
Characteristic Photoprotective Molecules from the Sphagnum World: A Solution-Phase Ultrafast Study of Sphagnic Acid


Abstract
:1. Introduction
2. Results
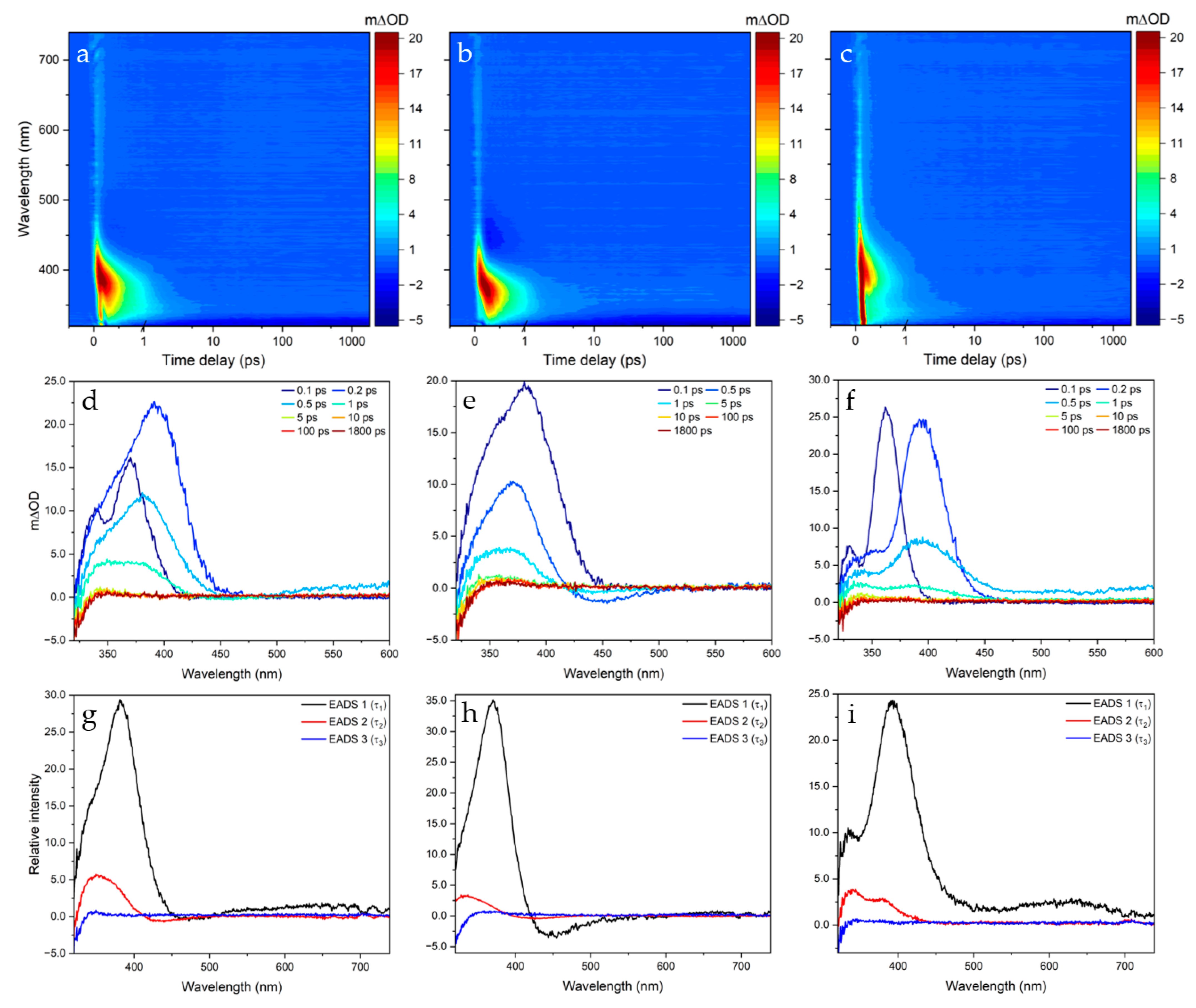
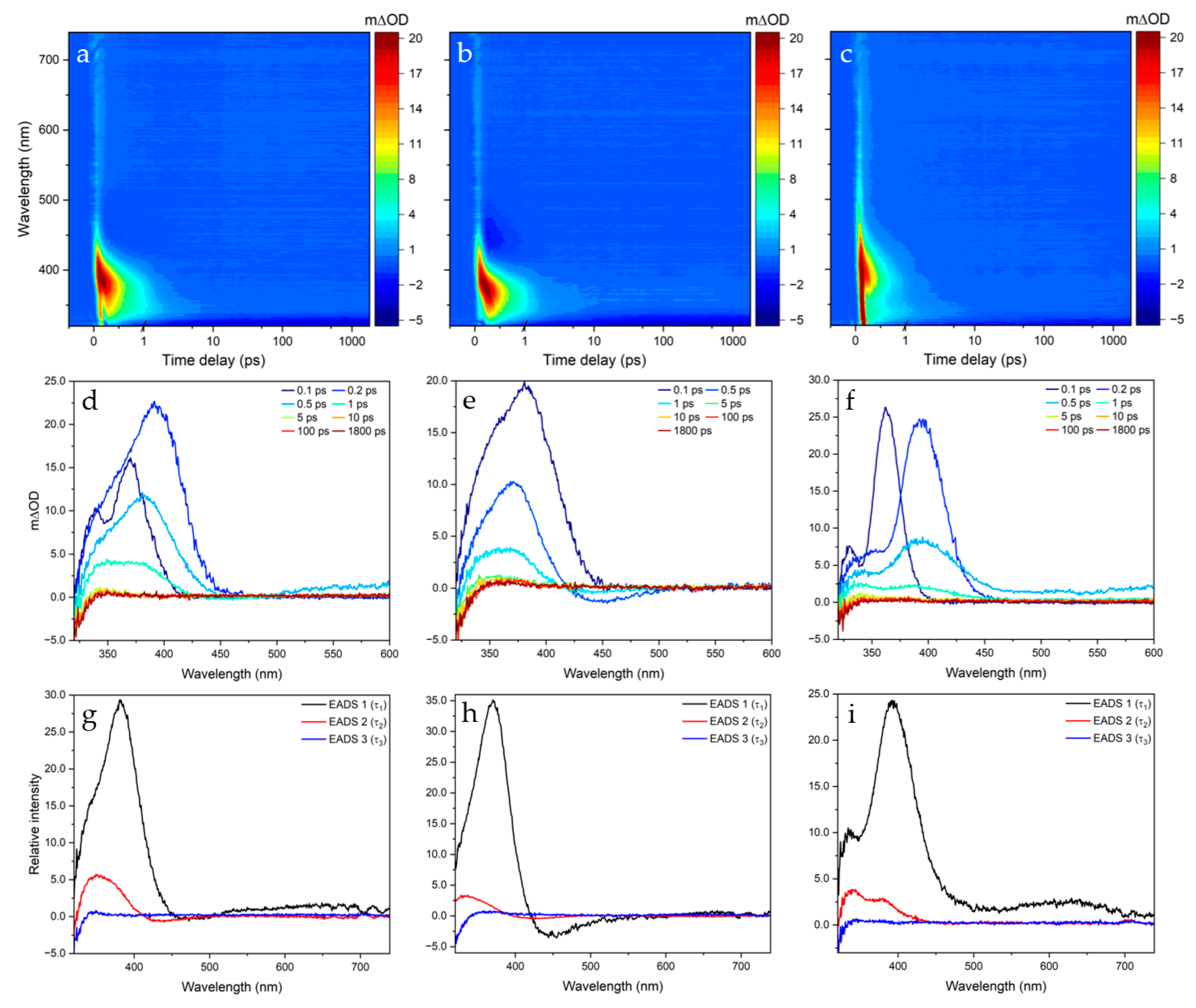
2.1. Transient Electronic Absorption Spectroscopy (TEAS)
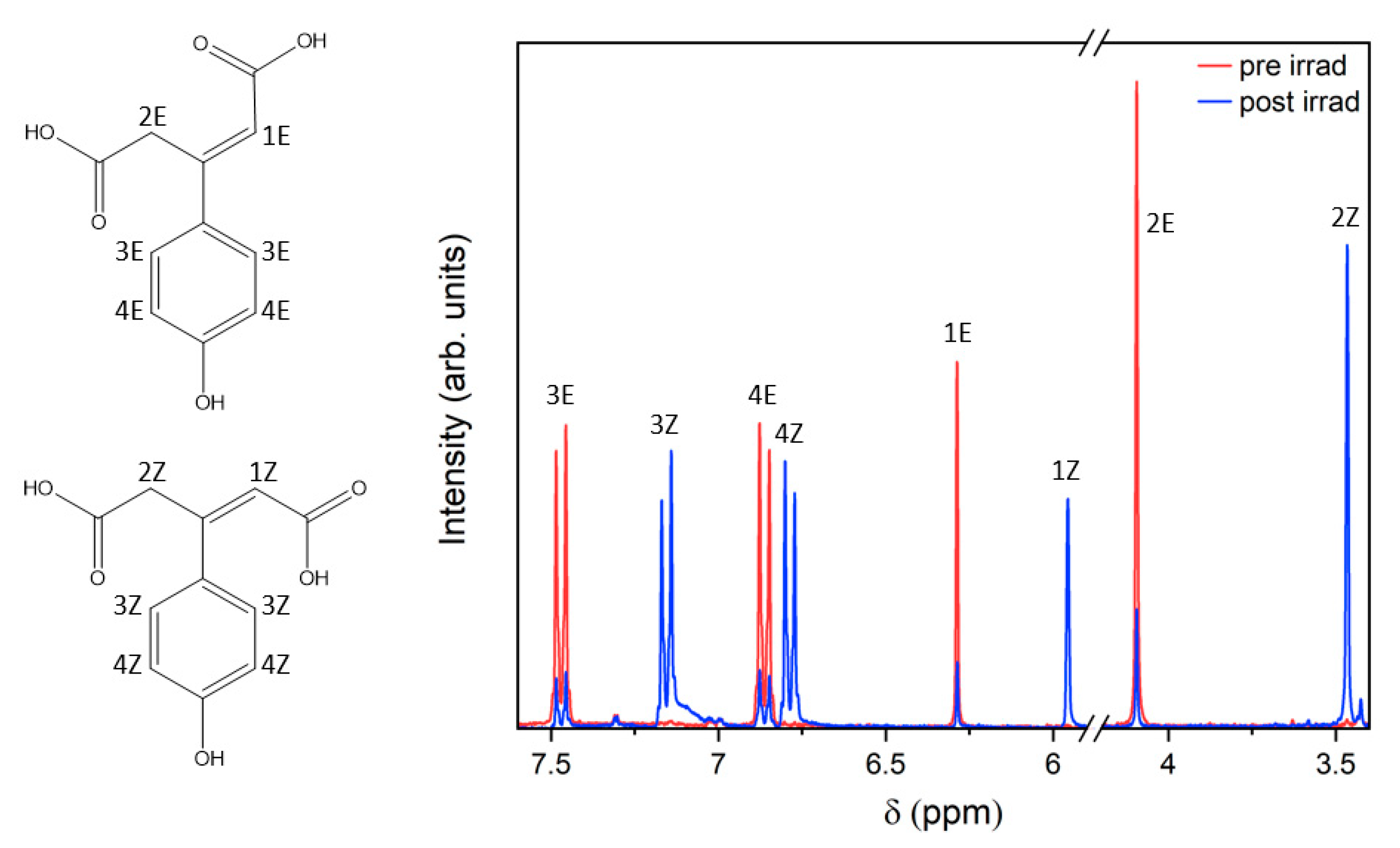
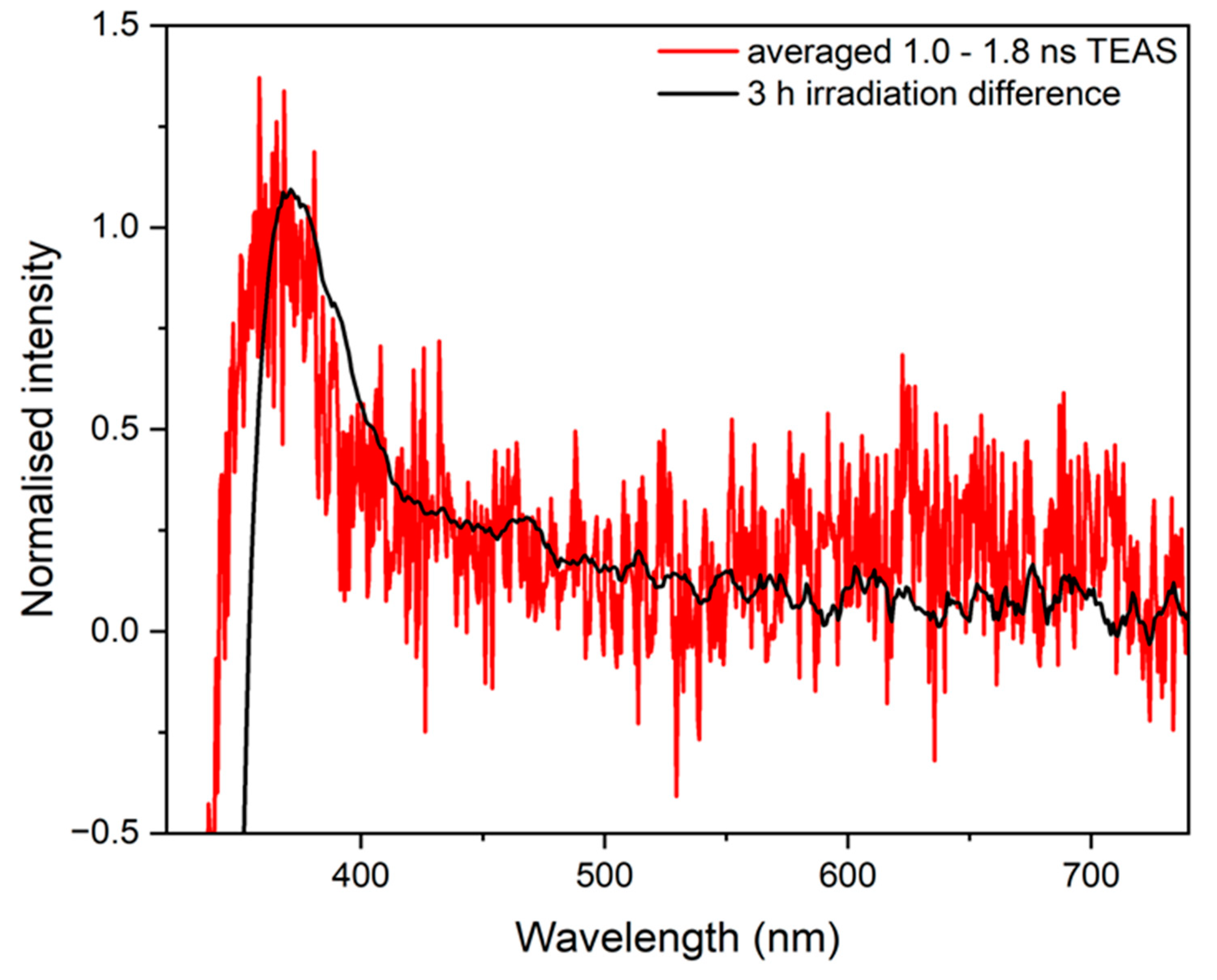
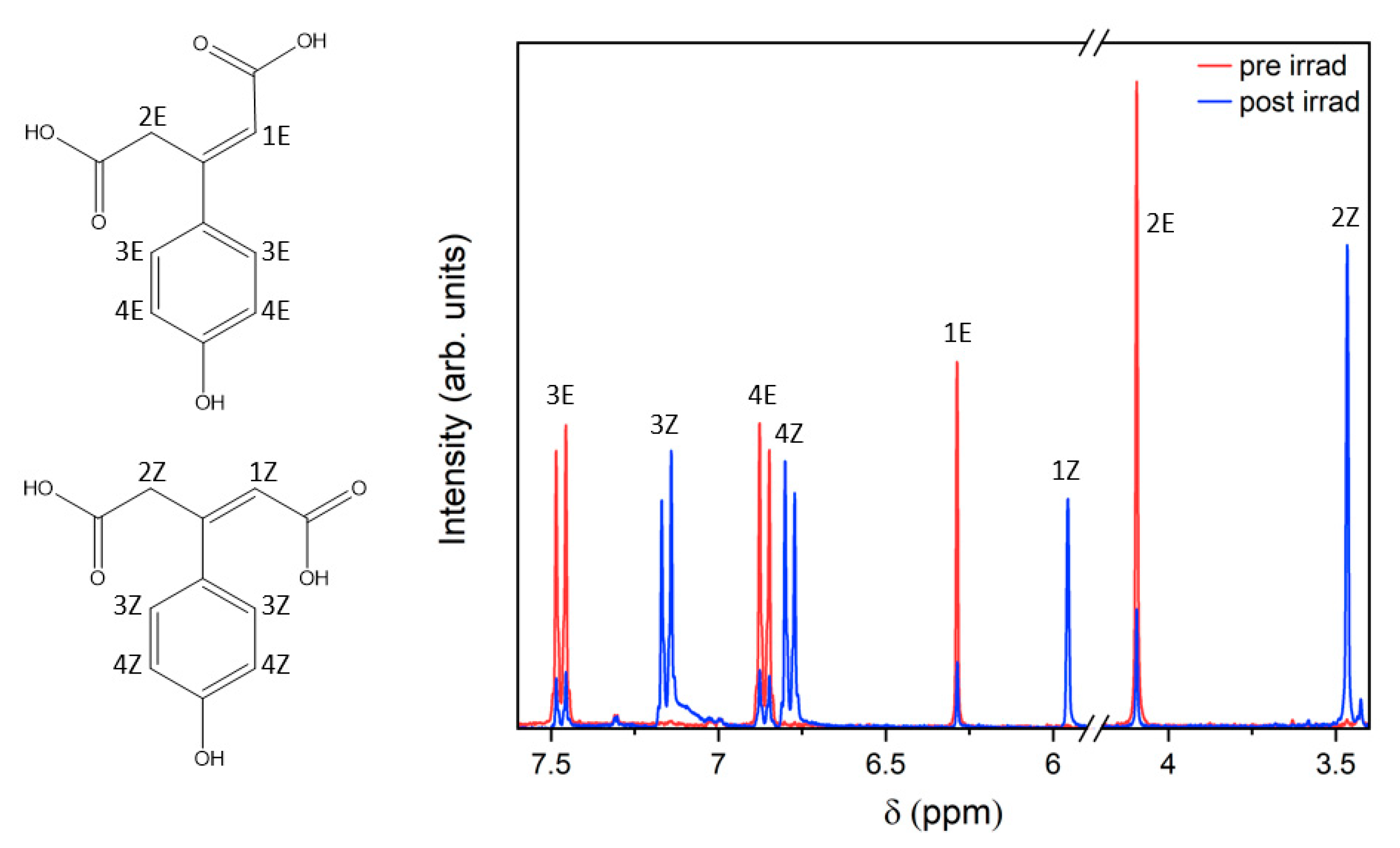
2.2. Steady-State Spectroscopy
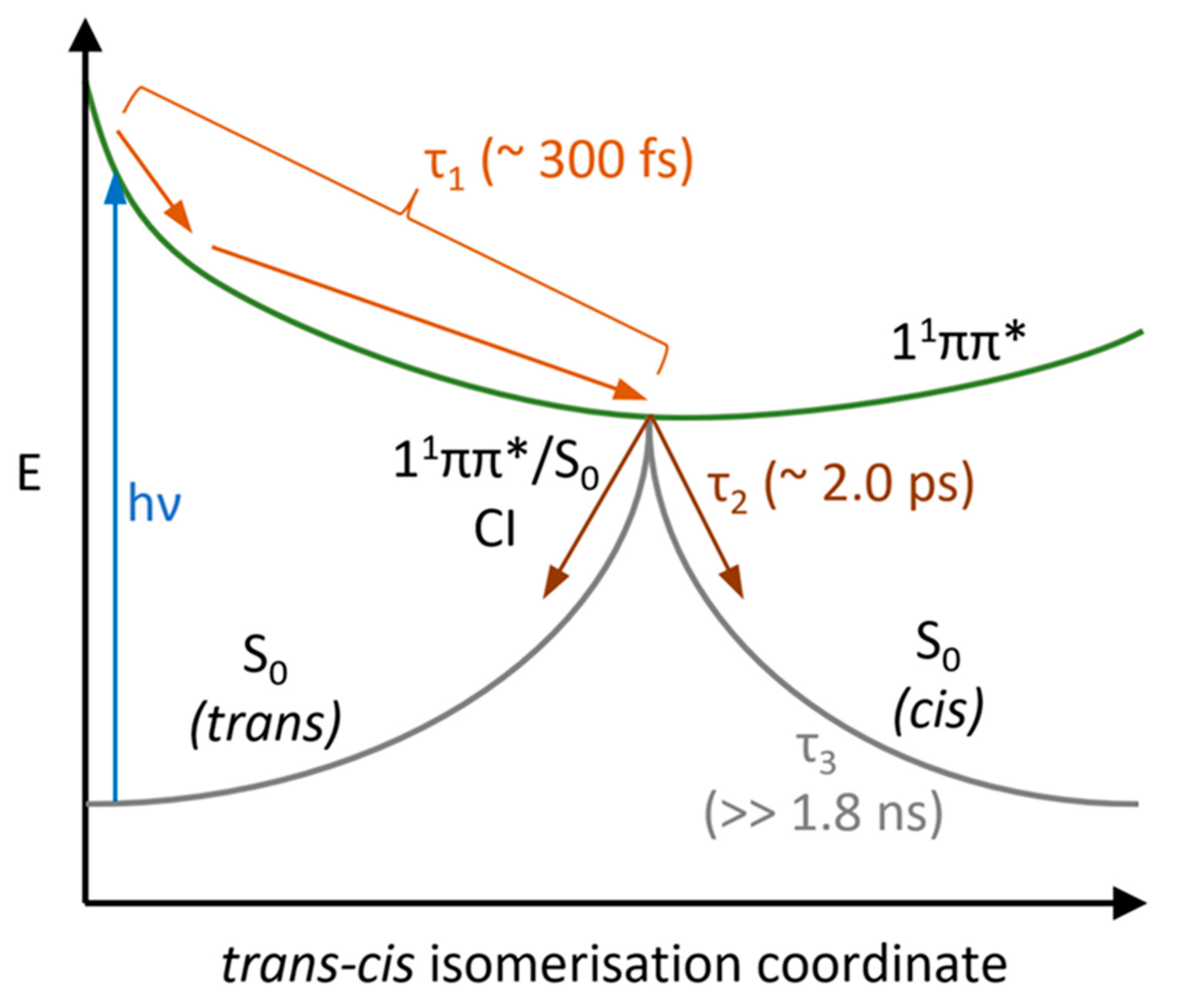
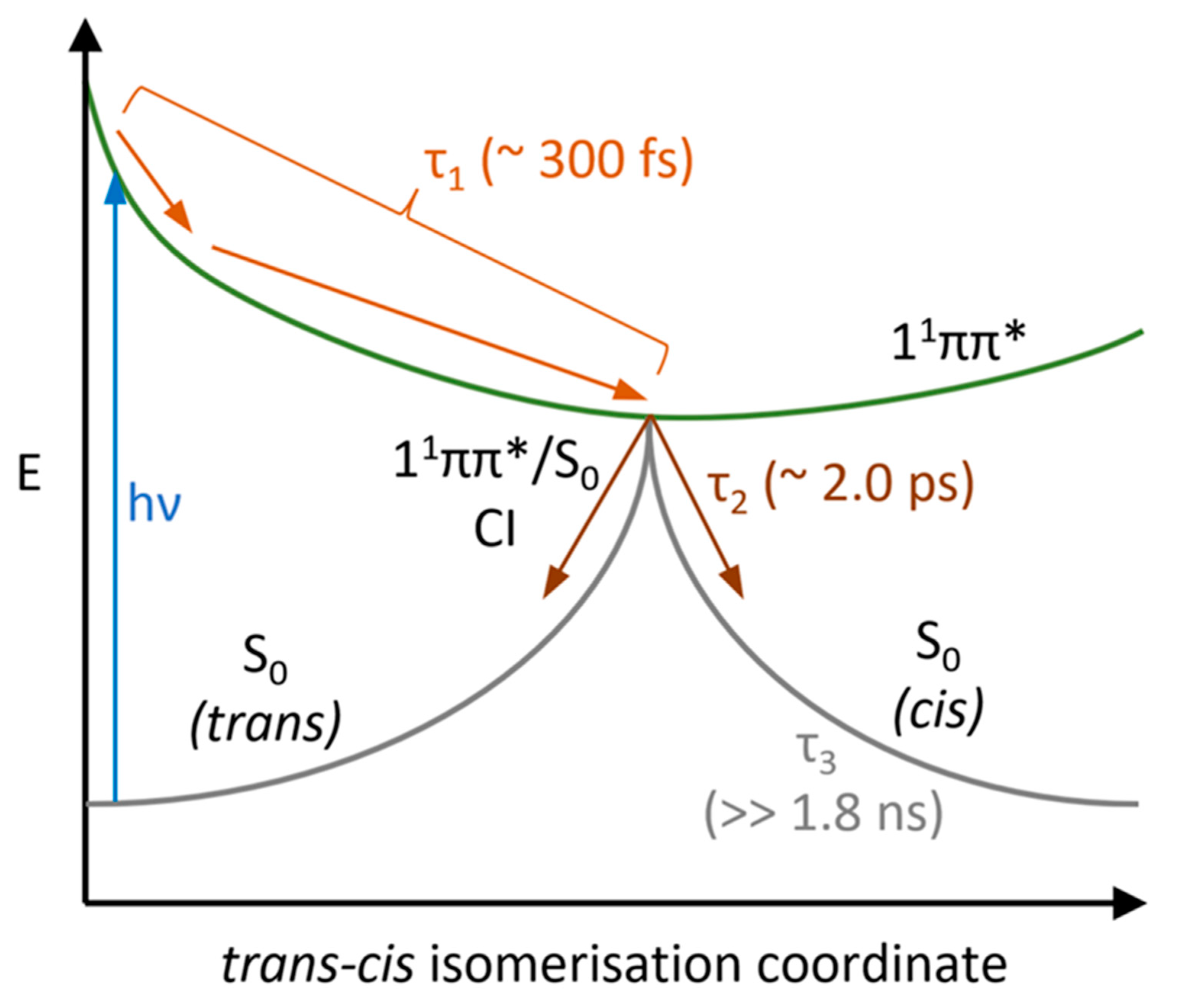
3. Discussion
4. Materials and Methods
4.1. Transient Electronic Absorption Spectroscopy (TEAS)
4.2. Steady-State Spectroscopy
4.3. Computational Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Frederick, J.E.; Snell, H.E.; Haywood, E.K. Solar ultraviolet radiation at the earth’s surface. Photochem. Photobiol. 1989, 50, 443–450. [Google Scholar] [CrossRef]
