

Tris(pentafluorophenyl)borane-catalyzed Hydride Transfer Reactions in Polysiloxane Chemistry—Piers–Rubinsztajn Reaction and Related Processes



Abstract
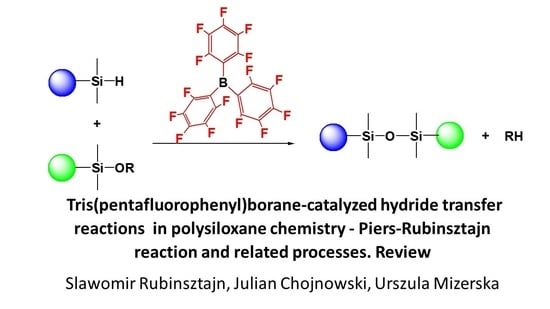
1. Introduction
2. Mechanism of Piers–Rubinsztajn Reaction
3. Hydride Transfer from Si to C: Piers–Rubinsztajn Reaction Leading to SiOSi
3.1. Cross-Linking of Linear Polysiloxanes in the Preparation of Silicone Elastomers, Coatings and Foams
3.2. Synthesis of Functionalized Polysiloxanes
3.3. Synthesis of Polysiloxanes Functionalized with Thermolabile Groups and Novel Thermoset Materials
3.4. Synthesis of Branched and Hyperbranched Polysiloxanes and MDTQ Resins
3.5. Preparation of Polysiloxanes with a Regular Complex Structure with Cyclic or Cage Groups in the Backbone
3.6. Modification of Nature-Based Materials Using the P-R Reaction
3.7. Synthesis of Polysiloxanes with Organic Fragments in Main Chains
4. Hydride Transfer from Si to C: P-R Reaction Leading to SiOC
5. Hydride Transfer from Si to H: Dehydrogenative Condensation Leading to SiOSi
6. Hydride Transfer from Si to H: Dehydrogenative Condensation Leading to SiOC
7. Hydride Transfer from Si to Si
8. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Butts, M.; Cella, J.; Wood, C.D.; Gillette, G.; Kerboua, R.; Leman, J.; Lewis, L.; Rubinsztajn, S.; Schattenmann, F.; Stein, J. Silicones. In Encyclopedia of Polymer Science and Technology, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; Volume 12, pp. 464–541. [Google Scholar]
- Mordor Intelligence Research & Advisory. Silicone Market Size & Share Analysis—Growth Trends & Forecasts (2023–2028). Mordor Intelligence, 2023. Available online: https://www.mordorintelligence.com/industry-reports/silicone-market (accessed on 22 July 2023).
- Kipping, F. Organic derivative of silicon. Preparation of alkyl silicon chlorides. Proc. Chem. Soc. 1904, 20, 15. [Google Scholar]
- Liebhafsky, H.A.; Liebhafsky, S.S.; Wise, G. Silicones under the Monogram: A Story of Industrial Research; John Wiley & Sons: Hoboken, NJ, USA, 1978. [Google Scholar]
- Noll, W. Chemistry and Technology of Silicones; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Li, Y.; Kawakami, Y. Efficient Synthesis of Poly(silyl ether)s by Pd/C and RhCl(PPh3)3-Catalyzed Cross-Dehydrocoupling Polymerization of Bis(hydrosilane)s with Diols. Macromolecules 1999, 32, 6871–6873. [Google Scholar] [CrossRef]
- Li, Y.; Seino, M.; Kawakami, Y. Asymmetric synthesis of optically active poly (silyl ether) s having reactive Si–H groups by stereoselective cross-dehydrocoupling polymerization of bis (silane) s with diols. Macromolecules 2000, 33, 5311–5314. [Google Scholar] [CrossRef]
- Zhang, R.; Mark, J.E.; Pinhas, A.R. Dehydrocoupling Polymerization of Bis-silanes and Disilanols to Poly(silphenylenesiloxane) As Catalyzed by Rhodium Complexes. Macromolecules 2000, 33, 3508–3510. [Google Scholar] [CrossRef]
- Massey, A.; Park, A. Perfluorophenyl derivatives of the elements: I. Tris(pentafluorophenyl)boron. J. Organomet. Chem. 1964, 2, 245–250. [Google Scholar] [CrossRef]
- Parks, D.J.; Blackwell, J.M.; Piers, W.E. Studies on the Mechanism of B(C6F5)3-Catalyzed Hydrosilation of Carbonyl Functions. J. Org. Chem. 2000, 65, 3090–3098. [Google Scholar] [CrossRef]
- Oestreich, M.; Hermeke, J.; Mohr, J. A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. [Google Scholar] [CrossRef]
- Hackel, T.; McGrath, N.A. Tris(pentafluorophenyl)borane-Catalyzed Reactions Using Silanes. Molecules 2019, 24, 432. [Google Scholar] [CrossRef]
- Fang, H.; Oestreich, M. Defunctionalisation catalysed by boron Lewis acids. Chem. Sci. 2020, 11, 12604–12615. [Google Scholar] [CrossRef]
- Parks, D.J.; Piers, W.E. Tris(pentafluorophenyl)boron-Catalyzed Hydrosilation of Aromatic Aldehydes, Ketones, and Esters. J. Am. Chem. Soc. 1996, 118, 9440–9441. [Google Scholar] [CrossRef]
- Gevorgyan, V.; Rubin, M.; Benson, S.; Liu, J.-X.; Yamamoto, Y. A Novel B(C6F5)3-Catalyzed Reduction of Alcohols and Cleavage of Aryl and Alkyl Ethers with Hydrosilanes. J. Org. Chem. 2000, 65, 6179–6186. [Google Scholar] [CrossRef]
- Gevorgyan, V.; Liu, J.-X.; Rubin, M.; Benson, S.; Yamamoto, Y. A novel reduction of alcohols and ethers with a HSiEt3catalytic B(C6F5)3 system. Tetrahedron Lett. 1999, 40, 8919–8922. [Google Scholar] [CrossRef]
- Blackwell, J.M.; Foster, K.L.; Beck, V.H.; Piers, W.E. B(C6F5)3-Catalyzed Silation of Alcohols: A Mild, General Method for Synthesis of Silyl Ethers. J. Org. Chem. 1999, 64, 4887–4892. [Google Scholar] [CrossRef]
- Piers, W.E. The Chemistry of Perfluoroaryl Boranes. Adv. Organomet. Chem. 2005, 52, 1–76. [Google Scholar] [CrossRef]
- Rubinsztajn, S.; Cella, J.A. Silicone Condensation Reaction. U.S. Patent 7064173B2, 20 June 2006. [Google Scholar]
