

Evaluation of 4-Aminoquinoline Hydrazone Analogues as Potential Leads for Drug-Resistant Malaria

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
2.1. Determination of IC50 Values for the Activity of 4-Aminoquinoline Hydrazones against the K1 Strain of P. falciparum
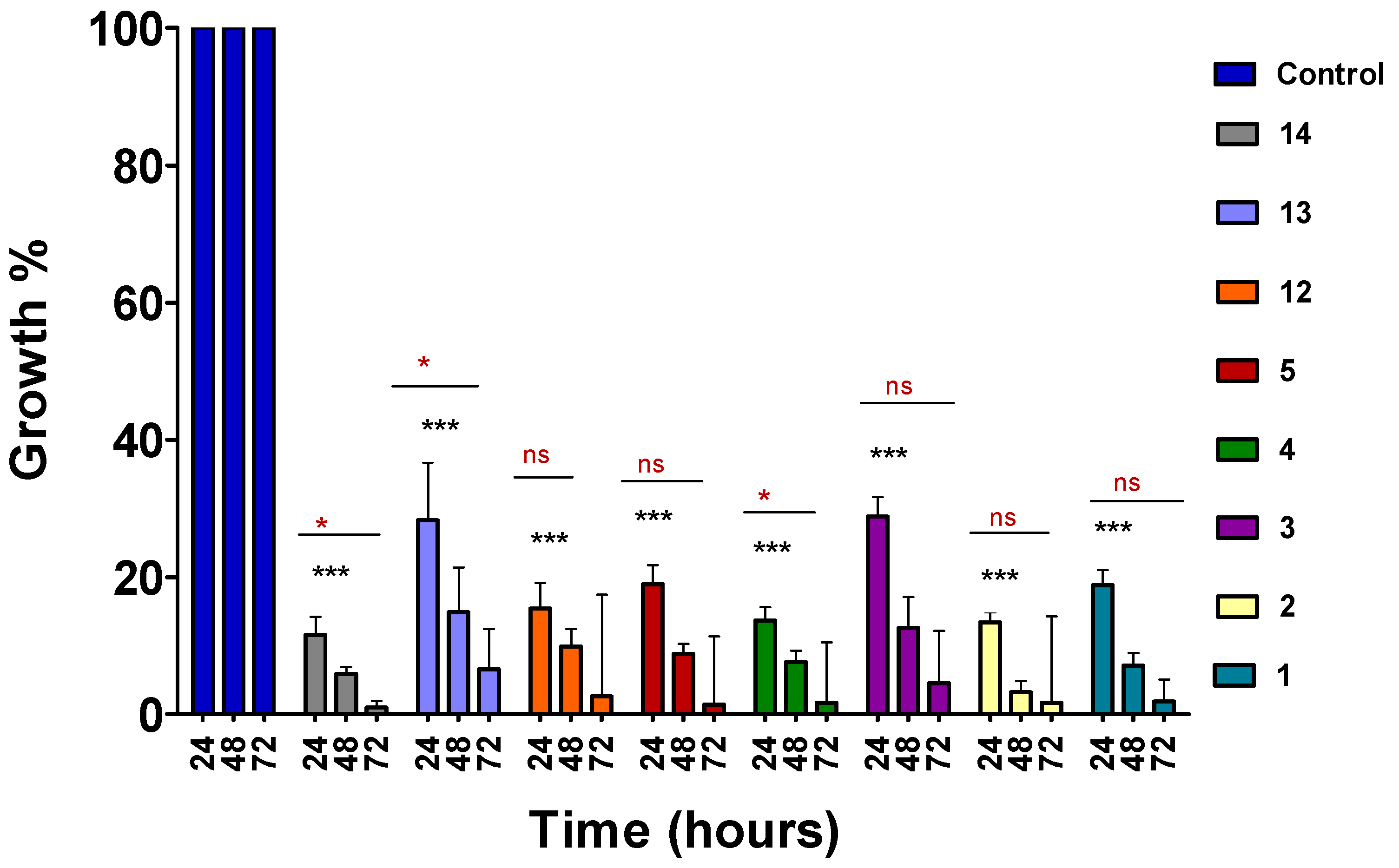
2.2. Time-Course Assay
2.3. Dose–Response of Lead Compounds after 72 h Incubation Period
2.4. MTT Cytotoxicity Assay
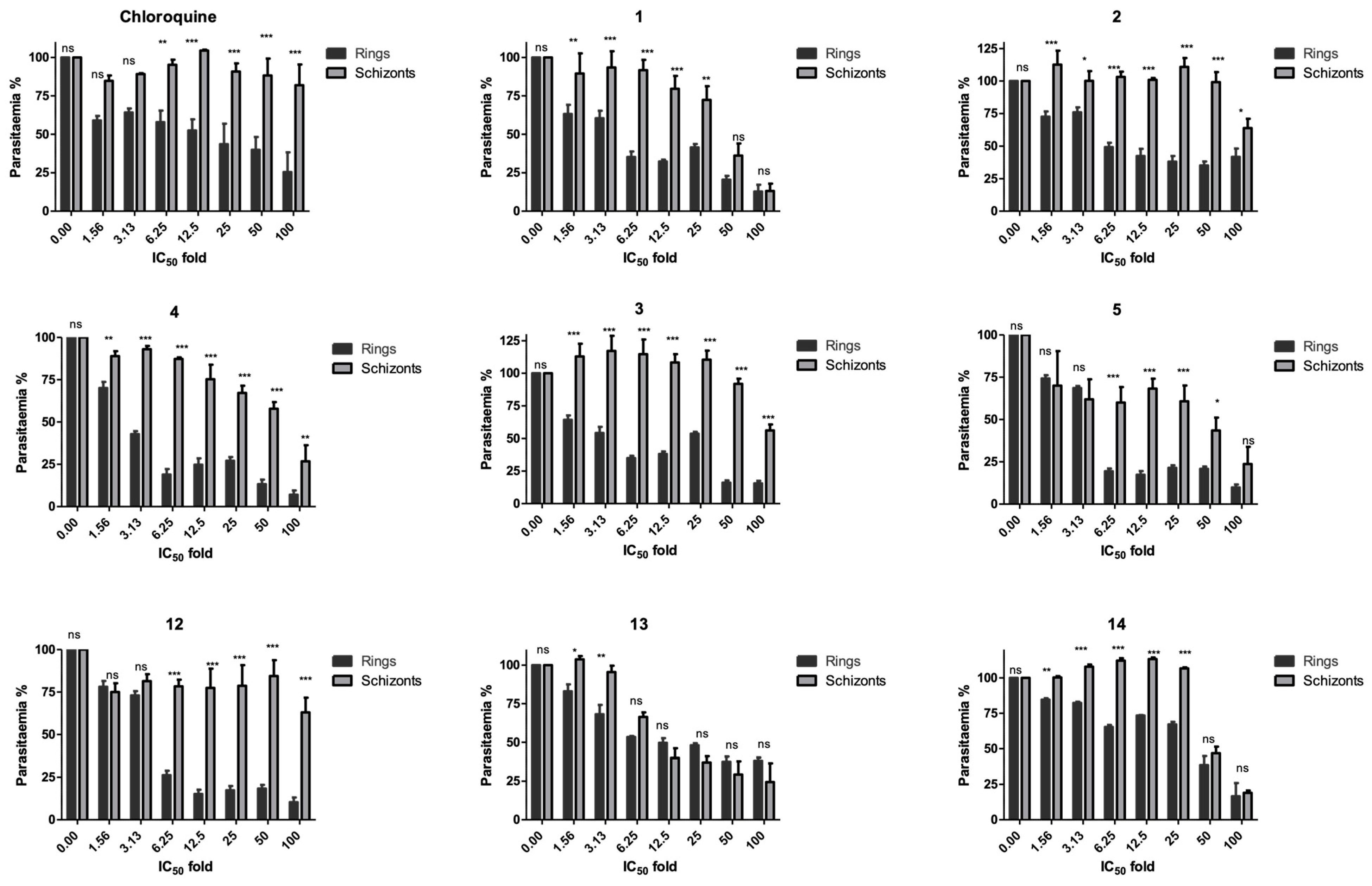
