
Human Triosephosphate Isomerase Is a Potential Target in Cancer Due to Commonly Occurring Post-Translational Modifications

 ,
,  ,
, 
Abstract
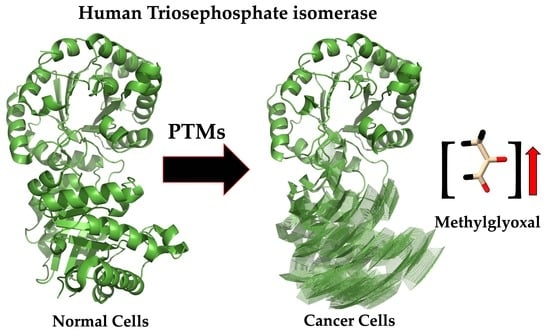
1. Introduction
2. Cancer Treatment
3. Cancer Cell Metabolism
4. The Glycolytic Pathway Could Play a Crucial Role in the Energy Metabolism of Cancer Cells
5. Triosephosphate Isomerase Is a Key Metabolic Enzyme
6. Beyond Being a Target to Impair Energy Metabolism in Cancer Cells, HsTIM Can Be a Methylglyoxal Factory
6.1. Triosephosphate Isomerase Is a Model Molecule for Understanding Structure–Function Relationships
6.2. Structural Alterations on the Deamidated Triosephosphate Isomerase Are the Base for Rational Drug Design
7. A Group of PTMs in HsTIM Could Be Considered a Target for Cancer Therapies
7.1. Phosphorylation in HsTIM
7.2. Heterodimers of HsTIM Phosphorylated at Ser21 Are Present in Cancer Cells
7.3. S-nitrosylation in HsTIM
7.4. S-glutathionylation in HsTIM
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Network GBoDC. Global Burden of Disease Study 2019 (GBD 2019) Results; Institute for Health Metrics and Evaluation (IHME): Seattle, WA, USA, 2020. [Google Scholar]
- Emole, J. Cancer Diagnosis and Treatment: An Overview for the General Practitioner; IntechOpen: London, UK, 2012. [Google Scholar]
- Sethi, S.; Ali, S.; Philip, P.A.; Sarkar, F.H. Clinical advances in molecular biomarkers for cancer diagnosis and therapy. Int. J. Mol. Sci. 2013, 14, 14771–14784. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhu, X.; Yan, T.; Yu, C.; Shen, C.; Hong, J.; Chen, H.; Fang, J.Y. Differentially Expressed lncRNAs in Gastric Cancer Patients: A Potential Biomarker for Gastric Cancer Prognosis. J. Cancer 2017, 8, 2575–2586. [Google Scholar] [CrossRef]
- Chrzanowska, N.M.; Kowalewski, J.; Lewandowska, M.A. Use of Fluorescence In Situ Hybridization (FISH) in Diagnosis and Tailored Therapies in Solid Tumors. Molecules 2020, 25, 1864. [Google Scholar] [CrossRef]
- Assumpção, A.L.; da Silva, R.C. Immuno-PCR in cancer and non-cancer related diseases: A review. Vet. Q. 2016, 36, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Olmedillas-López, S.; Olivera-Salazar, R.; García-Arranz, M.; García-Olmo, D. Current and Emerging Applications of Droplet Digital PCR in Oncology: An Updated Review. Mol. Diagn. Ther. 2022, 26, 61–87. [Google Scholar] [CrossRef]
- Paolillo, C.; Londin, E.; Fortina, P. Single-Cell Genomics. Clin. Chem. 2019, 65, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Suo, Y.; Gu, Z.; Wei, X. Advances of In Vivo Flow Cytometry on Cancer Studies. Cytom. Part A 2020, 97, 15–23. [Google Scholar] [CrossRef]
- Ito, E.; Iha, K.; Yoshimura, T.; Nakaishi, K.; Watabe, S. Early diagnosis with ultrasensitive ELISA. Adv. Clin. Chem. 2021, 101, 121–133. [Google Scholar] [CrossRef]
- Manne, U.; Srivastava, R.-G.; Srivastava, S. Keynote review: Recent advances in biomarkers for cancer diagnosis and treatment. Drug Discov. Today 2005, 10, 965–976. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Busch, H. A general concept for molecular biology of cancer. Cancer Res. 1976, 36, 4291–4294. [Google Scholar] [PubMed]
- Sun, M.; Zhao, W.; Zeng, Y.; Zhang, D.; Chen, Z.; Liu, C.; Wu, B. Fibrous sheath interacting protein 1 overexpression is associated with unfavorable prognosis in bladder cancer: A potential therapeutic target. OncoTargets Ther. 2017, 10, 3949–3956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xie, Y.; Tian, T.; Yang, Q.; Zhou, Y.; Qiu, J.; Xu, L.; Wen, N.; Lv, Q.; Du, Z. High expression levels of centromere protein A plus upregulation of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling pathway affect chemotherapy response and prognosis in patients with breast cancer. Oncol. Lett. 2021, 21, 410. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, W.; Ren, P.; Zhang, T. Upregulation of centromere protein M promotes tumorigenesis: A potential predictive target for cancer in humans. Mol. Med. Rep. 2020, 22, 3922–3934. [Google Scholar] [CrossRef]