- Cella, J.A.; Rubinsztajn, S. Silicone Condensation Reaction. U.S. Patent 7241851B2, 10 July 2007. [Google Scholar]
- Rubinsztajn, S.; Cella, J. Formation of siloxane bonds via new condensation process. Abstr. Pap. Am. Chem. Soc. 2004, 227, U452. [Google Scholar]
- Rubinsztajn, S.; Cella, J.A. A New Polycondensation Process for the Preparation of Polysiloxane Copolymers. Macromolecules 2005, 38, 1061–1063. [Google Scholar] [CrossRef]
- Brook, M.A.; Grande, J.B.; Ganachaud, F.; Muzafarov, A. New Synthetic Strategies for Structured Silicones Using B(C6F5)3. In Silicon Polymers; Springer: Berlin/Heidelberg, Germany, 2011; Volume 235, pp. 161–183. [Google Scholar] [CrossRef]
- Chen, X.; Yi, M.; Wu, S.; Tan, L.; Ge, X.; He, M.; Yin, G. Synthesis of Structurally Precise Polysiloxanes via the Piers–Rubinsztajn Reaction. Materials 2019, 12, 304. [Google Scholar] [CrossRef]
- Peng, J.; Bai, Y.; Li, J. Piers-Rubinsztajn Reaction and the Application in Siloxane/Polysiloxane Chemistry. Lett. Org. Chem. 2019, 16, 525–530. [Google Scholar] [CrossRef]
- Brook, M.A. New Control Over Silicone Synthesis using SiH Chemistry: The Piers-Rubinsztajn Reaction. Chem. A Eur. J. 2018, 24, 8458–8469. [Google Scholar] [CrossRef]
- Gao, H.; Battley, A.; Leitao, E.M. The ultimate Lewis acid catalyst: Using tris(pentafluorophenyl) borane to create bespoke siloxane architectures. Chem. Commun. 2022, 58, 7451–7465. [Google Scholar] [CrossRef]
- Zheng, S.; Brook, M.A. Reversible Redox Crosslinking of Thiopropylsilicones. Macromol. Rapid Commun. 2021, 42, e2000375. [Google Scholar] [CrossRef]
- Rubinsztajn, S. New Facile Process for Synthesis of Borosiloxane Resins. J. Inorg. Organomet. Polym. Mater. 2014, 24, 1092–1095. [Google Scholar] [CrossRef]
- Drozdov, F.V.; Milenin, S.A.; Gorodov, V.V.; Demchenko, N.V.; Buzin, M.I.; Muzafarov, A.M. Crosslinked polymers based on polyborosiloxanes: Synthesis and properties. J. Organomet. Chem. 2019, 891, 72–77. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. Silicon(II) Cation Cp*Si:+ X–: A New Class of Efficient Catalysts in Organosilicon Chemistry. Org. Process Res. Dev. 2019, 23, 2369–2377. [Google Scholar] [CrossRef]
- Fritz-Langhals, E.; Werge, S.; Kneissl, S.; Piroutek, P. Novel Si(II)+ and Ge(II)+ Compounds as Efficient Catalysts in Organosilicon Chemistry: Siloxane Coupling Reaction. Org. Process Res. Dev. 2020, 24, 1484–1495. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. Main Group Catalysis: Cationic Si(II) and Ge(II) Compounds as Catalysts in Organosilicon Chemistry. Reactions 2021, 2, 442–456. [Google Scholar] [CrossRef]
- Körte, L.A.; Schwabedissen, J.; Soffner, M.; Blomeyer, S.; Reuter, C.G.; Vishnevskiy, Y.V.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. Tris(perfluorotolyl)borane-A Boron Lewis Superacid. Angew. Chem. Int. Ed. 2017, 56, 8578–8582. [Google Scholar] [CrossRef]
- Sakata, K.; Fujimoto, H. Quantum Chemical Study of B(C6F5)3-Catalyzed Hydrosilylation of Carbonyl Group. J. Org. Chem. 2013, 78, 12505–12512. [Google Scholar] [CrossRef]
- Houghton, A.Y.; Hurmalainen, J.; Mansikkamäki, A.; Piers, W.E.; Tuononen, H.M. Direct observation of a borane–silane complex involved in frustrated Lewis-pair-mediated hydrosilylations. Nat. Chem. 2014, 6, 983–988. [Google Scholar] [CrossRef]
- Chojnowski, J.; Rubinsztajn, S.; Cella, J.A.; Fortuniak, W.; Cypryk, M.; Kurjata, J.; Kaźmierski, K. Mechanism of the B(C6F5)3-Catalyzed Reaction of Silyl Hydrides with Alkoxysilanes. Kinetic and Spectroscopic Studies. Organometallics 2005, 24, 6077–6084. [Google Scholar] [CrossRef]
- Beckett, M.A.; Brassington, D.S.; Coles, S.J.; Hursthouse, M.B. Lewis acidity of tris(pentafluorophenyl)borane: Crystal and molecular structure of B(C6F5)3·OPEt3. Inorg. Chem. Commun. 2000, 3, 530–533. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; Ugolotti, J.; White, A.J.P. From B(C6F5)3 to B(OC6F5)3: Synthesis of (C6F5)2BOC6F5 and C6F5B(OC6F5)2 and Their Relative Lewis Acidity. Organometallics 2005, 24, 1685–1691. [Google Scholar] [CrossRef]
- Rendler, S.; Oestreich, M. Conclusive Evidence for an SN2-Si Mechanism in the B(C6F5)3-Catalyzed Hydrosilylation of Carbonyl Compounds: Implications for the Related Hydrogenation. Angew. Chem. Int. Ed. 2008, 47, 5997–6000. [Google Scholar] [CrossRef]
- Mathew, J.; Eguchi, K.; Nakajima, Y.; Sato, K.; Shimada, S.; Choe, Y.-K. Tris(pentafluorophenyl)borane-Catalyzed Reactions of Siloxanes: A Combined Experimental and Computational Study. Eur. J. Org. Chem. 2017, 2017, 4922–4927. [Google Scholar] [CrossRef]