- Gallagher, R.P.; Lee, T.K. Adverse Effects of Ultraviolet Radiation: A Brief Review. Prog. Biophys. Mol. Biol. 2006, 92, 119–131. [Google Scholar] [CrossRef]
- De Gruijl, F.R.; Rebel, H. Early Events in UV Carcinogenesis—DNA Damage, Target Cells and Mutant P53 Foci. Photochem. Photobiol. 2008, 84, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Lautenschlager, S.; Wulf, H.C.; Pittelkow, M.R. Photoprotection. Lancet 2007, 370, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Serpone, N.; Dondi, D.; Albini, A. Inorganic and Organic UV Filters: Their Role and Efficacy in Sunscreens and Suncare Products. Inorganica Chim. Acta 2007, 360, 794–802. [Google Scholar] [CrossRef]
- Shaath, N.A. Ultraviolet Filters. Photochem. Photobiol. Sci. 2010, 9, 464–469. [Google Scholar] [CrossRef]
- Kockler, J.; Oelgemöller, M.; Robertson, S.; Glass, B.D. Photostability of Sunscreens. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 91–110. [Google Scholar] [CrossRef]
- Bonda, C.A.; Lott, D. Sunscreen Photostability. In Principles and Practice of Photoprotection; Springer: Cham, Switzerland, 2016; pp. 247–273. [Google Scholar] [CrossRef]
- Nguyen, V.P.T.; Stewart, J.D.; Ioannou, I.; Allais, F. Sinapic Acid and Sinapate Esters in Brassica: Innate Accumulation, Biosynthesis, Accessibility via Chemical Synthesis or Recovery From Biomass, and Biological Activities. Front. Chem. 2021, 9, 664602. [Google Scholar] [CrossRef]