- Ding, X.Q.; Zhao, S.; Yang, L.; Zhao, X.; Zhao, G.F.; Zhao, S.P.; Li, Z.J.; Zheng, H.C. The nucleocytoplasmic translocation and up-regulation of ING5 protein in breast cancer: A potential target for gene therapy. Oncotarget 2017, 8, 81953–81966. [Google Scholar] [CrossRef][Green Version]
- Földi, M.; Stickeler, E.; Bau, L.; Kretz, O.; Watermann, D.; Gitsch, G.; Kayser, G.; Zur Hausen, A.; Coy, J.F. Transketolase protein TKTL1 overexpression: A potential biomarker and therapeutic target in breast cancer. Oncol. Rep. 2007, 17, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.A.; Lee, G.; Kang, H.J.; Kim, Y.G.; Bae, S.H.; Lee, J.L.; Lee, K.H.; Hyun, M.S.; Kim, D.S. Overexpression of c-met Protein in Gastric Cancer and Role of uPAR as a Therapeutic Target. Cancer Res. Treat. 2003, 35, 9–15. [Google Scholar] [CrossRef]
- Bakir, M.A.; Eccles, S.; Babich, J.W.; Aftab, N.; Styles, J.; Dean, C.J.; Lambrecht, R.M.; Ott, R.J. c-erbB2 protein overexpression in breast cancer as a target for PET using iodine-124-labeled monoclonal antibodies. J. Nucl. Med. 1992, 33, 2154–2160. [Google Scholar]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef]
- Bolaños-Suárez, V.; Alfaro, A.; Espinosa, A.M.; Medina-Martínez, I.; Juárez, E.; Villegas-Sepúlveda, N.; Gudiño-Zayas, M.; Gutiérrez-Castro, A.; Román-Bassaure, E.; Salinas-Nieves, M.E.; et al. The mRNA and protein levels of the glycolytic enzymes lactate dehydrogenase A (LDHA) and phosphofructokinase platelet (PFKP) are good predictors of survival time, recurrence, and risk of death in cervical cancer patients. Cancer Med. 2023, 12, 14865–15762. [Google Scholar] [CrossRef] [PubMed]
- Da, Q.; Huang, L.; Huang, C.; Chen, Z.; Jiang, Z.; Huang, F.; Shen, T.; Sun, L.; Yan, Z.; Ye, X.; et al. Glycolytic regulatory enzyme PFKFB3 as a prognostic and tumor microenvironment biomarker in human cancers. Aging 2023, 15, 4533–4559. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Xavier, C.E.; Emaldi, M.; Mingo, J.; Øyjord, T.; Mælandsmo, G.M.; Fodstad, Ø.; Errarte, P.; Larrinaga, G.; Llarena, R.; López, J.I.; et al. The expression pattern of pyruvate dehydrogenase kinases predicts prognosis and correlates with immune exhaustion in clear cell renal cell carcinoma. Sci. Rep. 2023, 13, 7339. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, J.; Huang, B.; Fu, S.; Qu, S.; Yu, R.; Zhao, Y. ErbB2-upregulated HK1 and HK2 promote breast cancer cell proliferation, migration and invasion. Med. Oncol. 2023, 40, 154. [Google Scholar] [CrossRef]
- Spanò, D.P.; Bonelli, S.; Calligaris, M.; Carreca, A.P.; Carcione, C.; Zito, G.; Nicosia, A.; Rizzo, S.; Scilabra, S.D. High-Resolution Secretome Analysis of Chemical Hypoxia Treated Cells Identifies Putative Biomarkers of Chondrosarcoma. Proteomes 2022, 10, 25. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, H.; Chen, S.; Lu, Z.; Li, B.; Jiang, T.; Xuan, M.; Ye, R.; Liang, H.; Liu, X.; et al. SIRT1 regulated hexokinase-2 promoting glycolysis is involved in hydroquinone-enhanced malignant progression in human lymphoblastoid TK6 cells. Ecotoxicol. Environ. Saf. 2022, 241, 113757. [Google Scholar] [CrossRef]
- Oliveira, N.; Gomig, T.; Milioli, H.; Cordeiro, F.; Costa, G.; Urban, C.; Lima, R.; Cavalli, I.; Ribeiro, E. Comparative proteomic analysis of ductal and lobular invasive breast carcinoma. Genet. Mol. Res. 2016, 15, 1–10. [Google Scholar] [CrossRef]
- Linge, A.; Kennedy, S.; O’Flynn, D.; Beatty, S.; Moriarty, P.; Henry, M.; Clynes, M.; Larkin, A.; Meleady, P. Differential expression of fourteen proteins between uveal melanoma from patients who subsequently developed distant metastases versus those who did not. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4634–4643. [Google Scholar] [CrossRef]