2.5. Stage Specificity Analysis
2.6. In Vitro IC50 Values against 3D7 and Dd2 P. falciparum Strains
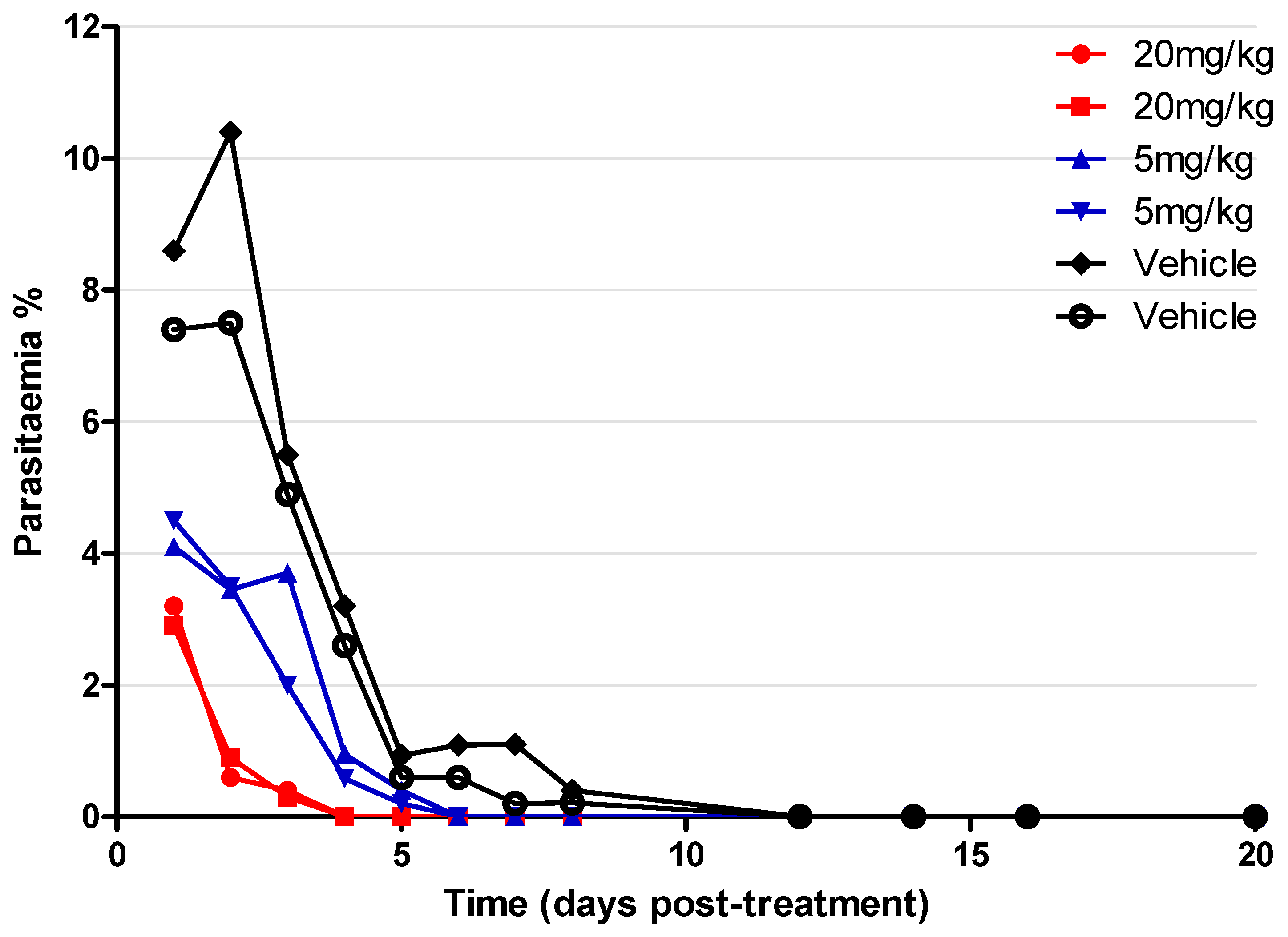
2.7. In Vivo Assay on P. yoelii Mouse Model
2.8. CalcuSyn Assay for Combination Therapy Assay
3. Discussion
4. Materials and Methods
4.1. Drug Preparation and Synthesis
4.1.1. Plasmodium falciparum Parasite Cultivation
4.1.2. Synchronization of K1 P. falciparum
4.1.3. Determination of IC50 in P. falciparum
4.1.4. Time-Course Assay
4.1.5. Flow Cytometry
4.1.6. MTT Assay for Testing Cell Cytotoxicity
4.1.7. Stage Specificity Assay
4.1.8. Derivation of the Dose–Response Curves and IC50 Values
4.1.9. Drug Interaction Assay for 4-Aminoquinoline Hydrazones
4.1.10. P. yoelii NL Mice Screening with Compound 2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. World Malaria Report 2022. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (accessed on 31 August 2022).