- Luo, J.; Liu, Y.; Yang, S.; Flourat, A.L.; Allais, F.; Han, K. Ultrafast Barrierless Photoisomerization and Strong Ultraviolet Absorption of Photoproducts in Plant Sunscreens. J. Phys. Chem. Lett. 2017, 8, 1025–1030. [Google Scholar] [CrossRef]
- Tanner, P.R. Sunscreen Product Formulation. Dermatol. Clin. 2006, 24, 53–62. [Google Scholar] [CrossRef]
- Forestier, S. Rationale for Sunscreen Development. J. Am. Acad. Dermatol. 2008, 58 (Suppl. 2), S133–S138. [Google Scholar] [CrossRef]
- Mejía-Giraldo, J.C.; Gallardo, C.; Puertas-Mejía, M.A. In Vitro Photoprotection and Antioxidant Capacity of Sphagnum Meridense Extracts, a Novel Source of Natural Sunscreen from the Mountains of Colombia. Pure Appl. Chem. 2015, 87, 961–970. [Google Scholar] [CrossRef]
- Drobnik, J.; Stebel, A. Tangled History of the European Uses of Sphagnum Moss and Sphagnol. J. Ethnopharmacol. 2017, 209, 41–49. [Google Scholar] [CrossRef] [PubMed]
