
The Synthesis and Base-Induced Breakdown of Triaryl 1,4-Oxathiins—An Experimental and DFT Study
 ,
,
Abstract
:1. Introduction
2. Results and Discussion
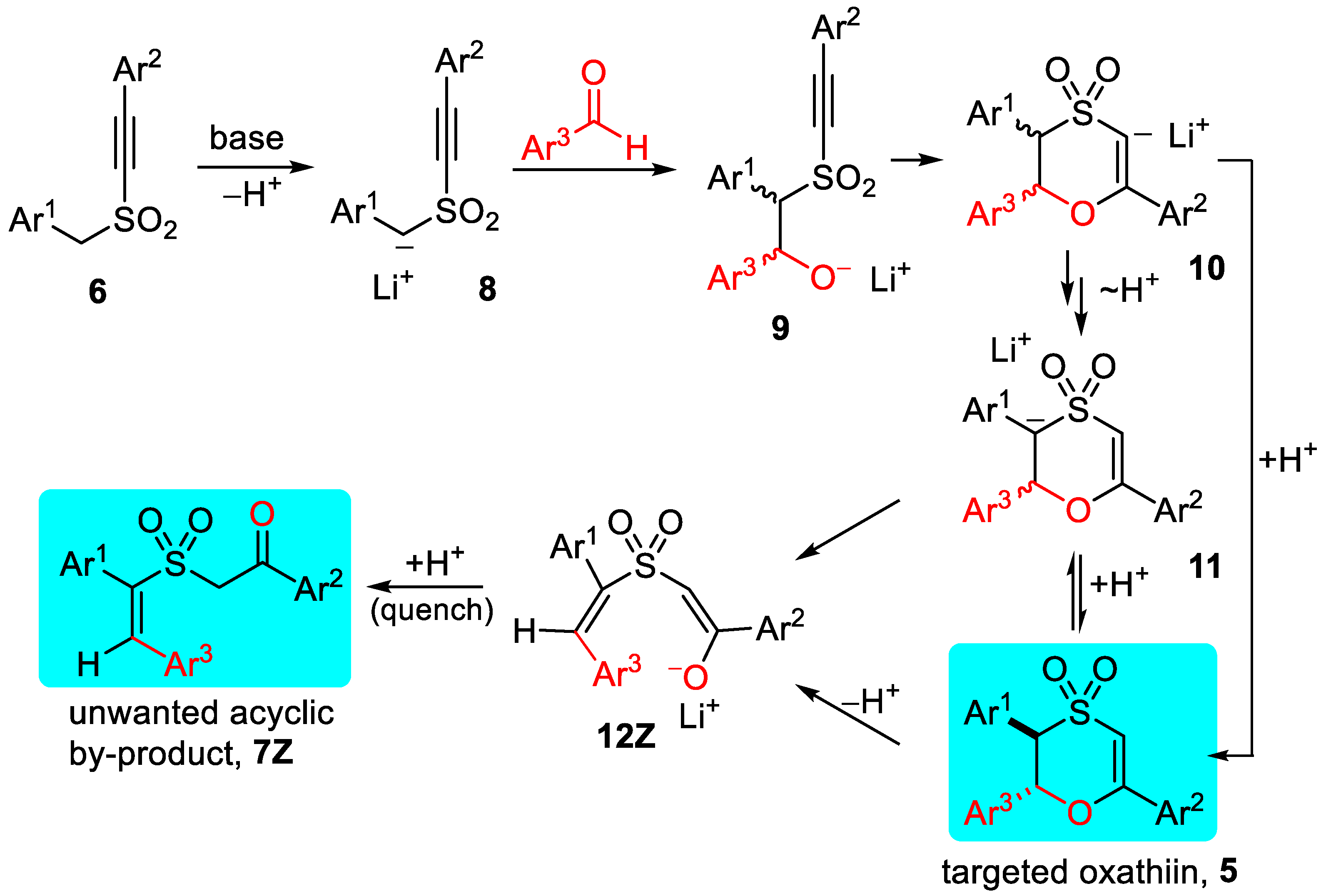
2.1. Preparation of Substituted 1,4-Oxathiins
2.2. Computational Chemistry
2.2.1. Oxathiin Ring Formation
2.2.2. Oxathiin Ring Opening
2.2.3. Proton Transfer Options and Overall Pathway
3. Experimental
3.1. General Reaction Procedures
3.2. Preparation of Trisubstituted 1,4-Oxathiins
General Method for Oxathiin-S,S-Dioxides
3.3. Oxathiins Ring Opening Experiments
3.3.1. Example Procedure for Base Catalyzed Ring Openings of Oxathiin 5a
3.3.2. Example Procedure for Isomerization of 12Z to 12E
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Guillaumet, G. 1,4-Dioxins, oxathiins, dithiins and their benzo derivatives. In Comprehensive Heterocyclic Chemistry II; Elsevier: Amsterdam, The Netherlands, 1996; Volume 6, pp. 447–481, 1177–1307. [Google Scholar]
- Caputo, R.; Ferreri, C.; Guaragna, A.; Palumbo, G.; Pedatella, S. New synthesis of carboxin and oxycarboxin pesticides: Application to the preparation of their new analogs substituted at the C-2 methyl group. J. Chem. Soc. Perkin Trans. 1 1995, 1971–1973. [Google Scholar] [CrossRef]
- Schmeling, B.V.; Kulka, M. Systemic fungicidal activity of 1,4-oxathiin derivatives. Science 1966, 152, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.A.; Puttock, M.A.; Felauer, E.E.; Neidermyer, R.W. Substituted 2,3-dihydro-1,4-oxathiin as Plant Growth Regulators. DE2513202A1, 23 October 1975. [Google Scholar]
- Jaiswal, A.K.; Faisal, M.; Tailor, S.P. Environment conscious control of Fusarium oxysporum F. sp. Lycopersici-induced tomato wilt using bio agents, phytochemicals and their combination in marked contrast to chemical. Pharma Innov. 2023, 12, 481–488. [Google Scholar] [CrossRef]
- Kaur, M.; Gupta, P.K.; Kushwaha, K. Potential of chemical fungicides against Rhizoctonia solani Kuhn. inciting web blight of mungbean [Vigna radiata (L.) Wilczek]. Pharma Innov. 2022, 11, 6304–6308. [Google Scholar]
- Nivedita; Mahajan, S.; Kaur, H.; Astha; Paswal, S. In vitro evaluation of fungicides and plant extracts against the mycelial growth of Neovossia indica. Pharma Innov. 2023, 12, 5017–5019. [Google Scholar]
- Bhagat, N.S.; Kamdi, T.S.; Dadmal, K.D. Effect of seed coating treatments on field performance of soybean (Glycine max (L.) Merrill). Pharma Innov. 2022, 11, 1118–1119. [Google Scholar]
- Pathan, A.K.; Cuddy, W.; Kimberly, M.O.; Adusei-Fosu, K.; Rolando, C.A.; Park, R.F. Efficacy of fungicides applied for protectant and curative activity against myrtle rust. Plant Dis. 2020, 104, 2123–2129. [Google Scholar] [CrossRef]
