
Synthesis and Biological Evaluation of Novel 2-Amino-1,4-Naphthoquinone Amide-Oxime Derivatives as Potent IDO1/STAT3 Dual Inhibitors with Prospective Antitumor Effects

Abstract
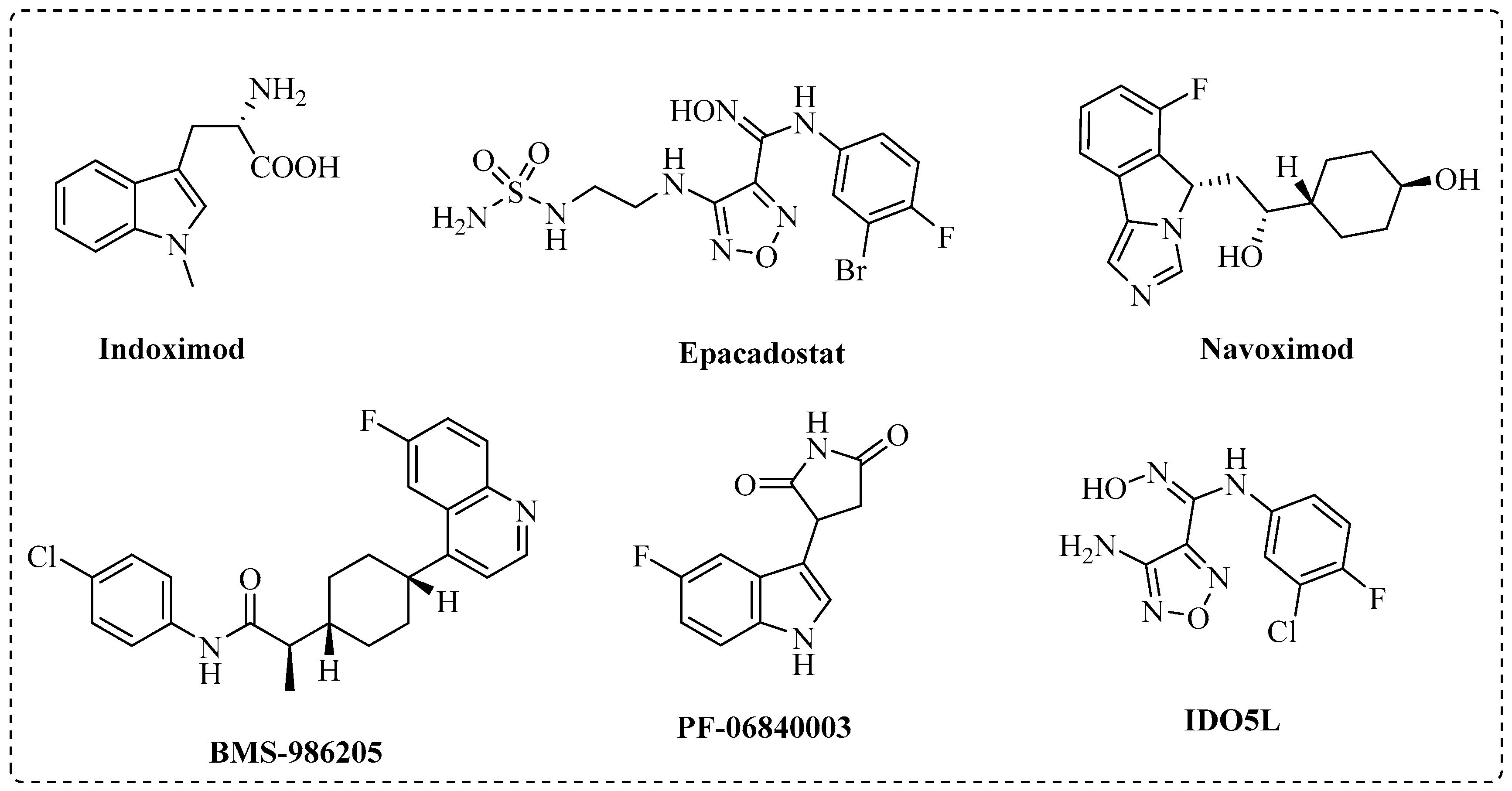
:1. Introduction
2. Results and Discussion
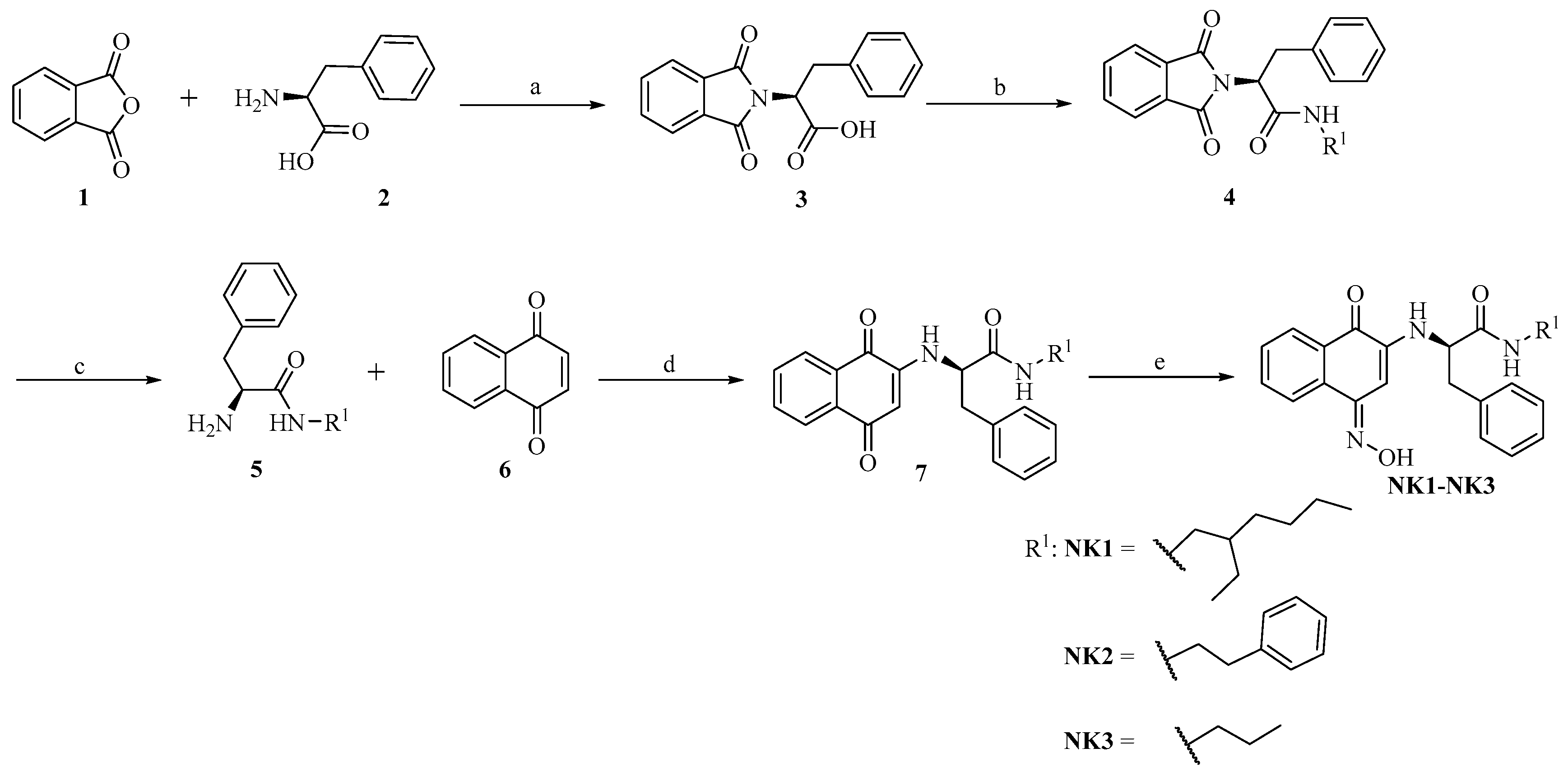
2.1. Chemistry
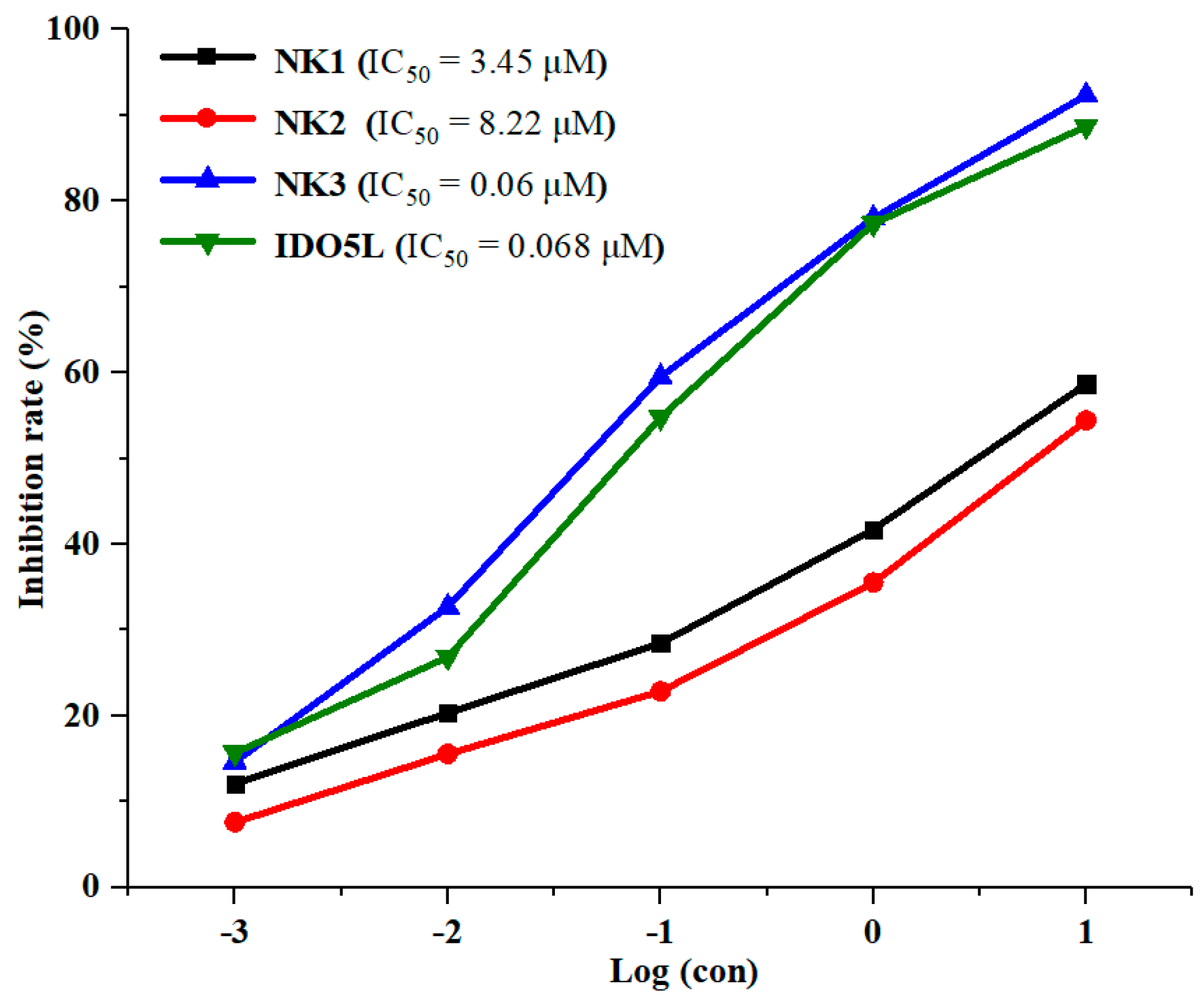
2.2. Inhibition of IDO1 Activity
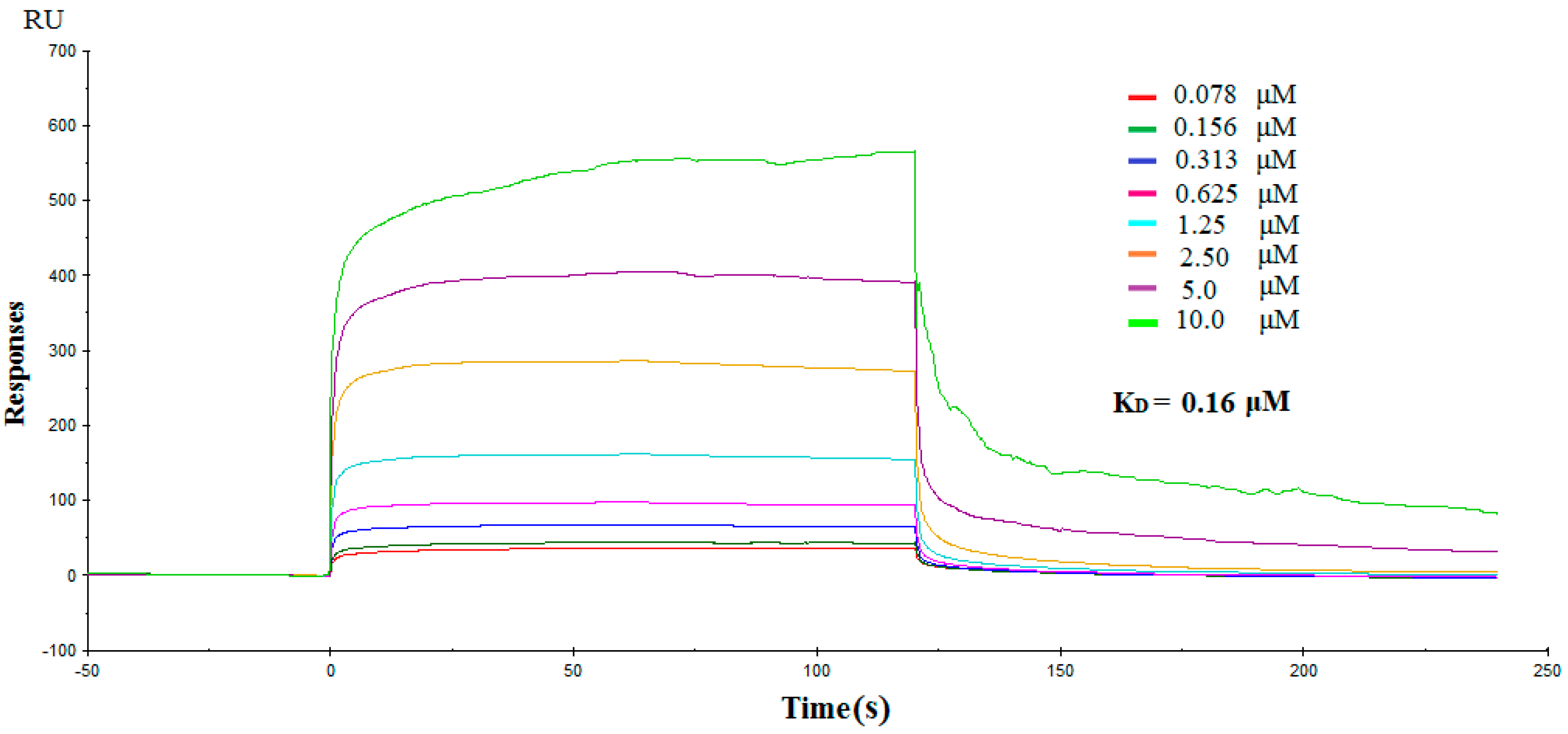
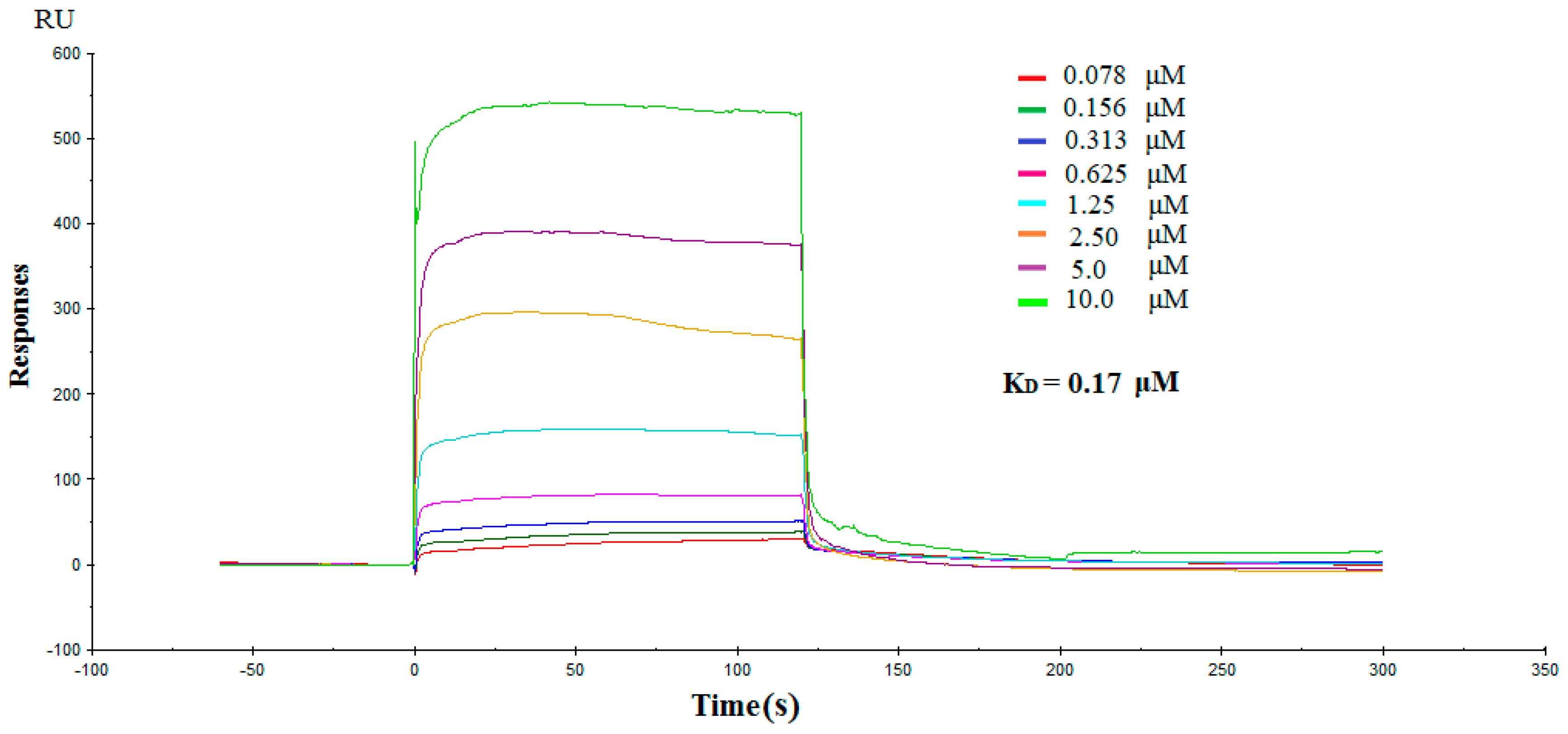
2.3. Direct Interactions between NK3 and IDO1
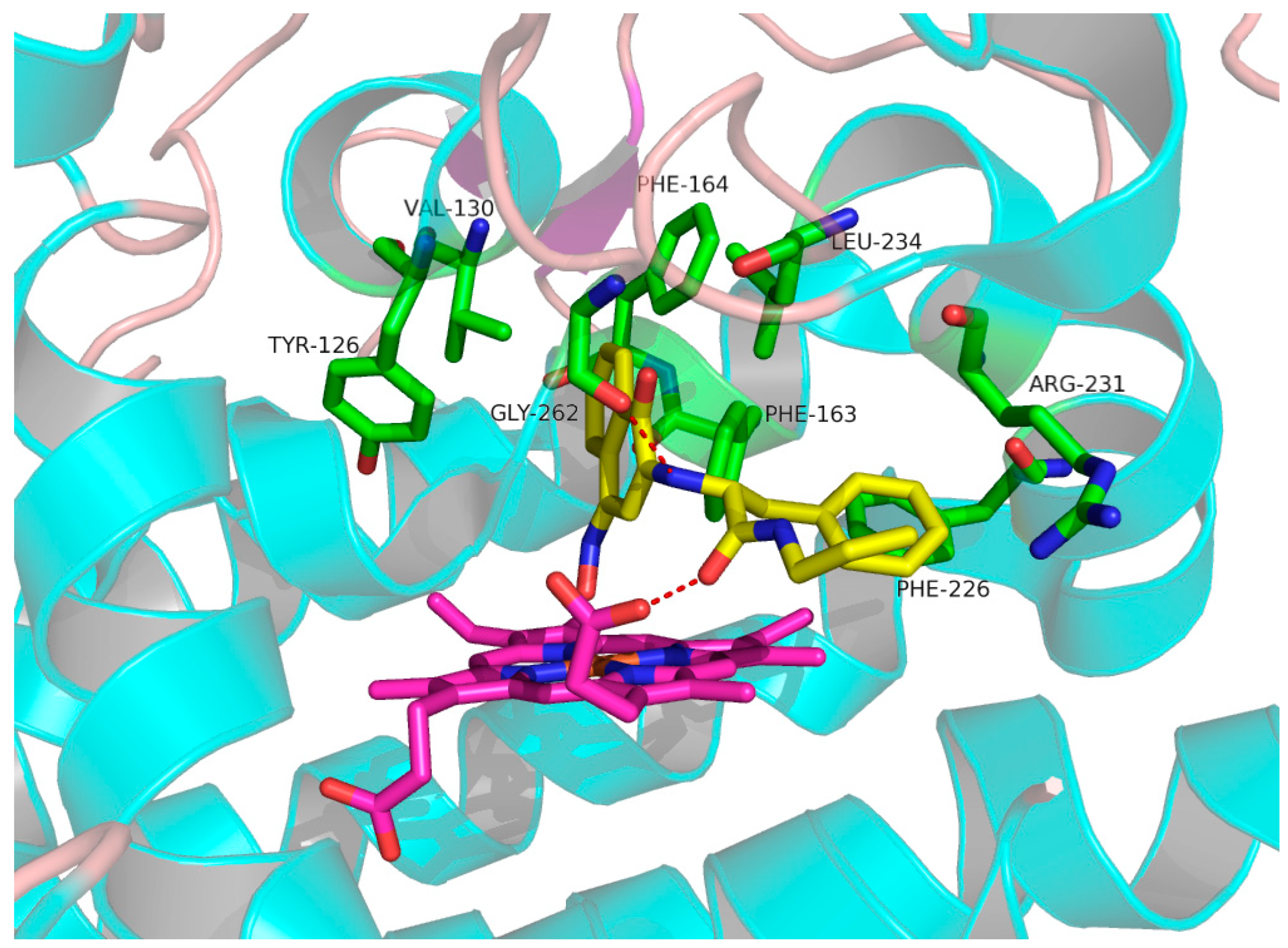
2.4. Molecular Docking Study of NK3 and IDO1
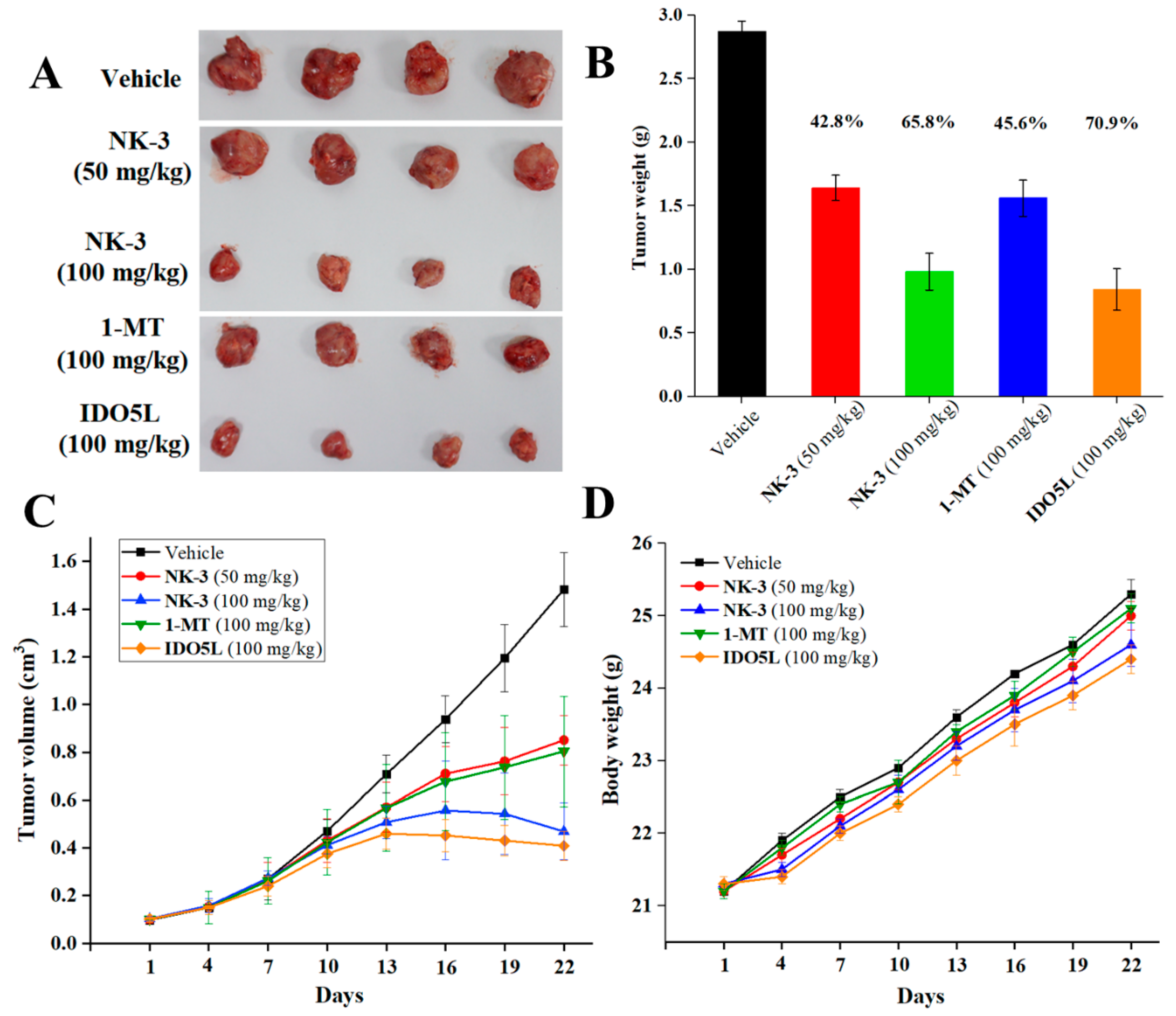
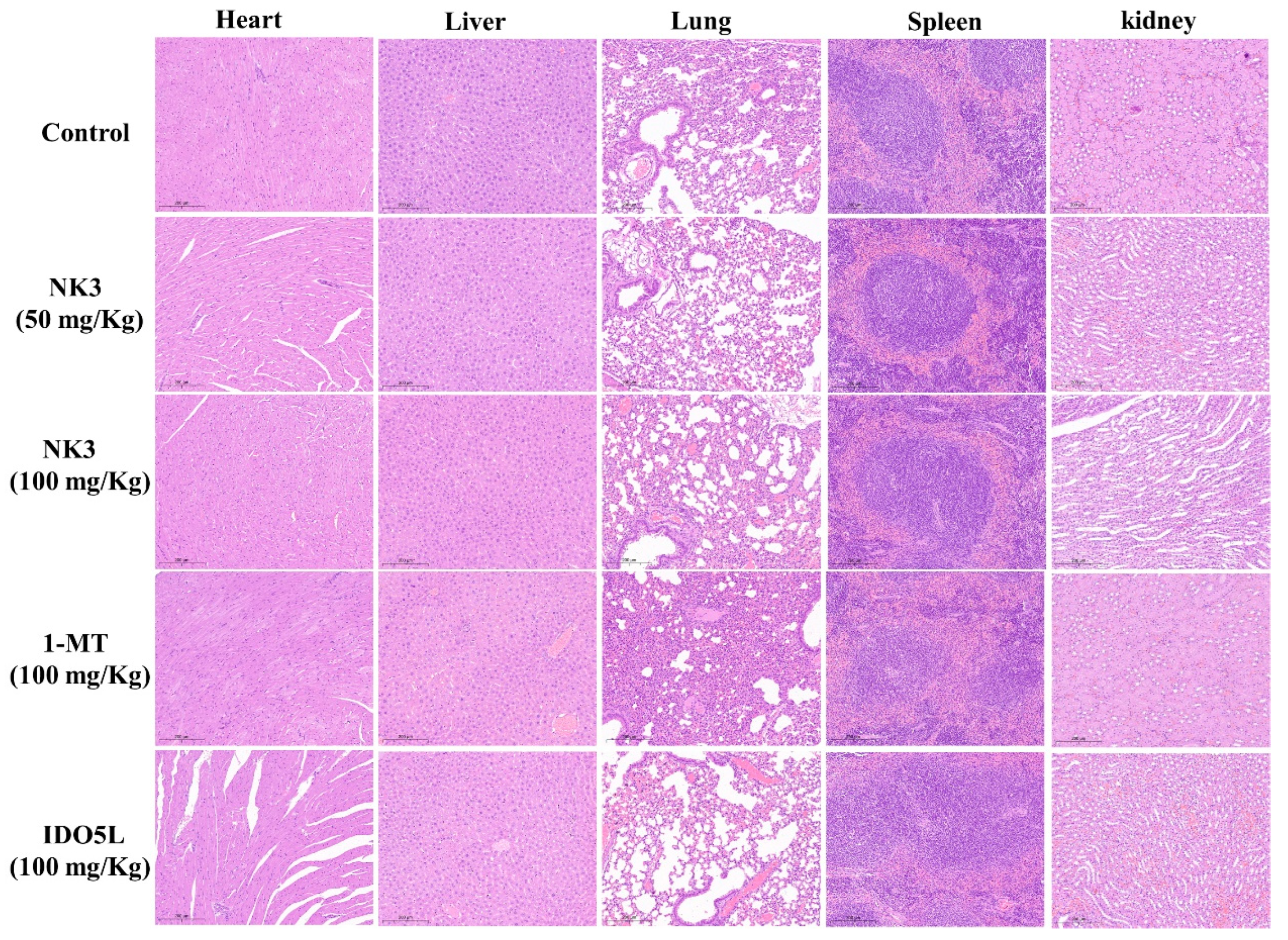
2.5. In Vivo Antitumor Efficacy of NK3 in CT26 Tumor-Bearing Mice
2.6. Cytotoxicity Assay
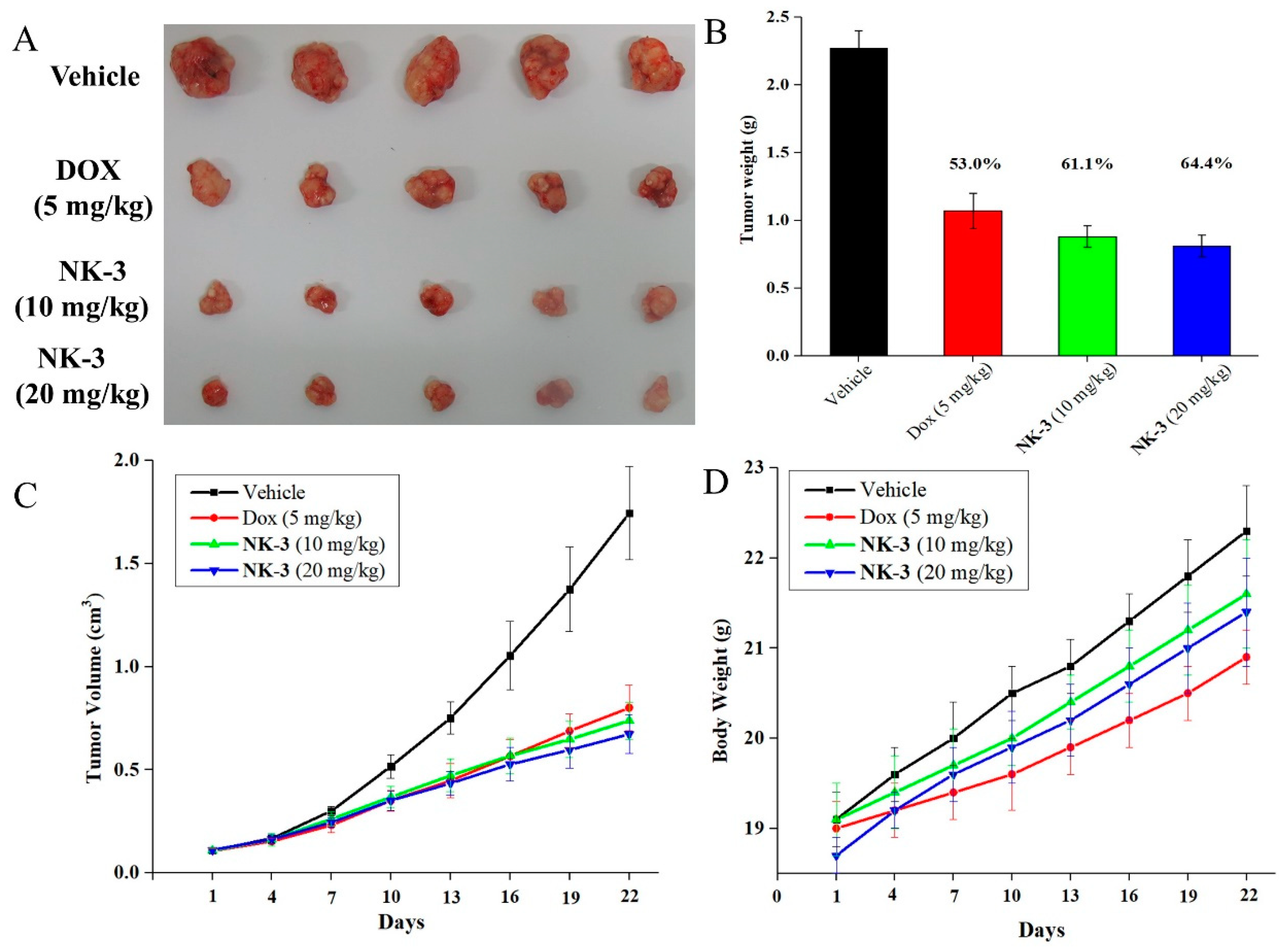
2.7. Antitumor Potency of NK3 in Nude Mice In Vivo
2.8. Compound NK3 Directly Bind with STAT3
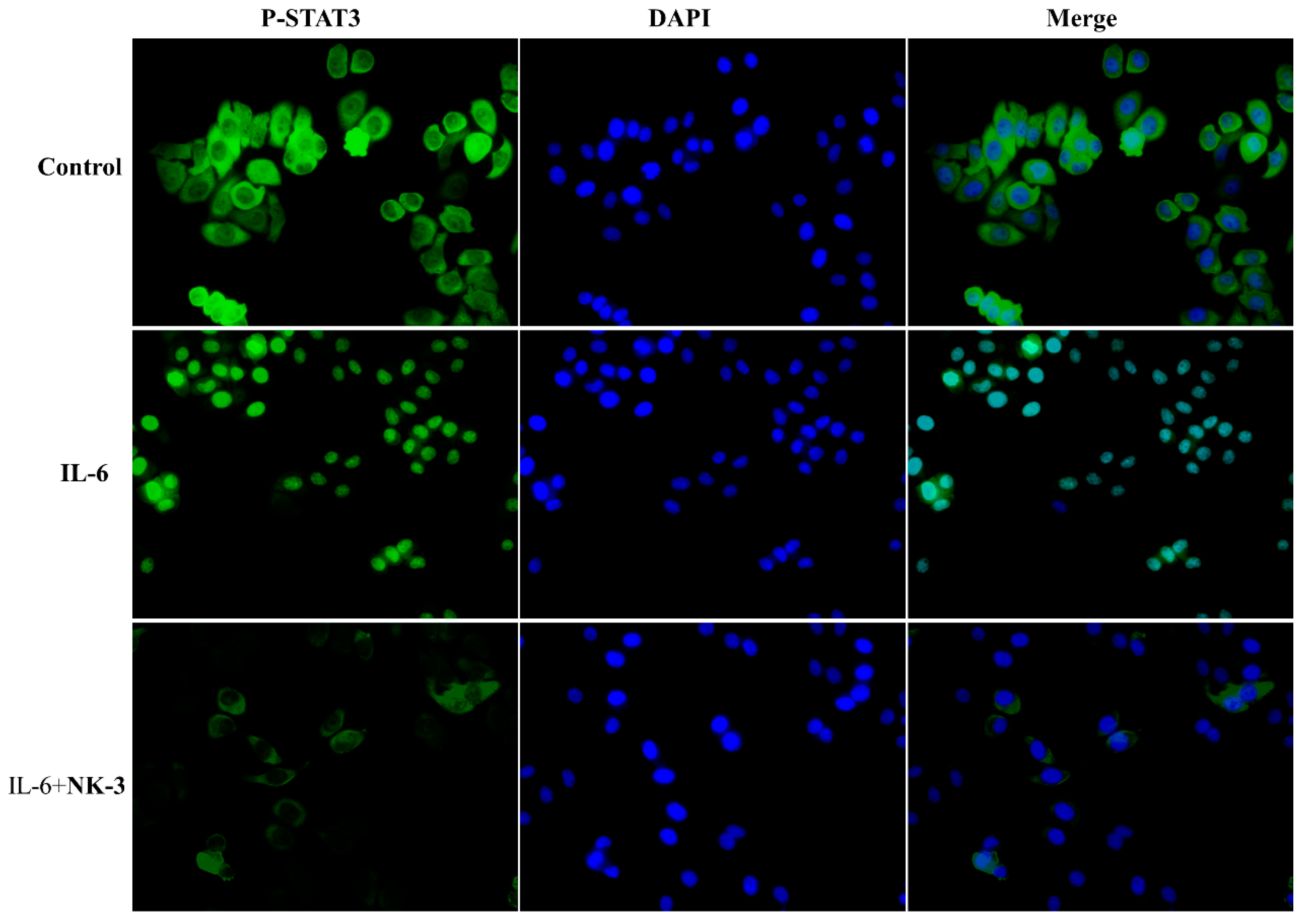
2.9. Compound NK3 Inhibited Nuclear Translocation of STAT3
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Preparation of Compounds NK1–NK3






3.3. Biological Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. New highlight of cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Patterson, L.F.S.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Brit. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Mondal, A.; Dey, S.; Laury-Kleintop, L.D.; Muller, A.J. Inflammatory reprogramming with IDO1 inhibitors: Turning immunologically unresponsive ‘cold’ tumors ‘hot’. Trends Cancer 2018, 4, 38–58. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Théta, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Eynde, B.J.V. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, R.; Li, S.; Liu, J. IDO1: An important immunotherapy target in cancer treatment. Int. Immunopharmacol. 2017, 47, 70–77. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Hu, F.; Li, Y.; Yang, Y.; Yan, J.; Kuang, C.; Yang, Q. Discovery of tryptanthrin derivatives as potent inhibitors of indoleamine 2,3-dioxygenase with therapeutic activity in lewis lung cancer (LLC) tumor-bearing mice. J. Med. Chem. 2013, 56, 8321–8331. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Heikkila, P.S.; Vaara, A.T.; von Smitten, K.A.J.; Vakkila, J.M.; Leidenius, M.H.K. Simultaneous Foxp3 and IDO expression is associated with sentinel lymph node metastases in breast cancer. BMC Cancer 2009, 9, 231–241. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR Pathway for Cancer Immunotherapy –Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; DuHadaway, J.B.; Jaller, D.; Curtis, P.; Metz, R.; Prendergast, G.C. Immunotherapeutic suppression of indoleamine 2,3-dioxygenase and tumor growth with ethyl pyruvate. Cancer Res. 2010, 70, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Leonardi, G.C.; Puccetti, P.; Fallarino, F.; Bianconi, V.; Sahebkar, A.; Baglivo, S.; Chiari, R.; Pirro, M. Targeting indoleamine-2,3-dioxygenase in cancer: Scientific rationale and clinical evidence. Pharmacol. Therapeut. 2019, 196, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, U.F.; Majjigapu, S.R.; Vogel, P.; Zoete, V.; Michielin, O. Challenges in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. J. Med. Chem. 2015, 58, 9421–9437. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Salunke, D.B. Diverse chemical space of indoleamine-2,3-dioxygenase 1 (Ido1) inhibitors. Eur. J. Med. Chem. 2021, 211, 113071. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liao, D.; Liu, D.; Ping, A.; Li, Z.; Bian, J. Development of Indoleamine 2,3-Dioxygenase 1 Inhibitors for Cancer Therapy and Beyond: A Recent Perspective. J. Med. Chem. 2020, 63, 15115–15139. [Google Scholar] [CrossRef]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Ravi, R.; Noonan, K.A.; Pham, V.; Bedi, R.; Zhavoronkov, A.; Ozerov, I.V.; Makarev, E.; Artemov, A.V.; Wysocki, P.T.; Mehra, R.; et al. Bifunctional immune checkpoint-targeted antibodyligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat. Commun. 