil, Air, and Atmospheric Bulk Deposition along the Plain to Mountain Transect in Hubei Province, Central China: Air-Soil Exchange and Long-Range Atmospheric Transport. Environ. Pollut. 2021, 291, 118139. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Chen, L.; Lu, Z.; Wang, J.; Yang, H.; Zhang, J.; Cai, M. Spatial Distribution and Source Apportionment of PAHs in Marine Surface Sediments of Prydz Bay, East Antarctica. Environ. Pollut. 2016, 219, 528–536. [Google Scholar] [CrossRef]
- Zelano, V.; Torazzo, A.; Berto, S.; Ginepro, M.; Prenesti, E.; Ferrari, A. Biomonitoring of Traffic Originated PAHs in the Air. Int. J. Environ. Anal. Chem. 2006, 86, 527–540. [Google Scholar] [CrossRef]
- Benedetti, B.; Caponigro, V.; Ardini, F. Experimental Design Step by Step: A Practical Guide for Beginners. Crit. Rev. Anal. Chem. 2022, 52, 1015–1028. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAHs | R2ADJ | Significant Coefficients |
|---|---|---|
| Acenaphthylene (ACL) | 63.06 | TEXP, TEXP2, TEXP × tEXP |
| Acenaphthene (AC) | 74.85 | TEXP, TEXP2, TEXP × tEXP |
| Fluorene (FL) | 90.34 | TEXP, tEXP, TEXP × tEXP |
| Phenanthrene (PH) | 96.21 | TEXP, tEXP, TEXP2, TEXP × tEXP |
| Anthracene (ANT) | 95.41 | TEXP, tEXP, TEXP2, TEXP × tEXP |
| Fluoranthene (FLT) | 77.71 | TEXP, tEXP, TEXP × tEXP |
| Pyrene (PY) | 70.05 | TEXP, tEXP, TEXP × tEXP |
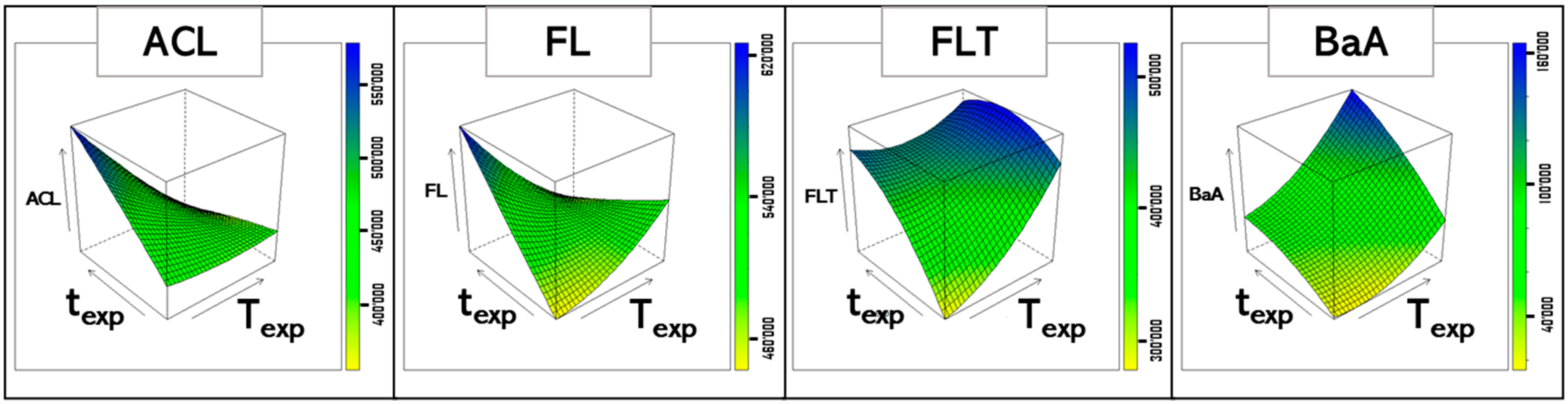
| PAHs | R2ADJ | Significant Coefficients |
|---|---|---|
| NAP | 65.78 | tEXP |
| ACL | 85.03 | TEXP, tEXP, TEXP × tEXP |
| AC | 75.03 | TEXP, TEXP × tEXP |
| FL | 75.01 | TEXP × tEXP |
| PH | 92.98 | tEXP, TEXP2, TEXP × tEXP |
| ANT | 90.16 | tEXP, TEXP × tEXP |
| FLT | 85.45 | TEXP, tEXP, TEXP × tEXP |
| PY | 70.52 | tEXP |
| BaA | 85.85 | TEXP, tEXP |
| CHR | 68.87 | TEXP, tEXP |
| BbF + BkF * | 60.63 | TEXP, tEXP |
| BaPY | 74.44 | TEXP, tEXP |
| Analyte | Linear Range (ng g−1) | R2 | LOD (ng g−1) | LOQ (ng g−1) | Inter-Day Precision (n = 6) | Trueness (n = 6) |
|---|---|---|---|---|---|---|