- Olah, G.A.; Li, X.-Y.; Wang, Q.; Rasul, G.; Prakash, G.S. Trisilyloxonium ions: Preparation, NMR spectroscopy, ab initio/IGLO studies, and their role in cationic polymerization of cyclosiloxanes. J. Am. Chem. Soc. 1995, 117, 8962–8966. [Google Scholar] [CrossRef]
- Cypryk, M.; Kurjata, J.; Chojnowski, J. Tertiary trisilyloxonium ion in cationic ring-opening polymerisation of a model cyclic siloxane, octamethyl-1, 4-dioxatetrasilacyclohexane. J. Organomet. Chem. 2003, 686, 373–378. [Google Scholar] [CrossRef]
- Sumerin, V.; Schulz, F.; Nieger, M.; Leskelä, M.; Repo, T.; Rieger, B. Facile Heterolytic H2Activation by Amines and B(C6F5)3. Angew. Chem. Int. Ed. 2008, 47, 6001–6003. [Google Scholar] [CrossRef]
- Bergquist, C.; Bridgewater, B.M.; Harlan, C.J.; Norton, J.R.; Friesner, R.A.; Parkin, G. Aqua, Alcohol, and Acetonitrile Adducts of Tris(perfluorophenyl)borane: Evaluation of Brønsted Acidity and Ligand Lability with Experimental and Computational Methods. J. Am. Chem. Soc. 2000, 122, 10581–10590. [Google Scholar] [CrossRef]
- Beringhelli, T.; Maggioni, D.; D’Alfonso, G. 1H and 19F NMR Investigation of the Reaction of B(C6F5)3 with Water in Toluene Solution. Organometallics 2001, 20, 4927–4938. [Google Scholar] [CrossRef]
- Rabanzo-Castillo, K.M.; Kumar, V.B.; Söhnel, T.; Leitao, E.M. Catalytic Synthesis of Oligosiloxanes Mediated by an Air Stable Catalyst, (C6F5)3B(OH2). Front. Chem. 2020, 8, 477. [Google Scholar] [CrossRef]
- Schneider, A.F.; Chen, Y.; Brook, M.A. Trace water affects tris(pentafluorophenyl)borane catalytic activity in the Piers–Rubinsztajn reaction. Dalton Trans. 2019, 48, 13599–13606. [Google Scholar] [CrossRef]
- Longuet, C.; Joly-Duhamel, C.; Ganachaud, F. Copolycondensation of Regular Functional Silane and Siloxane in Aqueous Emulsion Using B(C6F5)3 as a Catalyst. Macromol. Chem. Phys. 2007, 208, 1883–1892. [Google Scholar] [CrossRef]
- Longuet, C.; Ganachaud, F. Copolycondensation of Functional Silanes and Siloxanes in Solution Using tris (pentafluorophenyl) borane as a Catalyst in a View to Generate Hybrid Silicones. In Silicon Based Polymers: Advances in Synthesis and Supramolecular Organization; Springer: Berlin/Heidelberg, Germany, 2008; pp. 119–134. [Google Scholar] [CrossRef]
- Luo, L.; Marks, T.J. Ziegler–Natta catalyst activation. Thermodynamic and kinetic aspects of metallocenium ion-pair formation, dissociation, and structural reorganization. Top. Catal. 1999, 7, 97–106. [Google Scholar] [CrossRef]
- Fuller, A.-M.; Hughes, D.L.; Lancaster, S.J.; White, C.M. Synthesis and Structure of the Dimethyl Sulfide Adducts of Mono- and Bis(pentafluorophenyl)borane. Organometallics 2010, 29, 2194–2197. [Google Scholar] [CrossRef]
- Lorber, C.; Choukroun, R.; Vendier, L. Reactivity of B(C6F5)3 with Simple Early Transition Metal Alkoxides: Alkoxide-Aryl Exchange, THF Ring-Opening, or Acetonitrile CC Coupling. Organometallics 2008, 27, 5017–5024. [Google Scholar] [CrossRef]
- Janiak, C.; Braun, L.; Scharmann, T.G.; Girgsdies, F. A Water Adduct of Tris(pentafluorophenyl)borane: (C6F5)3B(OH2)–Dioxane–CH2Cl2(1/1/1). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 1722–1724. [Google Scholar] [CrossRef]
- Parks, D.J.; Piers, W.E.; Yap, G.P.A. Synthesis, Properties, and Hydroboration Activity of the Highly Electrophilic Borane Bis(pentafluorophenyl)borane, HB(C6F5)21. Organometallics 1998, 17, 5492–5503. [Google Scholar] [CrossRef]
- Rubinsztajn, S.; Chojnowski, J.; Cypryk, M.; Mizerska, U.; Fortuniak, W.; Bak-Sypien, I.I. Kinetic and mechanistic studies of the transformation of the catalyst, tris(pentafluorophenyl)borane, in the presence of silyl and germyl hydrides. J. Catal. 2019, 379, 90–99. [Google Scholar] [CrossRef]
- Wakabayashi, R.; Kuroda, K. Siloxane-Bond Formation Promoted by Lewis Acids: A Nonhydrolytic Sol-Gel Process and the Piers-Rubinsztajn Reaction. Chempluschem 2013, 78, 764–774. [Google Scholar] [CrossRef]
- Fawcett, A.S.; Grande, J.B.; Brook, M.A. Rapid, metal-free room temperature vulcanization produces silicone elastomers. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 644–652. [Google Scholar] [CrossRef]
- Grande, J.B.; Fawcett, A.S.; McLaughlin, A.J.; Gonzaga, F.; Bender, T.P.; Brook, M.A. Anhydrous formation of foamed silicone elastomers using the Piers–Rubinsztajn reaction. Polymer 2012, 53, 3135–3142. [Google Scholar] [CrossRef]
- Hickman, A.M.; Chmel, N.; Cameron, N.R.; Keddie, D.J.; Schiller, T.L. Influence of the tetraalkoxysilane crosslinker on the properties of polysiloxane-based elastomers prepared by the Lewis acid-catalysed Piers-Rubinsztajn reaction. Polym. Chem. 2021, 12, 4934–4941. [Google Scholar] [CrossRef]
- Khalimon, A.Y.; Piers, W.E.; Blackwell, J.M.; Michalak, D.J.; Parvez, M. A Photo Lewis Acid Generator (PhLAG): Controlled Photorelease of B(C6F5)3. J. Am. Chem. Soc. 2012, 134, 9601–9604. [Google Scholar] [CrossRef]