2018, 9, 741–755. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, dierentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Cheng, X.-D.; Zhang, W.-D.; Qin, J.-J. Recent Update on Development of Small-Molecule STAT3 Inhibitors for Cancer Therapy: From Phosphorylation Inhibition to Protein Degradation. J. Med. Chem. 2021, 64, 8884–8915. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Crowe, P.J.; Goldstein, D.; Yang, J.L. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers. Int. J. Oncol. 2012, 41, 1181–1191. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef]
- Awuah, S.G.; Zheng, Y.R.; Bruno, P.M.; Hemann, M.T.; Lippard, S.J. A Pt(IV) pro-drug preferentially targets indoleamine-2,3-dioxygenase, providing enhanced ovarian cancer immune-chemotherapy. J. Am. Chem. Soc. 2015, 137, 14854–14857. [Google Scholar] [CrossRef]
- Huang, R.; Jing, X.; Huang, X.; Pan, Y.; Fang, Y.; Liang, G.; Liao, Z.; Wang, H.; Chen, Z.; Zhang, Y. Bifunctional Naphthoquinone Aromatic Amide-Oxime Derivatives Exert Combined Immunotherapeutic and Antitumor Effects through Simultaneous Targeting of Indoleamine-2,3-dioxygenase and Signal Transducer and Activator of Transcription 3. J. Med. Chem. 2020, 63, 1544–1563. [Google Scholar] [CrossRef]