- Thongwatchara, P.; Promwikorn, W.; Srisomsap, C.; Chokchaichamnankit, D.; Boonyaphiphat, P.; Thongsuksai, P. Differential protein expression in primary breast cancer and matched axillary node metastasis. Oncol. Rep. 2011, 26, 185–191. [Google Scholar] [PubMed]
- Chen, W.-Z.; Pang, B.; Yang, B.; Zhou, J.-G.; Sun, Y.-H. Differential proteome analysis of conditioned medium of BPH-1 and LNCaP cells. Chin. Med. J. 2011, 124, 3806–3809. [Google Scholar]
- Roth, U.; Razawi, H.; Hommer, J.; Engelmann, K.; Schwientek, T.; Müller, S.; Baldus, S.E.; Patsos, G.; Corfield, A.P.; Paraskeva, C. Differential expression proteomics of human colorectal cancer based on a syngeneic cellular model for the progression of adenoma to carcinoma. Proteomics 2010, 10, 194–202. [Google Scholar] [CrossRef]
- Tamesa, M.S.; Kuramitsu, Y.; Fujimoto, M.; Maeda, N.; Nagashima, Y.; Tanaka, T.; Yamamoto, S.; Oka, M.; Nakamura, K. Detection of autoantibodies against cyclophilin A and triosephosphate isomerase in sera from breast cancer patients by proteomic analysis. Electrophoresis 2009, 30, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-W.; Peng, S.-Y.; Li, J.-T.; Wang, Y.; Zhang, Z.-P.; Cheng, Y.; Cheng, D.-Q.; Weng, W.-H.; Wu, X.-S.; Fei, X.-Z. Identification of metastasis-associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer Lett. 2009, 281, 71–81. [Google Scholar] [CrossRef]
- Kim, J.E.; Koo, K.H.; Kim, Y.H.; Sohn, J.; Park, Y.G. Identification of potential lung cancer biomarkers using an in vitro carcinogenesis model. Exp. Mol. Med. 2008, 40, 709–720. [Google Scholar] [CrossRef]
- Qi, Y.J.; He, Q.Y.; Ma, Y.F.; Du, Y.W.; Liu, G.C.; Li, Y.J.; Tsao, G.S.; Ngai, S.M.; Chiu, J.F. Proteomic identification of malignant transformation-related proteins in esophageal squamous cell carcinoma. J. Cell. Biochem. 2008, 104, 1625–1635. [Google Scholar] [CrossRef]
- Yang, F.; Xiao, Z.-Q.; Zhang, X.-Z.; Li, C.; Zhang, P.-F.; Li, M.-Y.; Chen, Y.; Zhu, G.-Q.; Sun, Y.; Liu, Y.-F. Identification of tumor antigens in human lung squamous carcinoma by serological proteome analysis. J. Proteome Res. 2007, 6, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Mikuriya, K.; Kuramitsu, Y.; Ryozawa, S.; Fujimoto, M.; Mori, S.; Oka, M.; Hamano, K.; Okita, K.; Sakaida, I.; Nakamura, K. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int. J. Oncol. 2007, 30, 849–855. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Katayama, M.; Nakano, H.; Ishiuchi, A.; Wu, W.; Oshima, R.; Sakurai, J.; Nishikawa, H.; Yamaguchi, S.; Otsubo, T. Protein pattern difference in the colon cancer cell lines examined by two-dimensional differential in-gel electrophoresis and mass spectrometry. Surg. Today 2006, 36, 1085–1093. [Google Scholar] [CrossRef]
- Zhang, D.; Tai, L.K.; Wong, L.L.; Chiu, L.-L.; Sethi, S.K.; Koay, E.S. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Mol. Cell. Proteom. 2005, 4, 1686–1696. [Google Scholar] [CrossRef]
- Chen, J.; He, Q.Y.; Yuen, A.P.W.; Chiu, J.F. Proteomics of buccal squamous cell carcinoma: The involvement of multiple pathways in tumorigenesis. Proteomics 2004, 4, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Lichtenfels, R.; Kellner, R.; Atkins, D.; Bukur, J.; Ackermann, A.; Beck, J.; Brenner, W.; Melchior, S.; Seliger, B. Identification of metabolic enzymes in renal cell carcinoma utilizing PROTEOMEX analyses. Biochim. Biophys. Acta-Proteins Proteom. 2003, 1646, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gharib, T.G.; Huang, C.-C.; Thomas, D.G.; Shedden, K.A.; Taylor, J.M.; Kardia, S.L.; Misek, D.E.; Giordano, T.J.; Iannettoni, M.D. Proteomic analysis of lung adenocarcinoma: Identification of a highly expressed set of proteins in tumors. Clin. Cancer Res. 2002, 8, 2298–2305. [Google Scholar] [PubMed]