| NAP | LOQ-200 | 0.9967 | 1.4 | 4.6 | 9.1% | 88 ± 8 |
| ACL | LOQ-200 | 0.9967 | 1.3 | 4.3 | 4.9% | 98 ± 5 |
| AC | LOQ-200 | 0.9966 | 1.1 | 3.6 | 4.5% | 98 ± 4 |
| FL | LOQ-200 | 0.9970 | 1.2 | 4.0 | 3.5% | 101 ± 3 |
| PH | LOQ-200 | 0.9951 | 0.9 | 3.1 | 3.4% | 95 ± 3 |
| ANT | LOQ-200 | 0.9954 | 1.3 | 4.3 | 7.3% | 105 ± 8 |
| FLT | LOQ-200 | 0.9912 | 1.4 | 4.8 | 7.9% | 95 ± 8 |
| PY | LOQ-200 | 0.9945 | 1.2 | 3.9 | 11.2% | 95 ± 11 |
| BaA | LOQ-200 | 0.9697 | 3.0 | 10.0 | 34.7% * | 130 ± 45 |
| CHR | LOQ-200 | 0.9782 | 1.7 | 5.8 | 20.9% | 103 ± 22 |
| BbF + BkF | LOQ-200 | 0.9167 | 4.4 | 14.7 | 43.6% | 137 ± 60 |
| Coded Values | Real Values | |||
|---|---|---|---|---|
| X1 | X2 | X3 | X4 | |
| TEXP (°C) | tINC (min) | tEXP (min) | tDES (min) | |
| −1 | 40 | 20 | 20 | 1 |
| 0 | 55 | 35 | 35 | 3 |
| +1 | 70 | 50 | 50 | 5 |
| Coded Values | Real Values | |
|---|---|---|
| X1 | X3 | |
| TEXP (°C) | tEXP (min) | |
| −1 | 80 | 60 |
| 0 | 90 | 75 |
| +1 | 100 | 90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetti, B.; Di Carro, M.; Scapuzzi, C.; Magi, E. Solvent-Free Determination of Selected Polycyclic Aromatic Hydrocarbons in Plant Material Used for Food Supplements Preparation: Optimization of a Solid Phase Microextraction Method. Molecules 2023, 28, 5937. https://doi.org/10.3390/molecules28165937
Benedetti B, Di Carro M, Scapuzzi C, Magi E. Solvent-Free Determination of Selected Polycyclic Aromatic Hydrocarbons in Plant Material Used for Food Supplements Preparation: Optimization of a Solid Phase Microextraction Method. Molecules. 2023; 28(16):5937. https://doi.org/10.3390/molecules28165937
Chicago/Turabian StyleBenedetti, Barbara, Marina Di Carro, Chiara Scapuzzi, and Emanuele Magi. 2023. "Solvent-Free Determination of Selected Polycyclic Aromatic Hydrocarbons in Plant Material Used for Food Supplements Preparation: Optimization of a Solid Phase Microextraction Method" Molecules 28, no. 16: 5937. https://doi.org/10.3390/molecules28165937