- Al, M.; Hassan, H.; Fahamb, A.E.; Mohamed, G.; Khalid, A.F. Microwave irradiation: A facile, scalable and convenient method for synthesis of N-phthaloylamino acids. Arab. J. Chem. 2012, 5, 285–289. [Google Scholar]
- Cheng, M.F.; Hung, M.S.; Song, J.S.; Lin, S.Y.; Liao, F.Y.; Wu, M.H.; Hsiao, W.; Hsieh, C.L.; Wu, J.S.; Chao, Y.S.; et al. Discovery and structure-activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2,3-dioxygenase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3403–3406. [Google Scholar] [CrossRef] [PubMed]
- Yue, E.W.; Douty, B.; Wayland, B.; Bower, M.; Liu, X.; Leffet, L.; Wang, Q.; Bowman, K.J.; Hansbury, M.J.; Liu, C.; et al. Discovery of potent competitive inhibitors of indoleamine 2,3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model. J. Med. Chem. 2009, 52, 7364–7367. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, H.; Ding, C.; Jiang, D.; Zhao, Z.; Li, Y.; Ding, X.; Gao, J.; Zhou, H.; Luo, C.; et al. Celastrol suppresses colorectal cancer via covalent targeting peroxiredoxin 1. Signal Transduct. Target. 2023, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Tojo, S.; Kohno, T.; Tanaka, T.; Kamioka, S.; Ota, Y.; Ishii, T.; Kamimoto, K.; Asano, S.; Isobe, Y. Crystal structures and structure–activity relationships of imidazothiazole derivatives as IDO1 inhibitors. ACS Med. Chem. Lett. 2014, 5, 1119–1123. [Google Scholar] [CrossRef]
