
Molecular Docking and Molecular Dynamics Studies Reveal the Anticancer Potential of Medicinal-Plant-Derived Lignans as MDM2-P53 Interaction Inhibitors

 , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
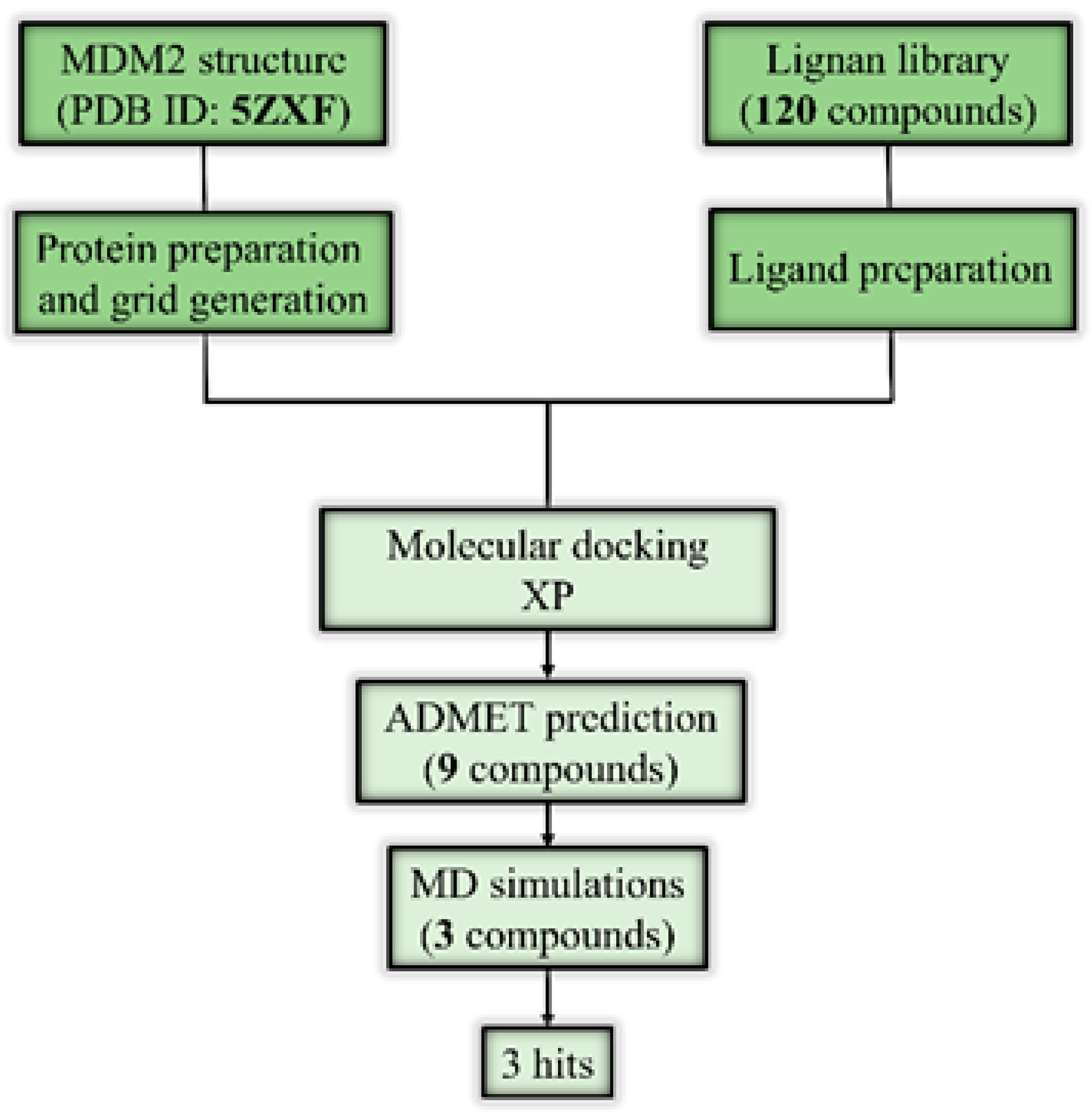
2. Results and Discussion