- Alaiya, A.; Roblick, U.; Egevad, L.; Carlsson, A.; Franzén, B.; Volz, D.; Huwendiek, S.; Linder, S.; Auer, G. Polypeptide expression in prostate hyperplasia and prostate adenocarcinoma. Anal. Cell. Pathol. 2000, 21, 1–9. [Google Scholar] [CrossRef]
- Skinner, K.; Dufour, R.; Haiderali, A.; Huang, M.; Schwartzberg, L. PCN131-assessing the real-world cost of care in patients with metastatic triple negative breast cancer (MTNBC) in the United States. Value Health 2018, 21, S36. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, H.; Huang, C.; Lei, Y. Cancer drug resistance: Redox resetting renders a way. Oncotarget 2016, 7, 42740. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Subramanian, S. Intrinsic resistance of solid tumors to immune checkpoint blockade therapy. Cancer Res. 2017, 77, 817–822. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Lin, H.; Zeng, J.; Xie, R.; Schulz, M.J.; Tedesco, R.; Qu, J.; Erhard, K.F.; Mack, J.F.; Raha, K.; Rendina, A.R.; et al. Discovery of a Novel 2,6-Disubstituted Glucosamine Series of Potent and Selective Hexokinase 2 Inhibitors. ACS Med. Chem. Lett. 2016, 7, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.; Zhang, Y.; Wu, C.; Yang, K.; Gao, S.; Zheng, M.; Li, X.; Li, H.; Chen, L. Structure based discovery of novel hexokinase 2 inhibitors. Bioorg Chem. 2020, 96, 103609. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, W.; Tian, H.; Li, B.; Yu, X.; Wang, G.; Sang, W.; Dai, Y. Metal-Phenolic Nanomedicines Regulate T-Cell Antitumor Function for Sono-Metabolic Cancer Therapy. ACS Nano 2023, 15, 14667–14677. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [PubMed]
- Marín-Hernández, Á.; Saavedra, E. Metabolic control analysis as a strategy to identify therapeutic targets, the case of cancer glycolysis. Biosystems 2023, 231, 104986. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Plancqueel, S.; Lazar, N.; Korri-Youssoufi, H.; Li de la Sierra-Gallay, I.; van Tilbeurgh, H.; Salmon, L. Novel N-substituted 5-phosphate-d-arabinonamide derivatives as strong inhibitors of phosphoglucose isomerases: Synthesis, structure-activity relationship and crystallographic studies. Bioorg Chem. 2020, 102, 104048. [Google Scholar] [CrossRef] [PubMed]
- Pekel, G.; Ari, F. Therapeutic targeting of cancer metabolism with triosephosphate isomerase. Chem. Biodivers. 2020, 17, e2000012. [Google Scholar] [CrossRef]
- López-Velázquez, G.; Fernández-Lainez, C.; de la Mora-de la Mora, J.I.; Caudillo de la Portilla, D.; Reynoso-Robles, R.; González-Maciel, A.; Ridaura, C.; García-Torres, I.; Gutiérrez-Castrellón, P.; Olivos-García, A.; et al. On the molecular and cellular effects of omeprazole to further support its effectiveness as an antigiardial drug. Sci. Rep. 2019, 9, 8922. [Google Scholar] [CrossRef]
- García-Torres, I.; De la Mora-De la Mora, I.; Hernández-Alcántara, G.; Molina-Ortiz, D.; Caballero-Salazar, S.; Olivos-García, A.; Nava, G.; López-Velázquez, G.; Enríquez-Flores, S. First characterization of a microsporidial triosephosphate isomerase and the biochemical mechanisms of its inactivation to propose a new druggable target. Sci. Rep. 2018, 8, 8591. [Google Scholar] [CrossRef] [PubMed]
- García-Torres, I.; De la Mora-De la Mora, I.; López-Velázquez, G.; Cabrera, N.; Flores-López, L.A.; Becker, I.; Herrera-López, J.; Hernández, R.; Pérez-Montfort, R.; Enríquez-Flores, S. Repurposing of rabeprazole as an anti-Trypanosoma cruzi drug that targets cellular triosephosphate isomerase. J. Enzyme Inhib. Med. Chem. 2023, 38, 2231169. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Mastrototaro, L.; Giardina, B. Glycolytic enzyme inhibitors in cancer treatment. Expert. Opin. Investig. Drugs 2008, 17, 1533–1545. [Google Scholar] [CrossRef]