- Balzarini, J.; Jonckheere, H.; Harrison, W.A.; Dao, D.C.; Anne, J.; De Clercq, E.; Karlsson, A. Oxathiin carboxanilide derivatives: A class of nonnucleoside HIV-1-specific reverse transcriptase inhibitors (NNRTIs) that are active against mutant HIV-1 strains resistant to other NNRTIs. Antivir. Chem. Chemother. 1995, 6, 169–178. [Google Scholar] [CrossRef]
- Miyauchi, H.; Tanio, T.; Ohashi, N. Synthesis and antifungal activity of new azole derivatives containing an oxathiane ring. Bioorg. Med. Chem. Lett. 1996, 6, 2377–2380. [Google Scholar] [CrossRef]
- Schroder Glad, S.; Birkebak Jensen, K.; Gron Noerager, N.; Sarvary, I.; Vestergaard, M.; Haahr Gouliaev, A.; Teuber, L.; Stasi, L.P. Preparation of Optionally Fused Heterocyclyl-Substituted Derivatives of Pyrimidine Useful for the Treatment of Inflammatory, Metabolic, Oncologic and Autoimmune Diseases. WO2016020295A1, 11 February 2016. [Google Scholar]
- Song, Z.J.; King, A.O.; Waters, M.S.; Lang, F.; Zewge, D.; Bio, M.; Leazer, J.L.; Javadi, G.; Kassim, A.; Tschaen, D.M.; et al. An efficient asymmetric synthesis of an estrogen receptor modulator by sulfoxide-directed borane reduction. Proc. Nat. Acad. Sci. USA 2004, 101, 5776–5781. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wu, J.; Yang, H. Synthesis, structure, and conformation of 2’,3’-fused oxathiane and thiomorpholine uridines. Helv. Chim. Acta 2007, 90, 1917–1924. [Google Scholar] [CrossRef]
- Caputo, R.; Giordano, F.; Guaragna, A.; Palumbo, G.; Pedatella, S. A facile stereospecific synthesis of chiral β-keto sulfoxides. Tetrahedron Asymmetry 1999, 10, 3463–3466. [Google Scholar] [CrossRef]
- Bułakowska, A.; Konieczny, M.T. Synthesis of Vinyl Sulfones by Ring Opening of 4,4-Dioxo-2,3-dihydrobenzo[b][1,4]oxathiines and Their In Situ Reactions with Nucleophilic or Electrophilic Agents. J. Heterocycl. Chem. 2015, 52, 440–444. [Google Scholar] [CrossRef]
- Capozzi, G.; Menichetti, S.; Nativi, C.; Provenzani, A. α-Oxosulfines, IV. Intramolecular hetero Diels-Alder reactions of α,α′-dioxosulfines—A new access to the [3.3.1]bicyclic skeleton. Eur. J. Org. Chem. 2000, 2000, 3721–3725. [Google Scholar] [CrossRef]
- Capozzi, G.; Fratini, P.; Menichetti, S.; Nativi, C. α-Oxosulfines. Part 1. Reactivity of α-oxosulfines obtained from retro Diels-Alder reaction of 1,4-oxathiin-S-oxides. Tetrahedron 1996, 52, 12233–12246. [Google Scholar] [CrossRef]
- Brewer, A.D.; Znotins, A.A. Reaction of dichlorocarbene with 2,3-dihydro-5,6-dimethyl-1,4-oxathiin and 2,3-dihydro-5,6-dimethyl-1,4-dithiin. J. Heterocycl. Chem. 1996, 33, 217–219. [Google Scholar] [CrossRef]