- He, X.; He, G.; Chu, Z.; Wu, H.; Wang, J.; Ge, Y.; Shen, H.; Zhang, S.; Shan, J.; Peng, K.; et al. Discovery of the First Potent IDO1/IDO2 Dual Inhibitors: A Promising Strategy for Cancer Immunotherapy. J. Med. Chem. 2021, 64, 17950–17968. [Google Scholar] [CrossRef]
- Wang, K.; Song, L.-H.; Liang, Q.-L.; Zhang, Y.; Ma, X.-L.; Wang, Q.; Zhang, H.-Y.; Jiang, C.-N.; Wei, J.-H.; Huang, R.-Z. Discovery of novel sulfonamide chromone-oxime derivatives as potent indoleamine 2,3-dioxygenase 1 inhibitors. Eur. J. Med. Chem. 2023, 254, 115349. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Li, C.; Zhang, W.; Xia, Y.; Li, S.; Lin, J.; Yu, K.; Liu, M.; Yang, L.; Luo, J.; et al. Discovery of an Orally Selective Inhibitor of Signal Transducer and Activator of Transcription 3 Using Advanced Multiple Ligand Simultaneous Docking. J. Med. Chem. 2017, 60, 2718–2731. [Google Scholar] [CrossRef]
- Fang, K.; Dong, G.; Li, Y.; He, S.; Wu, Y.; Wu, S.; Wang, W.; Sheng, C. Discovery of Novel Indoleamine 2,3-Dioxygenase 1 (IDO1) and Histone Deacetylase (HDAC) Dual Inhibitors. ACS Med. Chem. Lett. 2018, 9, 312–317. [Google Scholar] [CrossRef]