- Warburg, O.H. The classic: The chemical constitution of respiration ferment. Clin. Orthop. Relat. Res. 2010, 468, 2833–2839. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Über den stoffwechsel der carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Läsche, M.; Emons, G.; Gründker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope Between Life and Death. Front. Oncol. 2020, 10, 409. [Google Scholar] [CrossRef]
- Kato, Y.; Maeda, T.; Suzuki, A.; Baba, Y. Cancer metabolism: New insights into classic characteristics. Jpn. Dent. Sci. Rev. 2018, 54, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Qian, Y.; Yu, J.; Wong, C.C. Metabolic rewiring in the promotion of cancer metastasis: Mechanisms and therapeutic implications. Oncogene 2020, 39, 6139–6156. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Schwarz, R.; Zitzow, E.; Fiebig, A.; Hering, S.; Humboldt, Y.; Schoenwaelder, N.; Kämpfer, N.; Volkmar, K.; Hinz, B.; Kreikemeyer, B. PEGylation increases antitumoral activity of arginine deiminase of Streptococcus pyogenes. Appl. Microbiol. Biotechnol. 2022, 106, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Tran, Q.; Lee, H.; Park, J.; Kim, S.H.; Park, J. Targeting Cancer Metabolism—Revisiting the Warburg Effects. Toxicol. Res. 2016, 32, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Busk, M.; Horsman, M.R.; Kristjansen, P.E.; van der Kogel, A.J.; Bussink, J.; Overgaard, J. Aerobic glycolysis in cancers: Implications for the usability of oxygen-responsive genes and fluorodeoxyglucose-PET as markers of tissue hypoxia. Int. J. Cancer 2008, 122, 2726–2734. [Google Scholar] [CrossRef]
- Chang, G.-C.; Liu, K.-J.; Hsieh, C.-L.; Hu, T.-S.; Charoenfuprasert, S.; Liu, H.-K.; Luh, K.-T.; Hsu, L.-H.; Wu, C.-W.; Ting, C.-C. Identification of α-enolase as an autoantigen in lung cancer: Its overexpression is associated with clinical outcomes. Clin. Cancer Res. 2006, 12, 5746–5754. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.; Xu, R.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.-F.H. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Enríquez-Flores, S.; Flores-López, L.A.; García-Torres, I.; de la Mora-de la Mora, I.; Cabrera, N.; Gutiérrez-Castrellón, P.; Martínez-Pérez, Y.; López-Velázquez, G. Deamidated Human Triosephosphate Isomerase is a Promising Druggable Target. Biomolecules 2020, 10, 1050. [Google Scholar] [CrossRef] [PubMed]
- Enríquez-Flores, S.; Flores-López, L.A.; De la Mora-De la Mora, I.; García-Torres, I.; Gracia-Mora, I.; Gutiérrez-Castrellón, P.; Fernández-Lainez, C.; Martínez-Pérez, Y.; Olaya-Vargas, A.; de Vos, P.; et al. Naturally occurring deamidated triosephosphate isomerase is a promising target for cell-selective therapy in cancer. Sci. Rep. 2022, 12, 4028. [Google Scholar] [CrossRef]
- Ahmed, N.; Battah, S.; Karachalias, N.; Babaei-Jadidi, R.; Horányi, M.; Baróti, K.; Hollan, S.; Thornalley, P.J. Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochim. Biophys. Acta-Mol. Basis Dis. 2003, 1639, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Aissa, K.; Kamoun, F.; Sfaihi, L.; Ghedira, E.S.; Aloulou, H.; Kamoun, T.; Pissard, S.; Hachicha, M. Hemolytic anemia and progressive neurologic impairment: Think about triosephosphate isomerase deficiency. Fetal Pediatr. Pathol. 2014, 33, 234–238. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Marín-Hernández, Á.; Del Mazo-Monsalvo, I.; Saavedra, E.; Rodríguez-Enríquez, S. Assessment of the low inhibitory specificity of oxamate, aminooxyacetate and dichloroacetate on cancer energy metabolism. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 3221–3236. [Google Scholar] [CrossRef] [PubMed]