- Hahn, H.-G.; Mah, H.; Lee, S.-J. Synthesis of 1,4-thiazin-3-one by ring opening of 1,4-oxathiin. J. Korean Chem. Soc. 1995, 39, 878–880. [Google Scholar]
- Noland, W.E.; DeMaster, R.D. 4H-1,4-Thiazine 1,1-dioxide. Org. Syn. 1972, 52, 135–139. [Google Scholar] [CrossRef]
- Trost, B.M.; Shi, Z. A Concise Convergent Strategy to Acetogenins. (+)-Solamin and Analogs. J. Am. Chem. Soc. 1994, 116, 7459–7460. [Google Scholar] [CrossRef]
- Fjeldskaar, I.R.; Rongved, P.; Skatteboel, L. A convenient method for the preparation of bicyclic dihydro-1,4-dioxins, dihydro-1,4-oxathiins, dihydro-1,4-dithiins and related compounds. Acta Chem. Scand. Ser. B 1987, B41, 477–486. [Google Scholar] [CrossRef]
- Gharpure, S.J.; Anuradha, D.; Prasad, J.V.K.; Srinivasa Rao, P. Stereoselective Synthesis of cis-2,6-Disubstituted Morpholines and 1,4-Oxathianes by Intramolecular Reductive Etherification of 1,5-Diketones. Eur. J. Org. Chem. 2015, 2015, 86–90. [Google Scholar] [CrossRef]
- Selvaraj, S.; Dhanabalan, A.; Amaithirani, J.S.A.; Arumugam, N. Condensation of dimethyl sulfone with aromatic aldehydes using phase-transfer catalysis. Indian J. Chem. Sect. B 1991, 30B, 871–872. [Google Scholar]
- Carretero, J.C.; Garcia Ruano, J.L.; Rodriguez, J.H. Stereoespecific syntheses of 2,3-dimethyl-1,4-oxathiane S-oxides. Tetrahedron Lett. 1984, 25, 3029–3032. [Google Scholar] [CrossRef]
- Yamamoto, M.; Munakata, H.; Hussein, M.Z.; Kohmoto, S.; Yamada, K. Cyclization of alkynecarboxylic acids: Synthesis and reactions of 6-methylene-1,4-oxathian-2-ones and their 4,4-dioxides. J. Chem. Res. 1990, 12–13. [Google Scholar]
- Bosset, C.; Lefebvre, G.; Angibaud, P.; Stansfield, I.; Meerpoel, L.; Berthelot, D.; Guérinot, A.; Cossy, J. Iron-Catalyzed Synthesis of Sulfur-Containing Heterocycles. J. Org. Chem. 2017, 82, 4020–4036. [Google Scholar] [CrossRef] [PubMed]
- Bedford, S.T.; Grainger, R.S.; Steed, J.W.; Tisselli, P. Stereoselective synthesis of 2,5-disubstituted-1,4-oxathiane S-oxides. Org. Biomol. Chem. 2005, 3, 404–406. [Google Scholar] [CrossRef]
- Menichetti, S.; Nativi, C. Hetero Diels-Alder approach to oxathiins. Targets Heterocycl. Syst. 2003, 7, 108–139. [Google Scholar] [CrossRef]
- Yang, H.-B.; Yuan, Y.-C.; Wei, Y.; Shi, M. Amine-catalyzed tunable reactions of allenoates with dithioesters: Formal [4+2] and [2+2] cycloadditions for the synthesis of 2,3-dihydro-1,4-oxathiines and enantioenriched thietanes. Chem. Commun. 2015, 51, 6430–6433. [Google Scholar] [CrossRef]