- Kamau, A.; Mtaje, G.; Mataza, C.; Mwmbingu, G.; Mturi, N.; Mohammed, S.; Ong’ayo, G.; Nyutu, G.; Nyaguara, A.; Bejon, P.; et al. Malaria infection, disease and mortality among children and adults on the coast of Kenya. Malar J. 2020, 19, 210. [Google Scholar] [CrossRef]
- Sato, S. Plasmodium – A brief introduction to the parasites causing human malaria and their basic biology. J. Physiol. Antropol. 2021, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.S. Malaria. Clin. Lab. Med. 2010, 30, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Das, B.S. Renal failure in malaria. J. Vector Borne Dis. 2008, 45, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Laurens, M.B. RTS, S/AS01 vaccine (MosquirixTM): An overview. Hum Vaccin Immunother. 2020, 16, 48–489. [Google Scholar] [CrossRef]
- Bjorkman, A.; Benn, C.S.; Aaby, P.; Schapira, A. RTS,S/ AS01 malaria vaccine-proven safe and effective. Lancet Infect. Dis. 2023, 23, 318–322. [Google Scholar] [CrossRef]
- El-Moamly, A.A.; El-Sweify, M.A. Malaria vaccine: The 60-year journey of hope and final success-lessons learned and future prospect. Trop. Med. Health. 2023, 51, 29. [Google Scholar] [CrossRef]
- Rasmussen, C.; Alonso, R.; Ringwald, P. Current and emerging strategies to combat antimalarial resistance. Expert Rev Anti Infect Ther. 2022, 20, 353–372. [Google Scholar] [CrossRef]
- Imwong, M.; Hein, T.T.; Thuy-Nhien, N.; Dondorp, A.M.; White, N.J. Spread of a single multidrug resistant malaria parasite lineage (PfPailin) to Vietnam. Lancet Infect. Dis. 2017, 17, 1022–1023. [Google Scholar] [CrossRef]
- Okombo, J.; Chibale, K. Recent updates in the discovery and development of novel antimalarial drug candidates. Medchemcomm 2018, 9, 437–453. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Taylor, T.E.; Kamchonwongpaisan, S. Artemisinin and the antimalarial endoperoxides: From herbal remedy to targeted chemotherapy. Microbiol. Rev. 1996, 60, 301–315. [Google Scholar] [CrossRef]
- Noedl, H.; Se, Y.; Schaecher, K.; Smith, B.L.; Socheat, D.; Fukuda, M.M. Evidence of artemisinin-resistant malaria in Western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. [Google Scholar] [CrossRef]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Ph, D.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; et al. Artemisinin Resistance in. Drug Ther. 2009, 361, 455–467. [Google Scholar] [CrossRef]
- Panosian, C.B. Economic Access to Effective Drugs for Falciparum Malaria. Clin. Infect. Dis. 2005, 40, 713–717. [Google Scholar] [CrossRef]
- Matada, B.S.; Pattanashettar, R.; Yernale, N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021, 32, 115973. [Google Scholar] [CrossRef]
- Mustapha, D.; Hajiba, O.; Khadija, O.; Abderrafia, H.; Mostafa, K. Recent development of quinoline derivatives and their potential biological activities. Curr. Org. Synth. 2021, 18, 248–269. [Google Scholar]