- Khalimon, A.Y.; Shaw, B.K.; Marwitz, A.J.V.; Piers, W.E.; Blackwell, J.M.; Parvez, M. Photo Lewis acid generators: Photorelease of B(C6F5)3 and applications to catalysis. Dalton Trans. 2015, 44, 18196–18206. [Google Scholar] [CrossRef]
- Liao, M.; Chen, Y.; Brook, M.A. Spatially Controlled Highly Branched Vinylsilicones. Polymers 2021, 13, 859. [Google Scholar] [CrossRef]
- Madsen, F.B.; Javakhishvili, I.; Jensen, R.E.; Daugaard, A.E.; Hvilsted, S.; Skov, A.L. Synthesis of telechelic vinyl/allyl functional siloxane copolymers with structural control. Polym. Chem. 2014, 5, 7054–7061. [Google Scholar] [CrossRef]
- Lusterio, A.; Brook, M.A. Naturally Derived Silicone Surfactants Based on Saccharides and Cysteamine. Molecules 2021, 26, 4802. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Brook, M.A. High-Throughput Synthesis and Characterization of Aryl Silicones by Using the Piers–Rubinsztajn Reaction. Chem. Eur. J. 2019, 25, 15367–15374. [Google Scholar] [CrossRef]
- Schneider, A.F.; Lu, E.K.; Lu, G.; Brook, M.A. Facile synthesis of phenyl-rich functional siloxanes from simple silanes. J. Polym. Sci. 2020, 58, 3095–3106. [Google Scholar] [CrossRef]
- Temnikov, M.N.; Vasil’ev, V.G.; Buzin, M.I.; Muzafarov, A.M. Synthesis and comparison of the rheological and thermal properties of acyclic and polycyclic forms of polyphenylsilsesquioxane. Eur. Polym. J. 2020, 130, 109676. [Google Scholar] [CrossRef]
- Matsumoto, K.; Shimada, S.; Sato, K. Sequence-Controlled Catalytic One-Pot Synthesis of Siloxane Oligomers. Chem. Eur. J. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Oba, Y.; Nakajima, Y.; Shimada, S.; Sato, K. One-Pot Sequence-Controlled Synthesis of Oligosiloxanes. Angew. Chem. 2018, 130, 4727–4731. [Google Scholar] [CrossRef]
- Fan, W.; Hong, N.; Sun, Q.; Li, M.; Fu, W. Thermo-curable and photo-patternable polysiloxanes and polycarbosiloxanes by a facile Piers–Rubinsztajn polycondensation and post-modification. Polym. Chem. 2022, 13, 2187–2194. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Z.; Zhu, T.; Li, Z.; Wang, J.; Cheng, Y. Multi-benzocyclobutene functionalized siloxane monomers prepared by Piers-Rubinsztajn reaction for low-k materials. Eur. Polym. J. 2020, 126, 109562. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.; Jin, K.; Wang, L.; Sun, J.; Fang, Q. A New Fluorinated Polysiloxane with Good Optical Properties and Low Dielectric Constant at High Frequency Based on Easily Available Tetraethoxysilane (TEOS). Macromolecules 2017, 50, 9394–9402. [Google Scholar] [CrossRef]
- Chen, X.; Fang, L.; Wang, J.; He, F.; Chen, X.; Wang, Y.; Zhou, J.; Tao, Y.; Sun, J.; Fang, Q. Intrinsic High Refractive Index Siloxane–Sulfide Polymer Networks Having High Thermostability and Transmittance via Thiol–Ene Cross-Linking Reaction. Macromolecules 2018, 51, 7567–7573. [Google Scholar] [CrossRef]
- Liu, F.; Chen, X.; Fang, L.; Sun, J.; Fang, Q. An effective strategy for the preparation of intrinsic low-k and ultralow-loss dielectric polysiloxanes at high frequency by introducing trifluoromethyl groups into the polymers. Polym. Chem. 2020, 11, 6163–6170. [Google Scholar] [CrossRef]
- Liu, F.; Chen, X.; Hou, J.; Sun, J.; Fang, Q. A Fluorinated Thermocrosslinkable Organosiloxane: A New Low-k Material at High Frequency with Low Water Uptake. Macromol. Rapid Commun. 2021, 42, e2000600. [Google Scholar] [CrossRef]
- Thompson, D.B.; Brook, M.A. Rapid Assembly of Complex 3D Siloxane Architectures. J. Am. Chem. Soc. 2008, 130, 32–33. [Google Scholar] [CrossRef]
- Grande, J.B.; Urlich, T.; Dickie, T.; Brook, M.A. Silicone dendrons and dendrimers from orthogonal SiH coupling reactions. Polym. Chem. 2014, 5, 6728–6739. [Google Scholar] [CrossRef]
- Morgan, J.; Chen, T.; Hayes, R.; Dickie, T.; Urlich, T.; Brook, M.A. Facile synthesis of dendron-branched silicone polymers. Polym. Chem. 2017, 8, 2743–2746. [Google Scholar] [CrossRef]
- Melendez-Zamudio, M.; Chavda, K.; Brook, M.A. Chelating Silicone Dendrons: Trying to Impact Organisms by Disrupting Ions at Interfaces. Molecules 2022, 27, 1869. [Google Scholar] [CrossRef] [PubMed]
- Robeyns, C.; Picard, L.; Ganachaud, F. Synthesis, characterization and modification of silicone resins: An “Augmented Review”. Prog. Org. Coat. 2018, 125, 287–315. [Google Scholar] [CrossRef]
- Flagg, D.H.; McCarthy, T.J. Rediscovering Silicones: MQ Copolymers. Macromolecules 2016, 49, 8581–8592. [Google Scholar] [CrossRef]
- Kurjata, J.; Fortuniak, W.; Rubinsztajn, S.; Chojnowski, J. B(C6F5)3 catalyzed dehydrocarbon polycondensation of PhSiH3 with (MeO)4Si as model polyfunctional comonomers in new route to hydrophobic silicone TQ resins. Eur. Polym. J. 2009, 45, 3372–3379. [Google Scholar] [CrossRef]
