1. Introduction
Acid rain is a significant environmental concern that has been widely studied and discussed. It is caused by the release of sulfur dioxide (SO
2) and other acidic pollutants into the atmosphere, which react with water vapor to form sulfuric acid (H
2SO
4) and other acids [
1,
2,
3,
4,
5,
6,
7]. The natural pH of rain is close to 5.5. However, acid rain has a substantially lower pH, reaching 4.4. The low pH of this type of rain can cause structural problems in buildings and severe environmental damage, especially in primarily aquatic ecosystems [
8].
Among the main sources of sulfur dioxide are industrial processes of burning fossil fuels. Sulfur organic and inorganic compounds are present in fossil fuels, with almost 3% of sulfur by weight [
9]. However, sulfur supply comes from the desulfurization of fossil fuels of about 80%, which reduces the SO
2 emission [
10,
11,
12,
13]. Volcanoes, power plants, smelters, and the oil and gas industry are the primary sources of SO
2 [
14]. H
2S primary emission comes from organic matter in swamp areas [
15,
16]. Processes involving desulfurization reactions, such as the Claus reaction, are used to convert sulfur-based gases, such as H
2S and SO
2, into elemental sulfur. The reaction typically proceeds as follows:
where the first reaction consists of the oxidation of H
2S to SO
2 and the second reaction is a gaseous reaction between H
2S and SO
2 to form the expected product. However, the second reaction can be processed to form S
2O and H
2O so that two new side reactions can start to occur [
17]:
It is relevant to point out the greater facility presented by thiosulfurous acid to move to the H
2S∙∙∙SO
2 reagents, contrary to what would be expected to move to H
2O∙∙∙S
2O [
17].
In view of this problem, it becomes crucial to gain a comprehensive understanding of the interaction between H
2S and SO
2 as well as the associated energetics involved in forming the H
2S∙∙∙SO
2 complex. This understanding is particularly significant due to the influence of H
2S∙∙∙SO
2 in atmospheric chemistry and industrial contexts [
17].
Applying methods developed in theoretical chemistry enables a comprehensive analysis of the structure and energetics of molecular systems, relying on a fundamental understanding of intermolecular interactions [
18,
19,
20,
21,
22]. In the case of H
2S∙∙∙SO
2, there is evidence suggesting that the interactions occurring in this system are primarily associated with S∙∙∙S chalcogen–chalcogen interactions [
23,
24,
25,
26]. These interactions play a crucial role in determining the stability and behavior of the H
2S∙∙∙SO
2 complex, highlighting the significance of studying these specific intermolecular interactions at a molecular level.
Post–Hartree–Fock computational chemistry methods, such as second-order Møller–Plesset Perturbation Theory (MP2), can be employed to investigate dimers of H
2S∙∙∙H
2S, SO
2∙∙∙SO
2, and H
2S∙∙∙SO
2. These methods allow for determining interaction energies and the distribution of electronic density [
21,
27]. On the other hand, CCSD(T) (Coupled Cluster with Single and Double excitations and Triple excitations corrections) can be utilized to obtain data related to the transfer of electron density between atoms, as well as the energies involved in the process and the geometry of the system. Notably, a high degree of accuracy is observed in the calculated energies when compared with experimental data [
21,
23,
28]. An example is the recently reported results for H
2S∙∙∙H
2S, where CCSD(T) results agree with the geometry and vibrational frequencies [
21]. These computational methods provide valuable insights into the electronic structure, energetics, and geometries of H
2S∙∙∙H
2S, SO
2∙∙∙SO
2, and H
2S∙∙∙SO
2 dimers, aiding in the understanding of their properties and behavior.