- Pan, L.; Zheng, Q.; Chen, Y.; Yang, R.; Yang, Y.; Li, Z.; Meng, X. Design, synthesis and biological evaluation of novel naphthoquinone derivatives as IDO1 inhibitors. Eur. J. Med. Chem. 2018, 157, 423–436. [Google Scholar] [CrossRef]
- Nair, V.S.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Ther. Targets 2018, 22, 547–557. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Zhang, Z.; Guo, Y.; Wu, Y.; Wang, R.; Wang, L.; Mao, S.; Yao, X. Overexpression of Indoleamine 2,3-Dioxygenase 1 Promotes Epithelial-Mesenchymal Transition by Activation of the IL-6/STAT3/PD-L1 Pathway in Bladder Cancer. Transl. Oncol. 2019, 12, 485–492. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Z.; Guan, L.; Li, J.; Chen, S.; Yu, W.; Lai, M. LYW-6, a Novel Cryptotanshinone Derived STAT3 Targeting Inhibitor, Suppresses Colorectal Cancer Growth and Metastasis. Pharmacol. Res. 2020, 153, 104661. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) a | |||
|---|---|---|---|
| Compd. | HepG2 | Hct-116 | SKOV3 |
| NK1 | 1.28 ± 0.27 | 0.49 ± 0.14 | 0.95 ± 0.18 |
| NK2 | 5.08 ± 0.33 | 5.84 ± 1.34 | 4.25 ± 1.65 |
| NK3 | 0.16 ± 0.04 | 0.18 ± 0.04 | 0.48 ± 0.23 |
| DOX | 0.28 ± 0.03 | 0.37 ± 0.02 | 0.58 ± 0.28 |