- Chojnowski, J.; Rubinsztajn, S.; Fortuniak, W.; Kurjata, J. Synthesis of Highly Branched Alkoxysiloxane–Dimethylsiloxane Copolymers by Nonhydrolytic Dehydrocarbon Polycondensation Catalyzed by Tris(pentafluorophenyl)borane. Macromolecules 2008, 41, 7352–7358. [Google Scholar] [CrossRef]
- Yi, M.; Chen, X.; Wu, S.; Ge, J.; Zhou, X.; Yin, G. Fabrication of Reactive Poly(Phenyl-Substituted Siloxanes/Silsesquioxanes) with Si–H and Alkoxy Functional Groups via the Piers–Rubinsztajn Reaction. Polymers 2018, 10, 1006. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Liang, S.; Chen, Y.; Brook, M.A. Hyperbranched Silicone MDTQ Tack Promoters. Molecules 2019, 24, 4133. [Google Scholar] [CrossRef]
- Wong, M.Y.; Vishnu, I.L.; Bui, R.; Chen, Y.; Schneider, A.F.; Brook, M.A. T- and Q-rich Linear Silicones from the Piers-Rubinsztajn Reaction. Silicon 2023, 15, 887–895. [Google Scholar] [CrossRef]
- Shi, Y.; Cai, J.; Wu, X.; Cheng, Y. Benzocyclobutene-functionalized hyperbranched polysiloxane for low-k materials with good thermostability. Des. Monomers Polym. 2021, 24, 285–292. [Google Scholar] [CrossRef]
- Rajendra, V.; Sicard, C.; Brennan, J.D.; Brook, M.A. Printing silicone-based hydrophobic barriers on paper for microfluidic assays using low-cost ink jet printers. Analyst 2014, 139, 6361–6365. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Liu, Y. Cyclic Polysiloxanes with Linked Cyclotetrasiloxane Subunits. Angew. Chem. Int. Engl. 2017, 56, 8706–8710. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Han, Y.; Liu, Y. Synthesis of spirocyclosiloxanes for transparent copolymer thermosets. J. Appl. Polym. Sci. 2018, 135, 16370. [Google Scholar] [CrossRef]
- Li, G.; Liu, Y. Cyclosiloxane-containing Polymers and the Formation of Highly Stable Elastomer. Chem. Lett. 2020, 49, 299–302. [Google Scholar] [CrossRef]
- Kawatsu, T.; Fuchise, K.; Takeuchi, K.; Choi, J.-C.; Sato, K.; Matsumoto, K. Well-defined hydrogen and organofunctional polysiloxanes with spiro-fused siloxane backbones. Polym. Chem. 2021, 12, 2222–2227. [Google Scholar] [CrossRef]
- Sodkhomkhum, R.; Ervithayasuporn, V. Synthesis of poly(siloxane/double-decker silsesquioxane) via dehydrocarbonative condensation reaction and its functionalization. Polymer 2016, 86, 113–119. [Google Scholar] [CrossRef]
- Zhu, H.; Hiruta, S.; Demirci, A.; Kim, S.; Hoshino, N.; Akutagawa, T.; Mitsuishi, M. Effects of Hydride Transfer Ring-Opening Reaction on B(C6F5)3 Catalyzed Polymerization of D4H Cyclosiloxane and Dialkoxysilanes toward Thermally Stable Silsesquioxane–Siloxane Hybrid Materials. Macromolecules 2022, 55, 10134–10144. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Shiba, H.; Wada, H.; Shimojima, A.; Kuroda, K. Polymerization of Cyclododecasiloxanes with Si-H and Si-OEt Side Groups by the Piers-Rubinsztajn Reaction. Bull. Chem. Soc. Jpn. 2018, 91, 747–753. [Google Scholar] [CrossRef]
- Liu, N.; Yu, J.; Meng, Y.; Liu, Y. Hyperbranched Polysiloxanes Based on Polyhedral Oligomeric Silsesquioxane Cages with Ultra-High Molecular Weight and Structural Tuneability. Polymers 2018, 10, 496. [Google Scholar] [CrossRef]
- Pan, D.; Yi, E.; Doan, P.H.; Furgal, J.C.; Schwartz, M.; Clark, S.; Goodson, T.; Laine, R.M. Microporous inorganic/organic hybrids via oxysilylation of a cubic symmetry nanobuilding block [(HMe2SiOSiO1.5)8] with RxSi(OEt)4−x. J. Ceram. Soc. Jpn. 2015, 123, 756–763. [Google Scholar] [CrossRef]
- Gale, C.; Chin, B.; Tambe, C.; Graiver, D.; Brook, M.A. Silicone Structurants for Soybean Oil: Foams, Elastomers, and Candles. ACS Sustain. Chem. Eng. 2018, 7, 1347–1352. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Sewell, P.; Brook, M.A. Utilization of softwood lignin as both crosslinker and reinforcing agent in silicone elastomers. Green Chem. 2015, 17, 1811–1819. [Google Scholar] [CrossRef]
- Tao, Y.; Zhou, J.; Fang, L.; Wang, Y.; Chen, X.; Chen, X.; Hou, J.; Sun, J.; Fang, Q. Fluoro-containing Polysiloxane Thermoset with Good Thermostability and Acid Resistance Based on the Renewable Multifunctional Vanillin. ACS Sustain. Chem. Eng. 2019, 7, 7304–7311. [Google Scholar] [CrossRef]
- Xunjun, C.; Yingde, C.; Guoqiang, Y.; Liewen, L. Synthesis of vinyl substitute poly(silphenylene-siloxane) via silyl hydride-dialkoxysilane process. J. Appl. Polym. Sci. 2007, 106, 1007–1013. [Google Scholar] [CrossRef]
- Tian, S.; Li, J.; Cai, Z.; Shi, B.; Chen, W.; Cheng, Y. Piers-Rubinsztajn reaction for BCB functionalized silphenylene/silbiphenylene siloxane oligomers to highly crosslinked low-k thermosets. Eur. Polym. J. 2018, 108, 373–379. [Google Scholar] [CrossRef]