- Chu, X.; Wang, C.; Liu, W.; Liang, L.; Gong, K.; Zhao, C.; Sun, K. Quinoline and quinolone dimers and their biological activities: An overview. Eur. J. Med. Chem. 2019, 161, 101–117. [Google Scholar] [CrossRef]
- Loeb, F.; Clark, W.M.; Coatney, G.R.; Coggeshall, L.T.; Dieuaide, F.R.; Dochez, A.R.; Hakansson, E.G.; Marshall, E.K., Jr.; Marvel, C.S.; McCoy, O.R.; et al. Activity of a new antimalarial agent, chloroquine (SN 7618): Statement approved by the board for coordination of malarial studies. J. Am. Med. Assoc. 1946, 130, 1069–1070. [Google Scholar] [CrossRef]
- Winstanley, P.A.; Ward, S.A.; Snow, R.W. Clinical status and implications of antimalarial drug resistance. Microbes Infect. 2002, 4, 157–164. [Google Scholar] [CrossRef]
- Valluri, H.; Bhanot, A.; Shah, S.; Bhandaru, N.; Sundriyal, S. Basic Nitrogen (BaN) is a key property of antimalarial chemical space. J. Med. Chem. 2023, 66, 8382–8406. [Google Scholar] [CrossRef]
- Kaur, K.; Jain, M.; Reddy, R.P.; Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 2010, 45, 3245–3264. [Google Scholar] [CrossRef] [PubMed]
- Hussein, B.; Ikhmais, B.; Kadirvel, M.; Magwaza, R.N.; Halbert, G.; Bryce, R.A.; Stratford, I.J.; Freeman, S. Discovery of potent 4-aminoquinoline hydrazone inhibitors of NRH:quinoneoxidoreductase-2 (NQO2). Eur. J. Med. Chem. 2019, 182, 111649. [Google Scholar] [CrossRef] [PubMed]
- Fisher, N.; Bray, P.G.; Ward, S.A.; Biagini, G.A. The malaria parasite type II NADH: Quinone oxidoreductase: An alternative enzyme for an alternative lifestyle. Trends Parasitol. 2007, 23, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Chowdhary, S.; Legac, J.; Rosenthal, P.; Kumar, V. Quinoline-based heterocyclic hydrazones: Design, synthesis, ant-plasmodial assessment, and mechanistic insights. Chem. Biol. Drug Des. 2023, 101, 829–836. [Google Scholar] [CrossRef]
- Gemms, S.; Kukreja, G.; Fattorusso, C.; Persico, M.; Romano, M.P.; Altarelli, M.; Campiani, L.S.G.; Fattorusso, E.; Basilico, N.; Taramelli, D.; et al. Synthesis of N1-arylidene-N2-quinolyl- and N2-acrydinylhydrazones as potent antimalarial agents active against CQ-resistant P. falciparum strains. Bioorg. Med. Chem. Lett. 2006, 16, 5384–5388. [Google Scholar] [CrossRef]
- Kondaparla, S.; Agarwal, P.; Srivastava, K.; Puri, S.K.; Katti, S.B. Design, synthesis and in vitro antiplasmodial activity of some bisquinolines against chloroquine-resistant strain. Chem. Biol. Drug Des. 2017, 89, 901–906. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.-Y.; Liou, J.-P. Nitro-group containing drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef]
- Matthews, H.; Deakin, J.; Rajab, M.; Idris-Usman, M.; Nirmalan, N.J. Investigating antimalarial drug interactions of emetine dihydrochloride hydrate using CalcuSyn-based interactivity calculations. PLoS ONE 2017, 12, e0173303. [Google Scholar] [CrossRef]