- Orosz, F.; Oláh, J.; Ovádi, J. Triosephosphate isomerase deficiency: New insights into an enigmatic disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Brandhorst, T.T.; Kean, I.R.L.; Lawry, S.M.; Wiesner, D.L.; Klein, B.S. Phenylpyrrole fungicides act on triosephosphate isomerase to induce methylglyoxal stress and alter hybrid histidine kinase activity. Sci. Rep. 2019, 9, 5047. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhuo, Y.; Bian, X.; Li, J.; Zhang, Y.; Ma, L.; Lu, G.; Guo, M.Q.; Wu, J.L.; Li, N. Integrated Proteomics, Biological Functional Assessments, and Metabolomics Reveal Toosendanin-Induced Hepatic Energy Metabolic Disorders. Chem. Res. Toxicol. 2019, 32, 668–680. [Google Scholar] [CrossRef]
- Marsh, L.; Shah, K. A novel inhibitor of Mammalian triosephosphate isomerase found by an in silico approach. Int. J. Med. Chem. 2014, 2014, 469125. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Stroppolo, M.E.; Falconi, M.; Caccuri, A.M.; Desideri, A. Superefficient enzymes. Cell Mol. Life Sci. 2001, 58, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Wang, Z.; Guo, Y.; Yang, J.; Xing, Z.; Mu, L.; Zhang, X.; Ding, Z. Overexpression of triosephosphate isomerase inhibits proliferation of chicken embryonal fibroblast cells. Asian Pac. J. Cancer Prev. 2011, 12, 3479–3482. [Google Scholar] [PubMed]
- Jiang, H.; Ma, N.; Shang, Y.; Zhou, W.; Chen, T.; Guan, D.; Li, J.; Wang, J.; Zhang, E.; Feng, Y.; et al. Triosephosphate isomerase 1 suppresses growth, migration and invasion of hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2017, 482, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Adav, S.S.; Gallart-Palau, X.; Sze, S.K. Recent advances in mass spectrometric analysis of protein deamidation. Mass. Spectrom. Rev. 2017, 36, 677–692. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Gallart-Palau, X.; Wei, J.; Sze, S.K. Characterization of Glutamine Deamidation by Long-Length Electrostatic Repulsion-Hydrophilic Interaction Chromatography-Tandem Mass Spectrometry (LERLIC-MS/MS) in Shotgun Proteomics. Anal. Chem. 2016, 88, 10573–10582. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Molecular clocks. Proc. Natl. Acad. Sci. USA 2001, 98, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Decker, R.S.; Mohrenweiser, H.W. Cell proliferation-associated expression of a recently evolved isozyme of triosephosphate isomerase. Biochem. Genet. 1985, 23, 267–280. [Google Scholar] [CrossRef] [PubMed]
- de la Mora-de la Mora, I.; Torres-Larios, A.; Enriquez-Flores, S.; Mendez, S.-T.; Castillo-Villanueva, A.; Gomez-Manzo, S.; Lopez-Velazquez, G.; Marcial-Quino, J.; Torres-Arroyo, A.; Garcia-Torres, I. Structural effects of protein aging: Terminal marking by deamidation in human triosephosphate isomerase. PLoS ONE 2015, 10, e0123379. [Google Scholar]
- Yates, J., III; Gomes, F.; Durbin, K.; Schauer, K.; Nwachukwu, J.; Russo, R.; Njeri, J.; Saviola, A.; McClatchy, D.; Diedrich, J. Native top-down proteomics reveals EGFR–ERα signaling crosstalk in breast cancer cells dissociates NUTF2 dimers to modulate ERα signaling and cell growth. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, D.; Jarnuczak, A.F.; Viéitez, C.; Gehre, M.; Soucheray, M.; Mateus, A.; Kleefeldt, A.A.; Hill, A.; Garcia-Alonso, L.; Stein, F.; et al. The functional landscape of the human phosphoproteome. Nat. Biotechnol. 2020, 38, 365–373. [Google Scholar] [CrossRef]
- Duan, Y.; Li, J.; Wang, F.; Wei, J.; Yang, Z.; Sun, M.; Liu, J.; Wen, M.; Huang, W.; Chen, Z.; et al. Protein modifications throughout the lung cancer proteome unravel the cancer-specific regulation of glycolysis. Cell Rep. 2021, 37, 110137. [Google Scholar] [CrossRef]