- hard Tutschek, R. Isolierung Und Charakterisierung Der P-Hydroxy-β-(Carboxy-Methyl)-Zimtsäure (Sphagnumsäure) Aus Der Zellwand von Sphagnum MagellanicumBrid. Z. Pflanzenphysiol. 1975, 76, 353–365. [Google Scholar] [CrossRef]
- Hyyryläinen, A.; Turunen, M.; Rautio, P.; Huttunen, S. Sphagnum Mosses in a Changing UV-B Environment: A Review. Perspect. Plant Ecol. Evol. Syst. 2018, 33, 1–8. [Google Scholar] [CrossRef]
- Rasmussen, S.; Rudolph, H. Isolation, Purification and Characterization of UDP-Glucose: CIS-p-Coumaric Acid-β-D-Glucosyltransferase from Sphagnum Fallax. Phytochemistry 1997, 46, 449–453. [Google Scholar] [CrossRef]
- Kinoshita, S.N.; Harabuchi, Y.; Inokuchi, Y.; Maeda, S.; Ehara, M.; Yamazaki, K.; Ebata, T. Substitution Effect on the Nonradiative Decay and Trans → Cis Photoisomerization Route: A Guideline to Develop Efficient Cinnamate-Based Sunscreens. Phys. Chem. Chem. Phys. 2021, 23, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.M.M.; Hilbers, M.; Buma, W.J. Excited-State Dynamics of Isolated and Microsolvated Cinnamate-Based UV-B Sunscreens. J. Phys. Chem. Lett. 2014, 5, 2464–2468. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Gaspar, A.; Garrido, E.M.; Garrido, J.; Borges, F. Hydroxycinnamic Acid Antioxidants: An Electrochemical Overview. Biomed. Res. Int. 2013, 2013, 251754. [Google Scholar] [CrossRef]
- Sofw Journal 9-2018—SOFW—Verlag für Chemische Industrie. Available online: https://www.sofw.com/en/sofw-journal/archive/archive-2/124-sofw-journal-9-2018 (accessed on 9 May 2023).