- Panwar, P.; Burusco, K.K.; Abubaker, M.; Matthews, H.; Gutnov, A.; Fernández-Álvaro, E.; Bryce, R.A.; Wilkinson, J.; Nirmalan, N. Lead optimization of dehydroemetine for repositioned use in malaria. Antimicrob. Agents Chemother. 2020, 64, 1–22. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human Malaria Parasites in Continuous Culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Basco, L.K. Cultivation of asexual intraerythrocytic stages of Plasmodium falciparum. Pathogens 2023, 12, 900. [Google Scholar] [CrossRef] [PubMed]
- Le Manach, C.; Scheurer, C.; Sax, S.; Schleiferböck, S.; Cabrera, D.G.; Younis, Y.; Paquet, T.; Street, L.; Smith, P.; Ding, X.C.; et al. Fast in vitro methods to determine the speed of action and the stage-specificity of anti-malarials in Plasmodium falciparum. Malar. J. 2013, 12, 424. [Google Scholar] [CrossRef]
- Matthews, H.; Usman-idris, M.; Khan, F.; Read, M.; Nirmalan, N. Drug repositioning as a route to anti-malarial drug discovery: Preliminary investigation of the in vitro anti-malarial efficacy of emetine dihydrochloride hydrate. Malar. J. 2013, 12, 359. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Biamonte, M.A.; Wanner, J.; Le Roch, K.G. Recent advances in malaria drug discovery. Bioorganic Med. Chem. Lett. 2013, 23, 2829–2843. [Google Scholar] [CrossRef]
- Burrows, J.N.; Hooft Van Huijsduijnen, R.; Möhrle, J.J.; Oeuvray, C.; Wells, T.N. Designing the next generation of medicines for malaria control and eradication. Malar. J. 2013, 12, 187. [Google Scholar] [CrossRef]
- Fidock, D.A.; Rosenthal, P.J.; Croft, S.L.; Brun, R.; Nwaka, S. Antimalarial drug discovery: Efficacy models for compound screening. Nat. Rev. Drug Discov. 2004, 3, 509–520. [Google Scholar] [CrossRef]
- Eastman, R.T.; Fidock, D.A. Artemisinin-based combination therapies: A vital tool in efforts to eliminate malaria. Nat. Rev. Microbiol. 2009, 7, 864–874. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
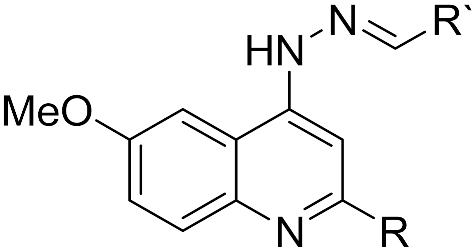 | |||||
|---|---|---|---|---|---|
| Plasmodium IC50(µM) ± SE | Cell Toxicity IC50(µM) ± SE | Cell Toxicity IC50(µM) ± SE | |||
| Compound | R | R’ | HepG2 | MDBK | |
| 1 | -Me | 4-Fluorophenyl | 0.612 ± 0.35 | 0.872 ± 0.79 | 3.77 ± 1.5 |
| 2 | -Me | Phenyl | 2.26 ± 2.2 | 1.90 ± 0.11 | 3.44 ± 2.7 |
| 3 | -Me | 3-Pyridinyl | 3.29 ± 1.6 | 3.00 ± 0.27 | 7.34 ± 7.6 |
| 4 | -Me | 2-Nitrofuranyl | 0.600 ± 0.84 | 1.59 ± 0.18 | 1.66 ± 0.21 |
| 5 | -Me | 2-Hydroxy-3-methoxyphenyl | 3.82 (wide) | 6.73 ± 0.73 | >25 |
| 6 | -Me | 4-Imidazoyl | 48.8 (wide) | 7.26 ± 1.00 | 11.7 ± 2.5 |
| 7 | -Me | 4-Nitrophenyl | 20.1 (wide) | 1.69 (wide) | 6.68 ± 1.7 |
| 8 | -Me | 4-Hydroxyphenyl | 22.3 (wide) | 1.97 ± 0.42 | 3.73 ± 1.2 |
| 9 | -Me | 4-Benzoic acid | >200 | >25 | >25 |