- Weinert, B.T.; Schölz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Fan, T.T.; Mao, Y.Z.; Hou, J.L.; Wang, M.; Zhang, M.; Lin, Y.; Zhang, L.; Yan, G.Q.; An, Y.P.; et al. Nuclear dihydroxyacetone phosphate signals nutrient sufficiency and cell cycle phase to global histone acetylation. Nat. Metab. 2021, 3, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Højlund, K.; Bowen, B.P.; Hwang, H.; Flynn, C.R.; Madireddy, L.; Geetha, T.; Langlais, P.; Meyer, C.; Mandarino, L.J.; Yi, Z. In vivo phosphoproteome of human skeletal muscle revealed by phosphopeptide enrichment and HPLC-ESI-MS/MS. J. Proteome Res. 2009, 8, 4954–4965. [Google Scholar] [CrossRef]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal 2016, 9, rs9. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Ill-Raga, G.; Bravo, R.; Nakaya, T.; de Fabritiis, G.; Coma, M.; Miscione, G.P.; Villà-Freixa, J.; Suzuki, T.; Fernàndez-Busquets, X.; et al. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain 2009, 132, 1335–1345. [Google Scholar] [CrossRef]
- Tsai, C.F.; Wang, Y.T.; Yen, H.Y.; Tsou, C.C.; Ku, W.C.; Lin, P.Y.; Chen, H.Y.; Nesvizhskii, A.I.; Ishihama, Y.; Chen, Y.J. Large-scale determination of absolute phosphorylation stoichiometries in human cells by motif-targeting quantitative proteomics. Nat. Commun. 2015, 6, 6622. [Google Scholar] [CrossRef]
- Deeb, S.J.; Cox, J.; Schmidt-Supprian, M.; Mann, M. N-linked glycosylation enrichment for in-depth cell surface proteomics of diffuse large B-cell lymphoma subtypes. Mol. Cell. Proteom. 2014, 13, 240–251. [Google Scholar] [CrossRef]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteomics 2014, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.M.; Carrizo, M.E.; Curtino, J.A. Characterization of human triosephosphate isomerase S-nitrosylation. Nitric Oxide 2018, 77, 26–34. [Google Scholar] [CrossRef]
- Chan, J.C.Y.; Soh, A.C.K.; Kioh, D.Y.Q.; Li, J.; Verma, C.; Koh, S.K.; Beuerman, R.W.; Zhou, L.; Chan, E.C.Y. Reactive Metabolite-induced Protein Glutathionylation: A Potentially Novel Mechanism Underlying Acetaminophen Hepatotoxicity. Mol. Cell. Proteom. 2018, 17, 2034–2050. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Li, J.; Yang, J.; Li, B.; Li, N.; Zhan, X. Quantitative Acetylomics Revealed Acetylation-Mediated Molecular Pathway Network Changes in Human Nonfunctional Pituitary Neuroendocrine Tumors. Front. Endocrinol. 2021, 12, 753606. [Google Scholar] [CrossRef]
- Lee, W.H.; Choi, J.S.; Byun, M.R.; Koo, K.T.; Shin, S.; Lee, S.K.; Surh, Y.J. Functional inactivation of triosephosphate isomerase through phosphorylation during etoposide-induced apoptosis in HeLa cells: Potential role of Cdk2. Toxicology 2010, 278, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Schachner, L.F.; Soye, B.D.; Ro, S.; Kenney, G.E.; Ives, A.N.; Su, T.; Goo, Y.A.; Jewett, M.C.; Rosenzweig, A.C.; Kelleher, N.L. Revving an Engine of Human Metabolism: Activity Enhancement of Triosephosphate Isomerase via Hemi-Phosphorylation. ACS Chem. Biol. 2022, 17, 2769–2780. [Google Scholar] [CrossRef]
- Sun, A.Q.; Yüksel, K.U.; Gracy, R.W. Relationship between the catalytic center and the primary degradation site of triosephosphate isomerase: Effects of active site modification and deamidation. Arch. Biochem. Biophys. 1992, 293, 382–390. [Google Scholar] [CrossRef]
- Tajes, M.; Eraso-Pichot, A.; Rubio-Moscardó, F.; Guivernau, B.; Ramos-Fernández, E.; Bosch-Morató, M.; Guix, F.X.; Clarimón, J.; Miscione, G.P.; Boada, M.; et al. Methylglyoxal produced by amyloid-β peptide-induced nitrotyrosination of triosephosphate isomerase triggers neuronal death in Alzheimer’s disease. J. Alzheimers Dis. 2014, 41, 273–288. [Google Scholar] [CrossRef]
- Tan, C.; Li, Y.; Huang, X.; Wei, M.; Huang, Y.; Tang, Z.; Huang, H.; Zhou, W.; Wang, Y.; Hu, J. Extensive protein S-nitrosylation associated with human pancreatic ductal adenocarcinoma pathogenesis. Cell Death Dis. 2019, 10, 914. [Google Scholar] [CrossRef]