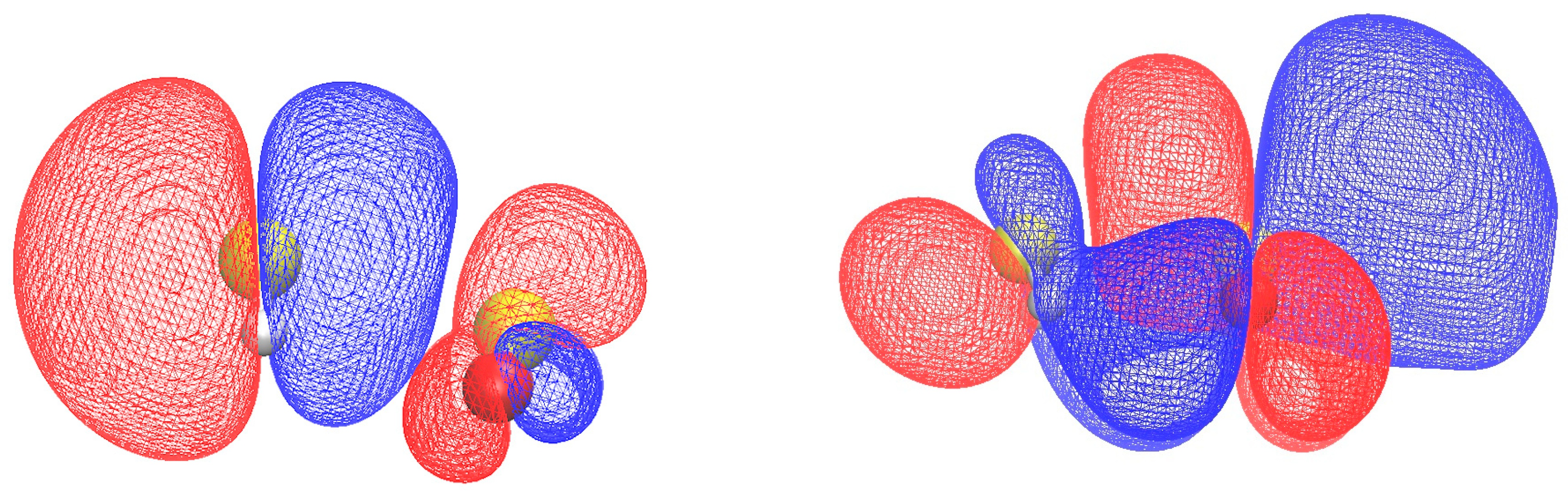
Another important aspect relates to the electronic properties of H
2S∙∙∙SO
2, particularly the energies of the frontier orbitals. These frontier orbitals play a significant role in ionization and electron attachment processes. By studying the energies of these orbitals, valuable information can be obtained regarding the reactivity and chemical behavior of the H
2S∙∙∙SO
2 system. Understanding the electronic properties and the energetics of the frontier orbitals provides insights into the potential for ionization or electron attachment events, which are relevant for various chemical and environmental processes involving H
2S∙∙∙SO
2. In this context, electron propagator theory (EPT) is reliable for obtaining accurate orbital energies. A detailed review on the methodology and applications of EPT was provided by Ortiz [
29].
The focus of the present study is to accurately determine the structure, interaction energy, and electronic properties of the H
2S∙∙∙SO
2 system. This aim is achieved by employing high-level ab initio methods, specifically CCSD(T) and EPT. Some emphasis was placed on the calculation of reactivity indexes such as the chemical potential, hardness, and electrophilicity, which are closely related to the ionization and electron attachment processes [
30].
3. Materials and Methods
The structures of H
2S, SO
2, and H
2S∙∙∙SO
2 were optimized using three different methods: MP2 (Møller–Plesset second-order perturbation theory) [
52,
53], CCSD (coupled cluster with single and double excitations) [
54,
55], and CCSD(T) (coupled cluster with single and double excitations, and correction to triple excitations) [
56].
The MP2 and CCSD calculations were performed using the Gaussian16 program package [
57]. On the other hand, the CCSD(T) calculation was carried out using the CFOUR program with analytical second derivatives [
58].
To perform these calculations, three different levels of basis sets were used: aug-cc-pVTZ (AVTZ), aug-cc-pVQZ (AVQZ), and aug-cc-pV5Z (AV5Z). These basis sets are known as the Dunning correlation-consistent basis sets [
59], and they provide increasingly accurate results including more basis functions from double-zeta to quintuple-zeta.