- Capozzi, G.; Fratini, P.; Menichetti, S.; Nativi, C. Generation and trapping of α,α′-dioxosulfines from 1,4-oxathiine-S-oxides. Tetrahedron Lett. 1995, 36, 5089–5092. [Google Scholar] [CrossRef]
- Capozzi, G.; Corti, A.; Menichetti, S.; Nativi, C. α-oxosulfines part 3. Generation and trapping of α-oxothioaldehyde S-oxides. Tetrahedron Lett. 1997, 38, 5041–5044. [Google Scholar] [CrossRef]
- Samzadeh-Kermani, A. Silver salt catalyzed synthesis of 1,4-oxathian-3-imine derivatives. Tetrahedron 2016, 72, 5301–5304. [Google Scholar] [CrossRef]
- Chumachenko, N.; Sampson, P. Synthesis of β-hydroxy sulfones via opening of hydrophilic epoxides with zinc sulfinates in aqueous media. Tetrahedron 2006, 62, 4540–4548. [Google Scholar] [CrossRef]
- McPhee, D.J. Synthesis of 1,3-oxathiolane Sulfoxide Compounds. CA2183714A1, 15 March 1997. [Google Scholar]
- Lee, W.S.; Hahn, H.G.; Chang, K.H. Synthesis of sulfur-oxygen-transposed dihydro-1,4-oxathiin derivative by unusual rearrangement of β-hydroxy-1,3-oxathiolanes. J. Org. Chem. 1989, 54, 2455–2457. [Google Scholar] [CrossRef]
- Ioannou, M.; Porter, M.J.; Saez, F. Conversion of 1,3-oxathiolanes to 1,4-oxathianes using a silylated diazo ester. Tetrahedron 2005, 61, 43–50. [Google Scholar] [CrossRef]
- Yang, W.; Sun, J. Organocatalytic Enantioselective Synthesis of 1,4-Dioxanes and Other Oxa-Heterocycles by Oxetane Desymmetrization. Angew. Chem. Int. Ed. 2016, 55, 1868–1871. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.U.; Shkoor, M.G.; Hossain, M.S.; Deen, M.C.; Soldatov, D.V.; Schwan, A.L. The base-mediated cyclization of selected benzyl alkynyl sulfones with aromatic aldehydes: Novel synthetic access to aryl-substituted 5,6-dihydro-1,4-oxathiin S,S-dioxides. J. Sulfur Chem. 2013, 34, 79–87. [Google Scholar] [CrossRef]
- Goehmann, P.; Schroeder, L.; Zschunke, A. Reaction of 5,6-dihydro-1,4-oxathiin 4,4-dioxides with nucleophiles. J. Prakt. Chem. 1986, 328, 380–388. [Google Scholar] [CrossRef]
- Ying, J.; Delaglio, F.; Torchia, D.A.; Bax, A. Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 2017, 68, 101–118. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision b.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Trinh, H.V.; Perrin, L.; Goekjian, P.G.; Gueyrard, D. Development of a Modified Julia Olefination of Imides for the Synthesis of Alkaloids. Eur. J. Org. Chem. 2016, 2016, 2944–2953. [Google Scholar] [CrossRef]
- Walker, M.; Harvey, A.J.A.; Sen, A.; Dessent, C.E.H. Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions. J.Phys. Chem. A 2013, 117, 12590–12600. [Google Scholar] [CrossRef] [PubMed]