- Biri-Kovács, B.; Kiss, B.; Vadászi, H.; Gógl, G.; Pálfy, G.; Török, G.; Homolya, L.; Bodor, A.; Nyitray, L. Ezrin interacts with S100A4 via both its N- and C-terminal domains. PLoS ONE 2017, 12, e0177489. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle 2010, 9, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Casadei, M.; Persichini, T.; Polticelli, F.; Musci, G.; Colasanti, M. S-glutathionylation of metallothioneins by nitrosative/oxidative stress. Exp. Gerontol. 2008, 43, 415–422. [Google Scholar] [CrossRef]
- Hameed, M.S. Allosteric inhibition of triose-phosphate isomerase by s-glutathionylation. J. Proteins Proteom. 2015, 6, 159–171. [Google Scholar]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.D.; Ferrarone, J.R.; Gardner, E.E.; Chang, J.W.; Wu, D.; Hollstein, P.E.; Liang, R.J.; Yuan, M.; Chen, Q.; Coukos, J.S.; et al. LKB1-Dependent Regulation of TPI1 Creates a Divergent Metabolic Liability between Human and Mouse Lung Adenocarcinoma. Cancer Discov. 2023, 13, 1002–1025. [Google Scholar] [CrossRef] [PubMed]
- Jandova, J.; Wondrak, G.T. Genomic GLO1 deletion modulates TXNIP expression, glucose metabolism, and redox homeostasis while accelerating human A375 malignant melanoma tumor growth. Redox Biol. 2021, 39, 101838. [Google Scholar] [CrossRef]
- Dube, G.; Tiamiou, A.; Bizet, M.; Boumahd, Y.; Gasmi, I.; Crake, R.; Bellier, J.; Nokin, M.J.; Calonne, E.; Deplus, R.; et al. Methylglyoxal: A novel upstream regulator of DNA methylation. J. Exp. Clin. Cancer Res. 2023, 42, 78. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Positions of Modified Amino Acid Residues | Post-Translational Modification | Reference |
|---|---|---|
| 14 | N6-acetyllysine | [101] |
| 16 | Deamidation | [79] |
| 21 | Phosphoserine | [102] |
| 28 | Phosphothreonine | [103] |
| 58 | Phosphoserine | [104] |
| 59 | Succinylation | [105] |
| 68 | 3′-nitrotyrosine | [103] |
| 69 | Ubiquitylation | [106] |
| 71, 76 | Phosphothreonine | [103] |
| 80 | Phosphoserine | [107] |
| 90 | Phosphothreonine | [103] |
| 97 | Phosphoserine | [103] |
| 106 | Phosphoserine | [108] |
| 135 | Methylation | [109] |
| 149, 156 * | N6-acetyllysine | [110] |
| 159 * | Phosphoserine | [111] |
| 165 ** | 3′-nitrotyrosine | [112] |
| 173 | Phosphothreonine | [113] |
| 194 | N6-acetyllysine | [105] |
| 196 | N-glycosylation | [114] |
| 204 | Phosphoserine | [108] |
| 209 ** | 3′-nitrotyrosine | [112] |
| 212 | Phosphoserine | [108] |
| 214 | Phosphothreonine | [115] |
| 217 | S-nitrosylation | [116] |
| 217 | Glutathionylation | [117] |
| 225 | N6-acetyllysine | [118] |
| 238 | N6-acetyllysine | [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enríquez-Flores, S.; De la Mora-De la Mora, I.; García-Torres, I.; Flores-López, L.A.; Martínez-Pérez, Y.; López-Velázquez, G. Human Triosephosphate Isomerase Is a Potential Target in Cancer Due to Commonly Occurring Post-Translational Modifications. Molecules 2023, 28, 6163. https://doi.org/10.3390/molecules28166163
Enríquez-Flores S, De la Mora-De la Mora I, García-Torres I, Flores-López LA, Martínez-Pérez Y, López-Velázquez G. Human Triosephosphate Isomerase Is a Potential Target in Cancer Due to Commonly Occurring Post-Translational Modifications. Molecules. 2023; 28(16):6163. https://doi.org/10.3390/molecules28166163
Chicago/Turabian StyleEnríquez-Flores, Sergio, Ignacio De la Mora-De la Mora, Itzhel García-Torres, Luis A. Flores-López, Yoalli Martínez-Pérez, and Gabriel López-Velázquez. 2023. "Human Triosephosphate Isomerase Is a Potential Target in Cancer Due to Commonly Occurring Post-Translational Modifications" Molecules 28, no. 16: 6163. https://doi.org/10.3390/molecules28166163
APA StyleEnríquez-Flores, S., De la Mora-De la Mora, I., García-Torres, I., Flores-López, L. A., Martínez-Pérez, Y., & López-Velázquez, G. (2023). Human Triosephosphate Isomerase Is a Potential Target in Cancer Due to Commonly Occurring Post-Translational Modifications. Molecules, 28(16), 6163. https://doi.org/10.3390/molecules28166163