The use of monoelectronic basis sets in these calculations can lead to an overestimation of interaction energies due to the finite size of the basis sets and the different variational spaces of the complex (H
2S∙∙∙SO
2) and fragments (H
2S and SO
2). To address this issue, it is necessary to apply the Counterpoise (CP) correction method, which helps estimate the influence of the Basis Set Superposition Error (BSSE) [
60]. The nature of the interaction between the H
2S and SO
2 monomers in H
2S∙∙∙SO
2 was discussed by non-covalent interactions (NCI) analysis [
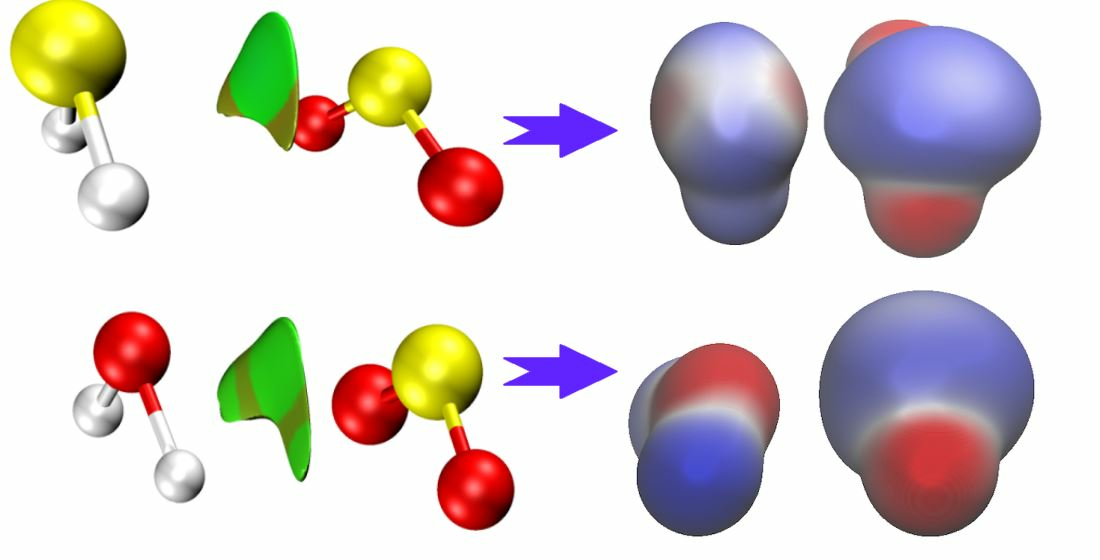
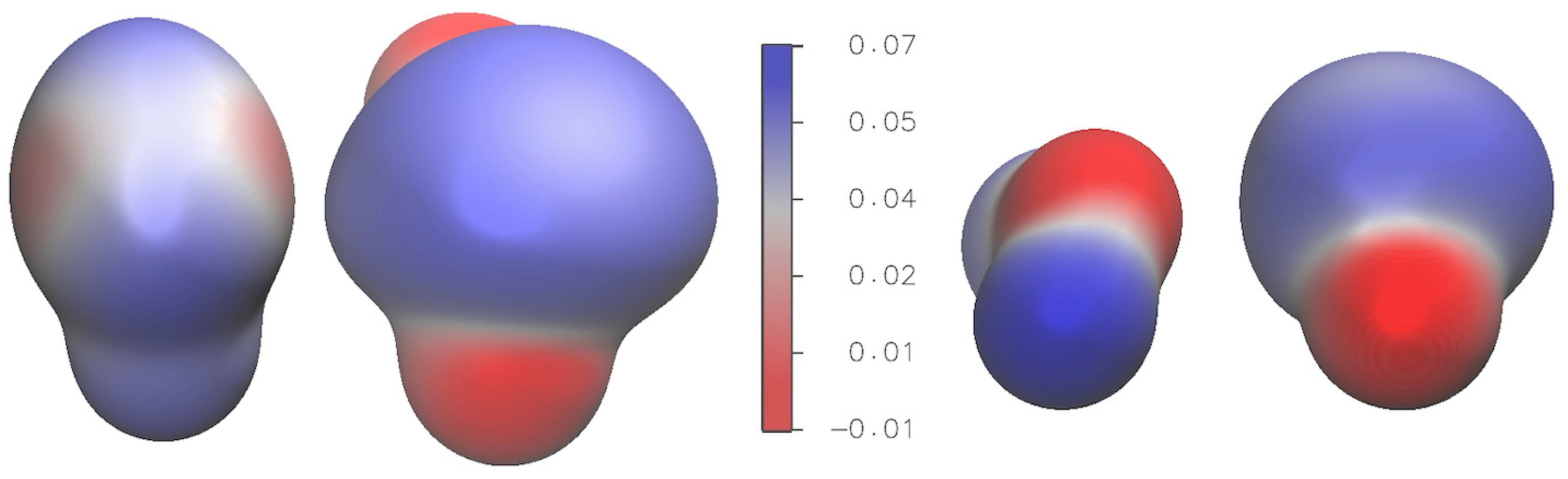
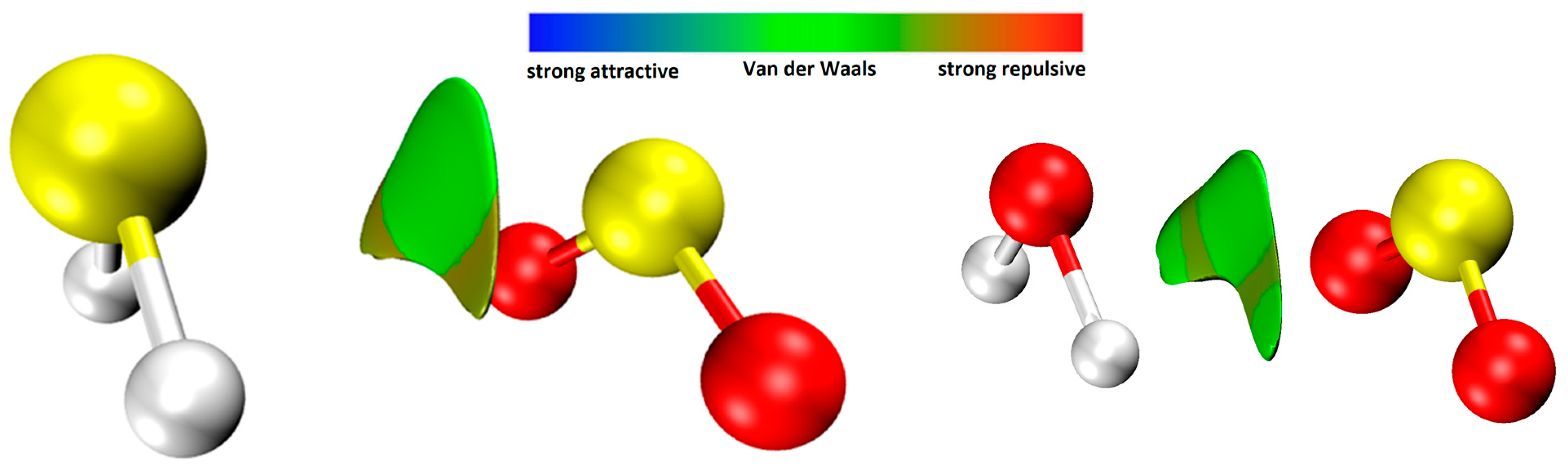
51].
Electron propagator theory (EPT) calculations were performed using the outer valence Green’s function (OVGF) and partial third-order (P3) approximations [
29]. EPT is known to provide accurate estimates of orbital energies. EPT calculations were carried out with the Gaussian16 program [
28].
The adiabatic ionization energies and electron affinities were predicted through ∆E calculations, where the energy difference between the neutral and charged species was calculated. Optimized geometries for the neutral, cationic, and ionic species were determined at the MP2/AVTZ level, and frequencies were calculated at the same level. Finally, energies were estimated at the CCSD/AVQZ//MP2/AVTZ level. Chemical reactivity indexes [
30] were calculated using ionization energies and electron affinities from ∆E(CCSD) calculations.
4. Conclusions