- Taskila, S.; Särkelä, R.; Tanskanen, J. Valuable Applications for Peat Moss. Biomass Convers. Biorefin. 2016, 6, 115–126. [Google Scholar] [CrossRef]
- Holt, E.L.; Rodrigues, N.D.N.; Cebrián, J.; Stavros, V.G. Determining the Photostability of Avobenzone in Sunscreen Formulation Models Using Ultrafast Spectroscopy. Phys. Chem. Chem. Phys. 2021, 23, 24439–24448. [Google Scholar] [CrossRef] [PubMed]
- Berenbeim, J.A.; Wong, N.G.K.; Cockett, M.C.R.; Berden, G.; Oomens, J.; Rijs, A.M.; Dessent, C.E.H. Unravelling the Keto-Enol Tautomer Dependent Photochemistry and Degradation Pathways of the Protonated UVA Filter Avobenzone. J. Phys. Chem. A 2020, 124, 2919–2930. [Google Scholar] [CrossRef] [PubMed]
- Moi, S.; Hosamani, B.; Kumar, K.; Gunaga, S.; Raghothama, S.; Gowd, K.H. Photochemical Studies of New Synthetic Derivatives of Avobenzone under Sunlight Using UV-Spectroscopy. J. Photochem. Photobiol. A Chem. 2021, 420, 113488. [Google Scholar] [CrossRef]
- Termer, M.; Carola, C.; Salazar, A.; Keck, C.M.; von Hagen, J. Methoxy-Monobenzoylmethane Protects Human Skin against UV-Induced Damage by Conversion to Avobenzone and Radical Scavenging. Molecules 2021, 26, 6141. [Google Scholar] [CrossRef]
- Cantrell, A.; Mcgarvey, D.J. Photochemical Studies of 4-Tert-Butyl-49-Methoxydibenzoylmethane (BM-DBM). J. Photochem. Photobiol. B 2001, 64, 117–122. [Google Scholar] [CrossRef]
- Horbury, M.D.; Baker, L.A.; Rodrigues, N.D.N.; Quan, W.D.; Stavros, V.G. Photoisomerization of Ethyl Ferulate: A Solution Phase Transient Absorption Study. Chem. Phys. Lett. 2017, 673, 62–67. [Google Scholar] [CrossRef]
- Krokidi, K.M.; Turner, M.A.P.; Pearcy, P.A.J.; Stavros, V.G. A Systematic Approach to Methyl Cinnamate Photodynamics. Mol. Phys. 2021, 119, e1811910. [Google Scholar] [CrossRef]
- Horbury, M.D.; Holt, E.L.; Mouterde, L.M.M.; Balaguer, P.; Cebrián, J.; Blasco, L.; Allais, F.; Stavros, V.G. Towards Symmetry Driven and Nature Inspired UV Filter Design. Nat. Commun. 2019, 10, 4748. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.; Seger, R.; Mullen, K.M.; van Stokkum, I.H.M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef]
- Berera, R.; van Grondelle, R.; Kennis, J.T.M. Ultrafast Transient Absorption Spectroscopy: Principles and Application to Photosynthetic Systems. Photosynth. Res. 2009, 101, 105–118. [Google Scholar] [CrossRef]
- Horbury, M.D.; Quan, W.D.; Flourat, A.L.; Allais, F.; Stavros, V.G. Elucidating Nuclear Motions in a Plant Sunscreen during Photoisomerization through Solvent Viscosity Effects. Phys. Chem. Chem. Phys. 2017, 19, 21127–21131. [Google Scholar] [CrossRef]
- Karsili, T.N.V.; Marchetti, B.; Ashfold, M.N.R.; Domcke, W. Ab Initio Study of Potential Ultrafast Internal Conversion Routes in Oxybenzone, Caffeic Acid, and Ferulic Acid: Implications for Sunscreens. J. Phys. Chem. A 2014, 118, 11999–12010. [Google Scholar] [CrossRef]
- Baker, L.A.; Greenough, S.E.; Stavros, V.G. A Perspective on the Ultrafast Photochemistry of Solution-Phase Sunscreen Molecules. J. Phys. Chem. Lett. 2016, 7, 4655–4665. [Google Scholar] [CrossRef] [PubMed]
- Abiola, T.T.; Whittock, A.L.; Stavros, V.G. Unravelling the Photoprotective Mechanisms of Nature-Inspired Ultraviolet Filters Using Ultrafast Spectroscopy. Molecules 2020, 25, 3945. [Google Scholar] [CrossRef] [PubMed]
- Abiola, T.T.; Rodrigues, N.D.N.; Ho, C.; Coxon, D.J.L.; Horbury, M.D.; Toldo, J.M.; Do Casal, M.T.; Rioux, B.; Peyrot, C.; Mention, M.M.; et al. New Generation UV-A Filters: Understanding Their Photodynamics on a Human Skin Mimic. J. Phys. Chem. Lett. 2021, 12, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Owrutsky, J.C.; Raftery, D.; Hochstrasser, R.M. Vibrational Relaxation Dynamics in Solutions. Annu. Rev. Phys. Chem. 1994, 45, 519–555. [Google Scholar] [CrossRef]