- Liang, S.; Wong, M.Y.; Schneider, A.; Liao, M.; Kräuter, G.; Tchoul, M.N.; Chen, Y.; Brook, M.A. Transparent silphenylene elastomers from highly branched monomers. Polym. Chem. 2021, 12, 209–215. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, L.; Li, J.; Ma, Q. Hyperbranched Polycarbosiloxanes: Synthesis by Piers-Rubinsztajn Reaction and Application as Precursors to Magnetoceramics. Polymers 2020, 12, 672. [Google Scholar] [CrossRef]
- Ai, L.; Chen, Y.; He, L.; Luo, Y.; Li, S.; Xu, C. Synthesis of structured polysiloxazanes via a Piers–Rubinsztajn reaction. Chem. Commun. 2019, 55, 14019–14022. [Google Scholar] [CrossRef]
- Grande, J.B.; Thompson, D.B.; Gonzaga, F.; Brook, M.A. Testing the functional tolerance of the Piers–Rubinsztajn reaction: A new strategy for functional silicones. Chem. Commun. 2010, 46, 4988–4990. [Google Scholar] [CrossRef]
- Tan, J.; Xiong, X.; He, Z.; Cao, F.; Sun, D. Aggregation Behavior of Polyether Based Siloxane Surfactants in Aqueous Solutions: Effect of Alkyl Groups and Steric Hindrance. J. Phys. Chem. B 2019, 123, 1390–1399. [Google Scholar] [CrossRef]
- Meng, L.; Wang, W.; Li, L.; Feng, S. Interface Adsorption and Aggregation Behavior of Sulfonate-Based Anionic Silicone Surfactants in Aqueous Solution. Chempluschem 2023, 88, e202300152. [Google Scholar] [CrossRef]
- Cella, J.; Rubinsztajn, S. Preparation of Polyaryloxysilanes and Polyaryloxysiloxanes by B(C6F5)3 Catalyzed Polyetherification of Dihydrosilanes and Bis-Phenols. Macromolecules 2008, 41, 6965–6971. [Google Scholar] [CrossRef]
- Schneider, A.F.; Laidley, E.; Brook, M.A. Facile Synthesis of Cx(AB)yCxTriblock Silicone Copolymers Utilizing Moisture Mediated Living-End Chain Extension. Macromol. Chem. Phys. 2019, 220, 1800575. [Google Scholar] [CrossRef]
- Laengert, S.E.; Schneider, A.F.; Lovinger, E.; Chen, Y.; Brook, M.A. Sequential Functionalization of a Natural Crosslinker Leads to Designer Silicone Networks. Chem. Asian J. 2017, 12, 1208–1212. [Google Scholar] [CrossRef]
- Yi, M.; Chen, X.; Shuttleworth, P.S.; Tan, L.; Ruan, Y.; Xu, Y.; Zheng, J.; Wu, S.; Hu, S.; Xie, S.; et al. Facile fabrication of eugenol-containing polysiloxane films with good optical properties and excellent thermal stability via Si–H chemistry. J. Mater. Chem. C 2021, 9, 8020–8028. [Google Scholar] [CrossRef]
- Heo, J.; Kang, T.; Jang, S.G.; Hwang, D.S.; Spruell, J.M.; Killops, K.L.; Waite, J.H.; Hawker, C.J. Improved Performance of Protected Catecholic Polysiloxanes for Bioinspired Wet Adhesion to Surface Oxides. J. Am. Chem. Soc. 2012, 134, 20139–20145. [Google Scholar] [CrossRef] [PubMed]
- Torrens, A.A.; Ly, A.L.; Fong, D.; Adronov, A. Rapid and Mild Cleavage of Aryl-Alkyl Ethers to Liberate Phenols. Eur. J. Org. Chem. 2022, 2022, e202200570. [Google Scholar] [CrossRef]
- Shi, M.; Huang, G.; Sun, J.; Fang, Q. Constructing low-k polymers at high frequency from two propenyl-containing biomasses through the grubbs reaction. Polym. Chem. 2023, 14, 999–1006. [Google Scholar] [CrossRef]
- Grande, J.B.; Gonzaga, F.; Brook, M.A. Rapid assembly of explicit, functional silicones. Dalton Trans. 2010, 39, 9369–9378. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, M.; Laine, R.M. An Approach to Epoxy Resins: Oxysilylation of Epoxides. Macromolecules 2020, 53, 2249–2263. [Google Scholar] [CrossRef]
- Chaiprasert, T.; Liu, Y.; Takeda, N.; Unno, M. Janus ring siloxane: A versatile precursor of the extended Janus ring and tricyclic laddersiloxanes. Dalton Trans. 2020, 49, 13533–13537. [Google Scholar] [CrossRef] [PubMed]
- Chaiprasert, T.; Liu, Y.; Takeda, N.; Unno, M. Vinyl-Functionalized Janus Ring Siloxane: Potential Precursors to Hybrid Functional Materials. Materials 2021, 14, 2014. [Google Scholar] [CrossRef] [PubMed]
- Sample, C.S.; Lee, S.-H.; Li, S.; Bates, M.W.; Lensch, V.; Versaw, B.A.; Bates, C.M.; Hawker, C.J. Metal-Free Room-Temperature Vulcanization of Silicones via Borane Hydrosilylation. Macromolecules 2019, 52, 7244–7250. [Google Scholar] [CrossRef]
- Kamino, B.A.; Grande, J.B.; Brook, M.A.; Bender, T.P. Siloxane–Triarylamine Hybrids: Discrete Room Temperature Liquid Triarylamines via the Piers–Rubinsztajn Reaction. Org. Lett. 2011, 13, 154–157. [Google Scholar] [CrossRef]
- Kamino, B.A.; Mills, B.; Reali, C.; Gretton, M.J.; Brook, M.A.; Bender, T.P. Liquid Triarylamines: The Scope and Limitations of Piers–Rubinsztajn Conditions for Obtaining Triarylamine–Siloxane Hybrid Materials. J. Org. Chem. 2012, 77, 1663–1674. [Google Scholar] [CrossRef]
- Fu, R.; Yu, L.; Zhang, J.; Yu, H.; Feng, S.; Xu, X.-D. Facile construction of aggregation-induced emission molecular liquids via Piers-Rubinsztajn reaction for green fluorescent ink. Chin. Chem. Lett. 2022, 33, 1993–1996. [Google Scholar] [CrossRef]