The present study employs high-level ab initio calculations to investigate the structure, vibrational frequencies, and electronic properties of the H2S∙∙∙SO2 complex. The analysis of vibrational frequencies reveals an intramolecular vibrational energy transfer phenomenon, where energy from the stretching modes of H2S is transferred to the ν1s mode of SO2. At the CCSD(T)/aug-cc-AVQZ level, the interaction energy between H2S and SO2 is predicted to be 2.78 kcal/mol. This provides insight into the strength of the interaction and the stability of the complex.
Electron propagator theory calculations yield a HOMO–LUMO gap of 8.24 eV for H2S∙∙∙SO2. This information sheds light on the electronic properties and the energy required for ionization and electron attachment processes. Furthermore, by utilizing ab initio results for the adiabatic ionization energy and electron affinity, the electrophilicity of H2S-SO2 is estimated to be 2.01 eV. This value is found to be similar to the electrophilicity of SO2, suggesting comparable reactivity and chemical behavior.
Overall, the combination of ab initio calculations and analysis provides valuable insights into the structure, vibrational dynamics, and electronic properties of the H2S∙∙∙SO2 complex, contributing to a deeper understanding of this system. The non-covalent interactions (NCI) analysis conducted on the H2S∙∙∙SO2 complex emphasizes the significant contribution of non-covalent van der Waals interactions in its energetic stabilization.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