- Kang, X.; Zhu, Y.; Zhang, J.; Xu, C.; Lan, Z. On-the-Fly Nonadiabatic Dynamics of Caffeic Sunscreen Compound. Chin. J. Chem. Phys. 2022. [Google Scholar] [CrossRef]
- Baker, L.A.; Horbury, M.D.; Greenough, S.E.; Allais, F.; Walsh, P.S.; Habershon, S.; Stavros, V.G. Ultrafast Photoprotecting Sunscreens in Natural Plants. J. Phys. Chem. Lett. 2016, 7, 56–61. [Google Scholar] [CrossRef]
- Zhao, X.; Li, J.; Luo, J.; Liu, J. Significant Acceleration of E-Z Photoisomerization Induced by Molecular Planarity Breaking. Chem. Phys. Lett. 2023, 822, 140480. [Google Scholar] [CrossRef]
- Becke, A.D.; Becke, D.A. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron Affinities of the First-row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate Evaluations of the Electrostatic Free Energy and Internal Energy Changes in Solution Processes. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Pascual-ahuir, J.L.; Silla, E.; Tuñon, I. GEPOL: An Improved Description of Molecular Surfaces. III. A New Algorithm for the Computation of a Solvent-Excluding Surface. J. Comput. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A3; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Cowden, A.M.; Whittock, A.L.; Holt, E.L.; Stavros, V.G.; Wills, M. Synthesis and Characterisation of Novel Composite Sunscreens Containing Both Avobenzone and Octocrylene Motifs. RSC Adv. 2023, 13, 17017–17027. [Google Scholar] [CrossRef]
- Marcin, S.; Aleksander, A. Acute Toxicity Assessment of Nine Organic UV Filters Using a Set of Biotests. Toxicol. Res. 2023, 1, 1–19. [Google Scholar] [CrossRef]
- Berardesca, E.; Zuberbier, T.; Sanchez Viera, M.; Marinovich, M. Review of the Safety of Octocrylene Used as an Ultraviolet Filter in Cosmetics. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 25–33. [Google Scholar] [CrossRef]
- Radice, M.; Manfredini, S.; Ziosi, P.; Dissette, V.; Buso, P.; Fallacara, A.; Vertuani, S. Herbal Extracts, Lichens and Biomolecules as Natural Photo-Protection Alternatives to Synthetic UV Filters. A Systematic Review. Fitoterapia 2016, 114, 144–162. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lifetime | Ethanol | Acetonitrile | Dioxane |
|---|---|---|---|
| τ1 (fs) | 280 ± 50 | 330 ± 60 | 236 ± 50 |
| τ2 (ps) | 1.31 ± 0.05 | 3.40 ± 0.06 | 1.37 ± 0.05 |
| τ3 (ns) | >>1.8 | >>1.8 | >>1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hymas, M.; Casademont-Reig, I.; Poigny, S.; Stavros, V.G. Characteristic Photoprotective Molecules from the Sphagnum World: A Solution-Phase Ultrafast Study of Sphagnic Acid. Molecules 2023, 28, 6153. https://doi.org/10.3390/molecules28166153
Hymas M, Casademont-Reig I, Poigny S, Stavros VG. Characteristic Photoprotective Molecules from the Sphagnum World: A Solution-Phase Ultrafast Study of Sphagnic Acid. Molecules. 2023; 28(16):6153. https://doi.org/10.3390/molecules28166153
Chicago/Turabian StyleHymas, Michael, Irene Casademont-Reig, Stéphane Poigny, and Vasilios G. Stavros. 2023. "Characteristic Photoprotective Molecules from the Sphagnum World: A Solution-Phase Ultrafast Study of Sphagnic Acid" Molecules 28, no. 16: 6153. https://doi.org/10.3390/molecules28166153
APA StyleHymas, M., Casademont-Reig, I., Poigny, S., & Stavros, V. G. (2023). Characteristic Photoprotective Molecules from the Sphagnum World: A Solution-Phase Ultrafast Study of Sphagnic Acid. Molecules, 28(16), 6153. https://doi.org/10.3390/molecules28166153