| IDO5L | >40 | >40 | >40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.-Z.; Liang, Q.-L.; Jing, X.-T.; Wang, K.; Zhang, H.-Y.; Wang, H.-S.; Ma, X.-L.; Wei, J.-H.; Zhang, Y. Synthesis and Biological Evaluation of Novel 2-Amino-1,4-Naphthoquinone Amide-Oxime Derivatives as Potent IDO1/STAT3 Dual Inhibitors with Prospective Antitumor Effects. Molecules 2023, 28, 6135. https://doi.org/10.3390/molecules28166135
Huang R-Z, Liang Q-L, Jing X-T, Wang K, Zhang H-Y, Wang H-S, Ma X-L, Wei J-H, Zhang Y. Synthesis and Biological Evaluation of Novel 2-Amino-1,4-Naphthoquinone Amide-Oxime Derivatives as Potent IDO1/STAT3 Dual Inhibitors with Prospective Antitumor Effects. Molecules. 2023; 28(16):6135. https://doi.org/10.3390/molecules28166135
Chicago/Turabian StyleHuang, Ri-Zhen, Qiao-Ling Liang, Xiao-Teng Jing, Ke Wang, Hui-Yong Zhang, Heng-Shan Wang, Xian-Li Ma, Jian-Hua Wei, and Ye Zhang. 2023. "Synthesis and Biological Evaluation of Novel 2-Amino-1,4-Naphthoquinone Amide-Oxime Derivatives as Potent IDO1/STAT3 Dual Inhibitors with Prospective Antitumor Effects" Molecules 28, no. 16: 6135. https://doi.org/10.3390/molecules28166135
APA StyleHuang, R.-Z., Liang, Q.-L., Jing, X.-T., Wang, K., Zhang, H.-Y., Wang, H.-S., Ma, X.-L., Wei, J.-H., & Zhang, Y. (2023). Synthesis and Biological Evaluation of Novel 2-Amino-1,4-Naphthoquinone Amide-Oxime Derivatives as Potent IDO1/STAT3 Dual Inhibitors with Prospective Antitumor Effects. Molecules, 28(16), 6135. https://doi.org/10.3390/molecules28166135