- Gale, C.B.; Yan, Z.B.; Fefer, M.; Goward, G.R.; Brook, M.A. Synthesis of Siliconized Photosensitizers for Use in 1O2-Generating Silicone Elastomers: An Electron Paramagnetic Resonance Study. Macromolecules 2021, 54, 4333–4341. [Google Scholar] [CrossRef]
- Gale, C.B.; Brook, M.A.; Skov, A.L. Compatibilization of porphyrins for use as high permittivity fillers in low voltage actuating silicone dielectric elastomers. RSC Adv. 2020, 10, 18477–18486. [Google Scholar] [CrossRef]
- Feng, M.; Wang, N.; Li, J.; Feng, S.; Xu, X.-D. Facile construction of luminescent silicone elastomers from the compatibilization of porphyrins via the Piers-Rubinsztajn reaction. Colloids Surf. A Physicochem. Eng. Asp. 2022, 642, 128646. [Google Scholar] [CrossRef]
- Chadwick, R.C.; Grande, J.B.; Brook, M.A.; Adronov, A. Functionalization of Single-Walled Carbon Nanotubes via the Piers–Rubinsztajn Reaction. Macromolecules 2014, 47, 6527–6530. [Google Scholar] [CrossRef]
- Deforth, T.; Mignani, G. Use of a Boron Derivative as Heat Activated Catalyst for Polymerization and/or Crosslinking of Silicone by Dehydrogenative Condensation. U.S. Patent 20030139287A1, 24 July 2003. [Google Scholar]
- Zhou, D.; Kawakami, Y. Tris(pentafluorophenyl)borane as a Superior Catalyst in the Synthesis of Optically Active SiO-Containing Polymers. Macromolecules 2005, 38, 6902–6908. [Google Scholar] [CrossRef]
- Xue, L.; Kawakami, Y. Precise Synthesis of Poly(silphenylenesiloxane)s with Epoxy Side Functional Groups by Tris(pentafluorophenyl)borane as a Catalyst. Polym. J. 2007, 39, 379–388. [Google Scholar] [CrossRef]
- Szawiola, A.M.; Lessard, B.H.; Raboui, H.; Bender, T.P. Use of Piers–Rubinsztajn Chemistry to Access Unique and Challenging Silicon Phthalocyanines. ACS Omega 2021, 6, 26857–26869. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, I.K.; Tukhvatshin, R.S.; Kholodkov, D.N.; Novikov, R.A.; Solodilov, V.I.; Arzumanyan, A.V. Dumbbell-Shaped, Graft and Bottlebrush Polymers with All-Siloxane Nature: Synthetic Methodology, Thermal, and Rheological Behavior. Macromol. Rapid Commun. 2021, 42, e2000645. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, J.; Lewandowski, D.; Hreczycho, G. B(C6F5)3-Catalyzed Dehydrocoupling of POSS Silanols with Hydrosilanes: A Metal-Free Strategy for Effecting Functionalization of Silsesquioxanes. Inorg. Chem. 2020, 59, 9206–9214. [Google Scholar] [CrossRef]
- Lou, P.; Zhang, X.; Tan, Y.; Zhang, Z.; Bian, W.; Ma, H.; Xie, Z. 1H NMR relaxation and theoretical calculation study on Tris(pentafluorophenyl)borane as a catalyst in preparation of Poly(carborane-siloxane) polymers. Polym. Test. 2019, 73, 412–417. [Google Scholar] [CrossRef]
- Moitra, N.; Ichii, S.; Kamei, T.; Kanamori, K.; Zhu, Y.; Takeda, K.; Nakanishi, K.; Shimada, T. Surface Functionalization of Silica by Si–H Activation of Hydrosilanes. J. Am. Chem. Soc. 2014, 136, 11570–11573. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, Z.; Guo, B.; Zhang, L. Concurrently improved dispersion and interfacial interaction in rubber/nanosilica composites via efficient hydrosilane functionalization. Compos. Sci. Technol. 2019, 169, 217–223. [Google Scholar] [CrossRef]
- Sweetman, M.J.; McInnes, S.J.P.; Vasani, R.B.; Guinan, T.; Blencowe, A.; Voelcker, N.H. Rapid, metal-free hydrosilanisation chemistry for porous silicon surface modification. Chem. Commun. 2015, 51, 10640–10643. [Google Scholar] [CrossRef]
- Escorihuela, J.; Pujari, S.P.; Zuilhof, H. Organic Monolayers by B(C6F5)3-Catalyzed Siloxanation of Oxidized Silicon Surfaces. Langmuir 2017, 33, 2185–2193. [Google Scholar] [CrossRef]
- Liao, M.; Schneider, A.F.; Laengert, S.E.; Gale, C.B.; Chen, Y.; Brook, M.A. Living synthesis of silicone polymers controlled by humidity. Eur. Polym. J. 2018, 107, 287–293. [Google Scholar] [CrossRef]
- Tran, J.-A.; Madsen, J.; Skov, A.L. Scalable Synthetic Route to PDMS Ring Polymers in High Yields from Commercially Available Materials Using the Piers-Rubinsztajn Reaction. ACS Omega 2022, 7, 46884–46890. [Google Scholar] [CrossRef]
- Melendez-Zamudio, M.; Silverthorne, K.E.C.; Brook, M.A. High Refractive Index, Enantiopure Silicones Based on BINOL. Macromol. Rapid Commun. 2022, 43, e2200022. [Google Scholar] [CrossRef]
- Szawiola, A.M.; Souza, N.d.M.; Lessard, B.H.; Bender, T.P. Phenoxylated siloxane-based polymers via the Piers–Rubinsztajn process. Polym. Int. 2017, 66, 1324–1328. [Google Scholar] [CrossRef]
- Zhang, Z.; Lyons, L.J.; Jin, J.J.; Amine, K.; West, R. Synthesis and Ionic Conductivity of Cyclosiloxanes with Ethyleneoxy-Containing Substituents. Chem. Mater. 2005, 17, 5646–5650. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Brook, M.A. Reductive Degradation of Lignin and Model Compounds by Hydrosilanes. ACS Sustain. Chem. Eng. 2014, 2, 1983–1991. [Google Scholar] [CrossRef]