- Ramig, K.; Subramaniam, G.; Karimi, S.; Szalda, D.J.; Ko, A.; Lam, A.; Li, J.; Coaderaj, A.; Cavdar, L.; Bogdan, L.; et al. Interplay of Nitrogen-Atom Inversion and Conformational Inversion in Enantiomerization of 1H-1-Benzazepines. J.Org. Chem. 2016, 81, 3313–3320. [Google Scholar] [CrossRef] [PubMed]
- Cadoni, E.; Arca, M.; De Montis, S.; Fattuoni, C.; Perra, E.; Cabiddu, M.G.; Usai, M.; Cabiddu, S. Lithium 2,3-dihydro-1-benzothiophene-1,1-dioxide: Synthesis, characterization, DFT calculations, and reactivity toward aldehydes and azomethines. Tetrahedron 2007, 63, 11122–11134. [Google Scholar] [CrossRef]
- Belostotskii, A.M.; Albeck, A.; Hassner, A. Asymmetric Induction by a Remote Chiral Substituent—Computationally Determined Stereodifferentiation in Michael Additions of α-Lithiated Allyl Sulfones. Eur. J. Org. Chem. 2007, 2007, 4837–4844. [Google Scholar] [CrossRef]
- Robiette, R.; Pospíšil, J. On the Origin of E/Z Selectivity in the Modified Julia Olefination—Importance of the Elimination Step. Eur. J. Org. Chem. 2013, 2013, 836–840. [Google Scholar] [CrossRef]
- Wei, W.; Khangarot, R.K.; Stahl, L.; Veresmortean, C.; Pradhan, P.; Yang, L.; Zajc, B. Generating Stereodiversity: Diastereoselective Fluorination and Highly Diastereoselective Epimerization of α-Amino Acid Building Blocks. Org. Lett. 2018, 20, 3574–3578. [Google Scholar] [CrossRef]
- Nakamura, S.; Hirata, N.; Yamada, R.; Kita, T.; Shibata, N.; Toru, T. Catalytic and Highly Enantioselective Reactions of α-Sulfonyl Carbanions with Chiral Bis(oxazoline)s. Chem. Eur. J. 2008, 14, 5519–5527. [Google Scholar] [CrossRef]
- Aggarwal, S.; Vu, A.; Eremin, D.B.; Persaud, R.; Fokin, V.V. Arenes participate in 1,3-dipolar cycloaddition with in situ-generated diazoalkenes. Nat. Chem. 2023, 15, 764–772. [Google Scholar] [CrossRef]
- Legnani, L.; Porta, A.; Caramella, P.; Toma, L.; Zanoni, G.; Vidari, G. Computational Mechanistic Study of the Julia–Kocieński Reaction. J. Org. Chem. 2015, 80, 3092–3100. [Google Scholar] [CrossRef]
- Rodrigo, E.; Alonso, I.; Cid, M.B. A Protocol To Transform Sulfones into Nitrones and Aldehydes. Org. Lett. 2018, 20, 5789–5793. [Google Scholar] [CrossRef]
- Gais, H.J. Asymmetric Reactions of α-Sulfonyl Carbanions. In Organosulfur Chemistry in Asymmetric Synthesis; Toru, T., Bolm, C., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2008; pp. 375–398. [Google Scholar]
- Hossain, M.S.; Schwan, A.L. Separate Deprotonation Reactions Converge Mechanistically for a New Cyclization of Benzyl 1-Alkynyl Sulfones. Org. Lett. 2011, 13, 5330–5333. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| # | Scale a | Yield b,c | ||
| 1 |  | Ar1 = C6H5, 5a | 1 | 74% |
| 2 | C6H5, 5a | 4 | 86% d | |
| 3 | C6H5, 5a | 4 | 75% e | |