| 10 | -Me | 3-Hydroxyphenyl | 18.9 (wide) | 2.64 ± 0.20 | 9.71 ± 3.5 |
| 11 | -Me | 3,5-Dihydroxylphenyl | 35.9 ± 43 | 11.1 ± 2.9 | >25 |
| 12 | -Ph | 2-Hydroxy-3-methoxyphenyl | 5.67 ± 13 | 1.80 ± 0.78 | 6.60 |
| 13 | -Ph | Benzyl | 3.33 ± 2.4 | >25 | >25 |
| 14 | -Ph | 4-Nitrophenyl | 2.20 ± 1.5 | 0.89 ± 0.13 | 4.24 ± 1.0 |
| 15 | -Ph | 4-Imidazoyl | 28.3 ± 29 | 2.21 ± 0.15 | 9.31 ± 3.5 |
| 16 | -Ph | 4-N,N-dimethylaniline | 8.06 ± 4.5 | 4.15 ± 2.4 | 6.24 (wide) |
| Chloroquine | 0.199 ± 0.04 | ||||
| Cisplatin | 3.92 ± 0.64 | 9.15 ± 3.5 | |||
| Compound | Plasmodium IC50 (µM) ± SE | Selectivity Indices (MDBK) | Selectivity Indices (HepG2) |
|---|---|---|---|
| 1 | 0.0257 (wide) | 147 | 34 |
| 2 | 0.0329 ± 0.007 | 101 | 58 |
| 3 | 0.175 ± 0.04 | 42 | 17 |
| 4 | 0.219 ± 0.02 | 7 | 7 |
| 5 | 0.174 ± 0.03 | >143 | 39 |
| 12 | 0.133 ± 0.06 | 50 | 14 |
| 13 | 1.61 (wide) | >15 | >15 |
| 14 | 0.176 ± 0.06 | 31 | 5 |
| Compounds | 3D7 IC50 ± SE (µM) | Dd2 IC50 ± SE (µM) | K1 IC50 ± SE (µM) |
|---|---|---|---|
| 1 | 0.183 ± 1.04 | 0.133 ± 1.18 | 0.0257 (wide) |
| 2 | 0.0554 (wide) | 0.0244 (wide) | 0.0329 ± 0.007 |
| Drug Combination | CI Values at | ||||
|---|---|---|---|---|---|
| IC50 | IC75 | IC90 | m | r | |
| Atovaquone | N/A | N/A | N/A | 0.652 | 0.917 |
| Proguanil | N/A | N/A | N/A | 0.565 | 0.983 |
| Combination | 0.215 | 0.388 | 0.698 | 0.483 | 0.880 |
| Drug Combination | CI Values at | ||||
|---|---|---|---|---|---|
| IC50 | IC75 | IC90 | m | r | |
| 2 + Artemisinin | 0.822 | 1.907 | 4.563 | 0.263 | 0.869 |
| 2 + Artemether | 2.484 | 1.148 | 0.599 | 0.698 | 0.883 |
| 2 + Doxycycline | 1.285 | 1.112 | 0.963 | 0.467 | 0.973 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magwaza, R.N.; Abubaker, M.; Hussain, B.; Haley, M.; Couper, K.; Freeman, S.; Nirmalan, N.J. Evaluation of 4-Aminoquinoline Hydrazone Analogues as Potential Leads for Drug-Resistant Malaria. Molecules 2023, 28, 6471. https://doi.org/10.3390/molecules28186471
Magwaza RN, Abubaker M, Hussain B, Haley M, Couper K, Freeman S, Nirmalan NJ. Evaluation of 4-Aminoquinoline Hydrazone Analogues as Potential Leads for Drug-Resistant Malaria. Molecules. 2023; 28(18):6471. https://doi.org/10.3390/molecules28186471
Chicago/Turabian StyleMagwaza, Rachael N., Muna Abubaker, Buthaina Hussain, Michael Haley, Kevin Couper, Sally Freeman, and Niroshini J. Nirmalan. 2023. "Evaluation of 4-Aminoquinoline Hydrazone Analogues as Potential Leads for Drug-Resistant Malaria" Molecules 28, no. 18: 6471. https://doi.org/10.3390/molecules28186471
APA StyleMagwaza, R. N., Abubaker, M., Hussain, B., Haley, M., Couper, K., Freeman, S., & Nirmalan, N. J. (2023). Evaluation of 4-Aminoquinoline Hydrazone Analogues as Potential Leads for Drug-Resistant Malaria. Molecules, 28(18), 6471. https://doi.org/10.3390/molecules28186471