- Chen, Y.; Valentini, D.A.; Brook, M.A. Starch/Silicone Elastomers and Foams. Sustainability 2023, 15, 9941. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, S.; Yu, L.; Skov, A.L.; Etmimi, H.M.; Mallon, P.E.; Adronov, A.; Brook, M.A. Silicone-modified graphene oxide fillers via the Piers-Rubinsztajn reaction. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2379–2385. [Google Scholar] [CrossRef]
- Liao, M.; Chen, Y.; Brook, M.A. When Attempting Chain Extension, Even Without Solvent, It Is Not Possible to Avoid Chojnowski Metathesis Giving D3. Molecules 2021, 26, 231. [Google Scholar] [CrossRef]
- Rubinsztajn, S.; Cella, J.A.; Chojnowski, J.; Fortuniak, W.; Kurjata, J. Process for Synthesis of Diorganosilanes by Dispropor-tionation of Hydridosiloxanes. U.S. Patent 7148370B1, 12 December 2006. [Google Scholar]
- Chojnowski, J.; Fortuniak, W.; Kurjata, J.; Rubinsztajn, S.; Cella, J.A. Oligomerization of Hydrosiloxanes in the Presence of Tris(pentafluorophenyl)borane. Macromolecules 2006, 39, 3802–3807. [Google Scholar] [CrossRef]
- Chojnowski, J.; Rubinsztajn, S.; Fortuniak, W.; Kurjata, J. Oligomer and Polymer Formation in Hexamethylcyclotrisiloxane (D3)—Hydrosilane Systems Under Catalysis by tris(pentafluorophenyl)borane. J. Inorg. Organomet. Polym. Mater. 2007, 17, 173–187. [Google Scholar] [CrossRef]
- Barnes, Q.; Longuet, C.; Ganachaud, F. Cationic Polymerization of Hexamethylcyclotrisiloxane in Excess Water. Molecules 2021, 26, 4402. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, J.; Kurjata, J.; Fortuniak, W.; Rubinsztajn, S.; Trzebicka, B. Hydride Transfer Ring-Opening Polymerization of a Cyclic Oligomethylhydrosiloxane. Route to a Polymer of Closed Multicyclic Structure. Macromolecules 2012, 45, 2654–2661. [Google Scholar] [CrossRef]
- Mizerska, U.; Rubinsztajn, S.; Chojnowski, J.; Cypryk, M.; Uznanski, P.; Walkiewicz-Pietrzykowska, A.; Fortuniak, W. Self-Restructuring of Polyhydromethylsiloxanes by the Hydride Transfer Process: A New Approach to the Cross-Linking of Polysiloxanes and to the Fabrication of Thin Polysiloxane Coatings. Materials 2022, 15, 6981. [Google Scholar] [CrossRef] [PubMed]

































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents | Products |
|---|---|
| ≡Si-X + HO-Si≡; where X = halogen | ≡Si-O-Si≡ + HX |
| ≡Si-X + RO-Si≡; where X = halogen | ≡Si-O-Si≡ + RX |
| ≡Si-X + RC(O)O-Si≡; where X = halogen | ≡Si-O-Si≡ + RC(O)X |
| ≡Si-X + MtO-Si≡; where X = halogen, Mt = Li, Na, K | ≡Si-O-Si≡ + MtX |
| ≡Si-H + HO-Si≡ | ≡Si-O-Si≡ + H2 |
| ≡Si-OR + HO-Si≡ | ≡Si-O-Si≡ + ROH |
| ≡Si-O(O)CR + HO-Si≡ | ≡Si-O-Si≡ + RC(O)OH |
| ≡Si-O(O)CR + RO-Si≡ | ≡Si-O-Si≡ + RC(O)OR |
| ≡Si-NH2 + HO-Si≡ | ≡Si-O-Si≡ + NH3 |
| ≡Si-NR2 + HO-Si≡ | ≡Si-O-Si≡ + HNR2 |
| ≡Si-O(O)CCH3 + MtO-Si≡; where Mt = Li, Na, K | ≡Si-O-Si≡ + CH3C(O)OMt |
| Substrates | Products | Hydride Transfer | |
|---|---|---|---|
| 1 | R3SiH + R′3SiOR″ | R3SiOSiR′3 + R″H | From Si to C |
| 2 | R3SiH + R′OR″ | R3SiOR′ + R″H | From Si to C |
| 3 | R3SiH + HOSiR′3 | R3SiOSiR′3 + H2 | From Si to H |
| 4 | R3SiH + HOR′ | R3SiOR′ + H2 | From Si to H |
| 5 | R3SiH + HOH | R3SiOH + H2 | From Si to H |
| 6 | R3Si′H + R′3SiOR″ | R3Si′OR″ + R′3SiH | From Si to Si |
| 7 | R2HSiOSiHR2 + R2HSi′OSi′HR2 | R2HSiOSi′HR2 + R2HSiOSi′HR2 | From Si to Si |
| 8 | R2HSiOSiHR2 + R2HSi′OSi′HR2 | R2HSiOSiR2OSi′HR2 + R2Si′H2 | From Si to Si |
| 9 | R2HSiOSiHROSiHR2 + R3Si′OSi′RH2 | R2HSiOSi(OSi′R3)OSiHR2 + RSi′H3 | From Si to Si |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubinsztajn, S.; Chojnowski, J.; Mizerska, U. Tris(pentafluorophenyl)borane-catalyzed Hydride Transfer Reactions in Polysiloxane Chemistry—Piers–Rubinsztajn Reaction and Related Processes. Molecules 2023, 28, 5941. https://doi.org/10.3390/molecules28165941
Rubinsztajn S, Chojnowski J, Mizerska U. Tris(pentafluorophenyl)borane-catalyzed Hydride Transfer Reactions in Polysiloxane Chemistry—Piers–Rubinsztajn Reaction and Related Processes. Molecules. 2023; 28(16):5941. https://doi.org/10.3390/molecules28165941
Chicago/Turabian StyleRubinsztajn, Slawomir, Julian Chojnowski, and Urszula Mizerska. 2023. "Tris(pentafluorophenyl)borane-catalyzed Hydride Transfer Reactions in Polysiloxane Chemistry—Piers–Rubinsztajn Reaction and Related Processes" Molecules 28, no. 16: 5941. https://doi.org/10.3390/molecules28165941
APA StyleRubinsztajn, S., Chojnowski, J., & Mizerska, U. (2023). Tris(pentafluorophenyl)borane-catalyzed Hydride Transfer Reactions in Polysiloxane Chemistry—Piers–Rubinsztajn Reaction and Related Processes. Molecules, 28(16), 5941. https://doi.org/10.3390/molecules28165941