| 4 | 2-I-C6H4, 5b | 2 | 78% | |
| 5 | 3-Cl-C6H4, 5c | 3 | 76% e | |
| 6 | 4-NO2-C6H4, 5d | 3 | 62% e | |
| 7 | 4-CH3-C6H4, 5e | 1 | 76% | |
| 8 | 4-CH3O-C6H4, 5f | 1 | 44% | |
| 9 | 4-Br-C6H4, 5g | 1 | 73% | |
| 10 | 3-CN-C6H4, 5h | 3 | 64% | |
| 11 |  | Ar2 = 4-CN-C6H4, 5i | 1 | 69% |
| 12 | 4-Br-C6H4, 5j | 1 | 34% | |
| 13 | 4-F-C6H4, 5k | 1 | 78% | |
| 14 | 4-NO2-C6H4, 5l | 1 | 62% | |
| 15 | 2-CF3-C6H4, 5m | 2 | 35% | |
| 16 | 4-CH3O-C6H4, 5n | 2 | 74% | |
| 17 | 2-furyl, 5o | 2 | 37% | |
| 18 |  | X = 4-Me; Y = 4-NO2; R = Ph, 5p | 1 | 55% |
| 19 | X = 2-I; Y = 4-NO2; R = Ph, 5q | 1 | 64% d,e | |
| 20 | X = 2-I; Y-Ar = 2-thienyl; R = Ph, 5r | 4 | 35% d,e | |
| 21 | X = 2-I; Y = H; R = tBu, 5s | 3 | 39% d | |
| 22 | X = 2-I; Y = H; R = Me, 5t | 3 | 38% | |
 | ||
|---|---|---|
| Relative Ratio of Isomers b | ||
| Duration (h) | % 7Z | % 7E |
| 0 c | 76 | 24 |
| 24 | 26 | 74 |
| 48 | 3 | 97 |
| 72 | 0 | 100 |
| C5-C6 (Å) | C5-S (Å) | S-O1 (Å) | S-O2 (Å) | S-C3 (Å) | C3-C2 (Å) | C2-Ob (Å) | O1-Li a (Å) | O2-Li a (Å) | Ob-Li a (Å) | Ob-C6 (Å) | PhC3-C2Ph Dihedral | HC3-C2H Dihedral | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 9cis | 1.206 | 1.717 | 1.457 | 1.473 | 1.819 | 1.577 | 1.353 | - | 1.971 | 1.783 | 3.969 | 55.0 | 39.2 |
| 9cis‡ | 1.250 | 1.715 | 1.460 | 1.484 | 1.839 | 1.574 | 1.379 | - | 1.977 | 1.848 | 2.152 | 38.7 | 23.7 |
| 10cis | 1.344 | 1.732 | 1.465 | 1.502 | 1.845 | 1.570 | 1.434 | - | 1.915 | 2.152 | 1.426 | 10.7 | 4.7 |
| 9trans | 1.206 | 1.718 | 1.473 | 1.456 | 1.822 | 1.577 | 1.351 | 1.979 | - | 1.774 | 3.819 | −60.8 | 176.5 |
| 9trans‡ | 1.250 | 1.716 | 1.486 | 1.459 | 1.831 | 1.574 | 1.376 | 1.974 | - | 1.852 | 2.158 | −76.0 | 161.7 |
| 10trans | 1.344 | 1.731 | 1.505 | 1.464 | 1.856 | 1.548 | 1.434 | 1.900 | - | 2.287 | 1.423 | −83.3 | 161.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicol, E.A.; Sing, M.; Luu, L.U.; Remigio, E.J.; Mills, M.B.; Schwan, A.L. The Synthesis and Base-Induced Breakdown of Triaryl 1,4-Oxathiins—An Experimental and DFT Study. Molecules 2023, 28, 6180. https://doi.org/10.3390/molecules28176180
Nicol EA, Sing M, Luu LU, Remigio EJ, Mills MB, Schwan AL. The Synthesis and Base-Induced Breakdown of Triaryl 1,4-Oxathiins—An Experimental and DFT Study. Molecules. 2023; 28(17):6180. https://doi.org/10.3390/molecules28176180
Chicago/Turabian StyleNicol, Eric A., Matthew Sing, Lilly U. Luu, Erwin J. Remigio, Michelle B. Mills, and Adrian L. Schwan. 2023. "The Synthesis and Base-Induced Breakdown of Triaryl 1,4-Oxathiins—An Experimental and DFT Study" Molecules 28, no. 17: 6180. https://doi.org/10.3390/molecules28176180