2.1. Molecular Docking and ADMET Profiling
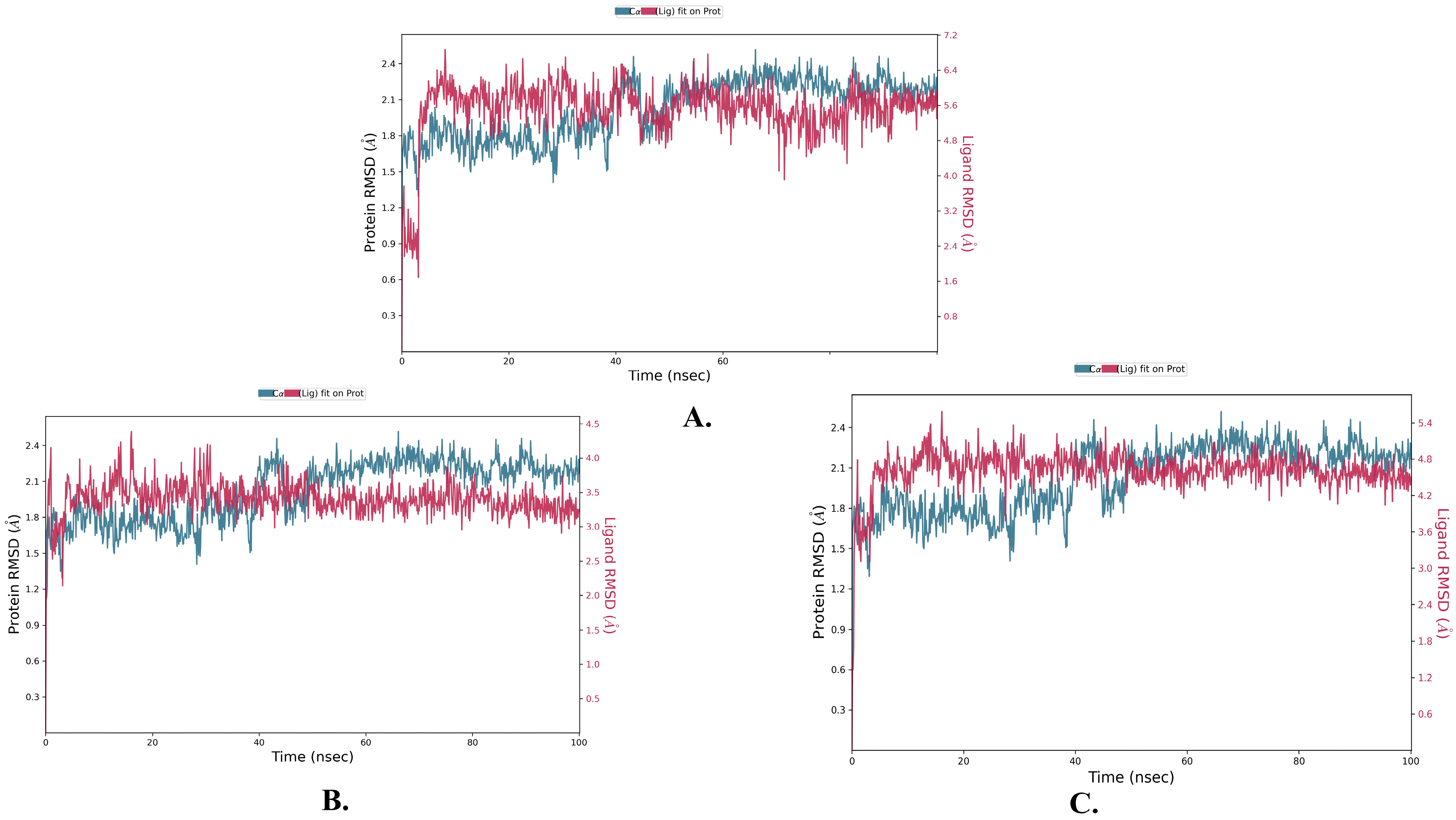
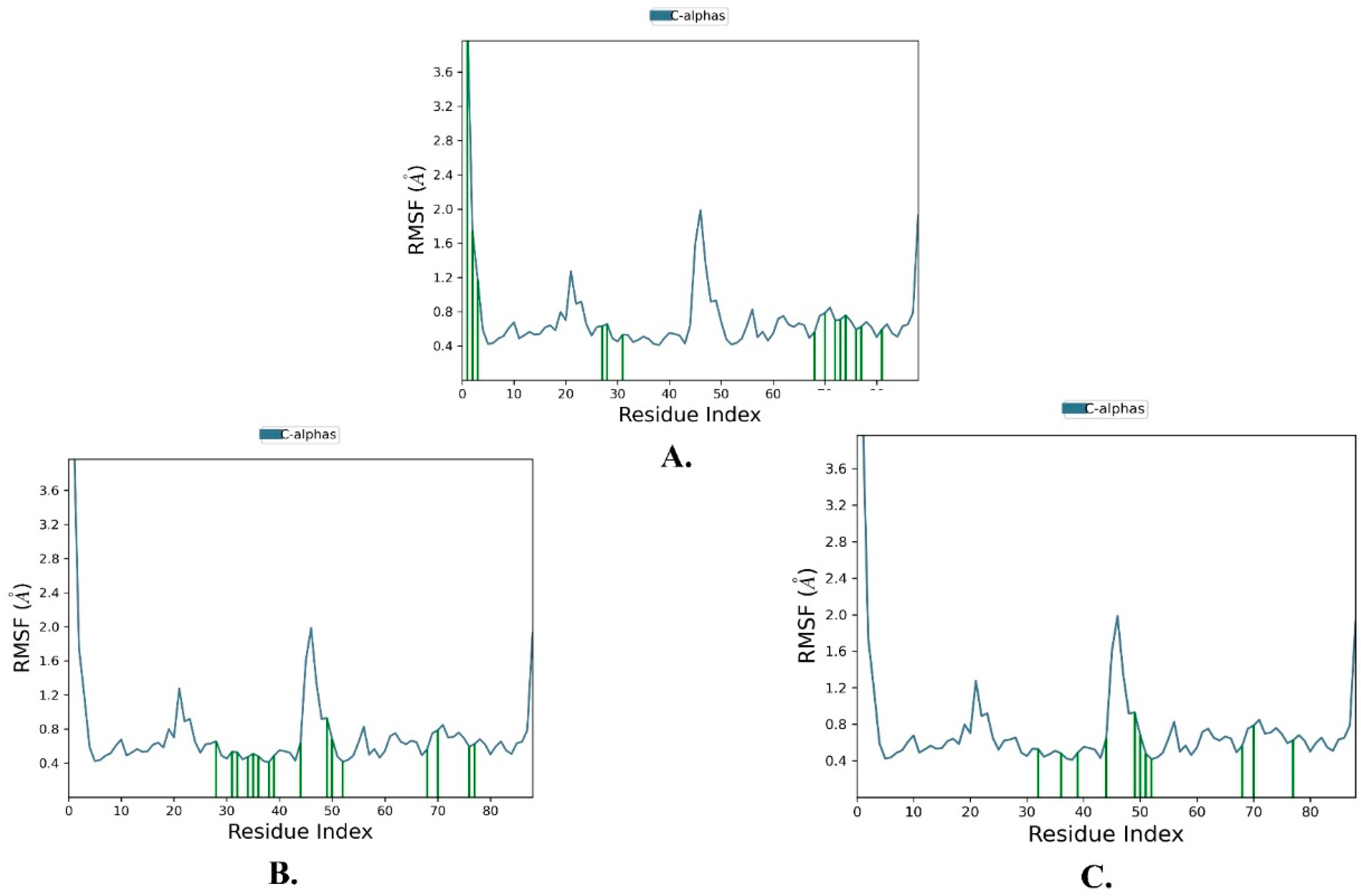
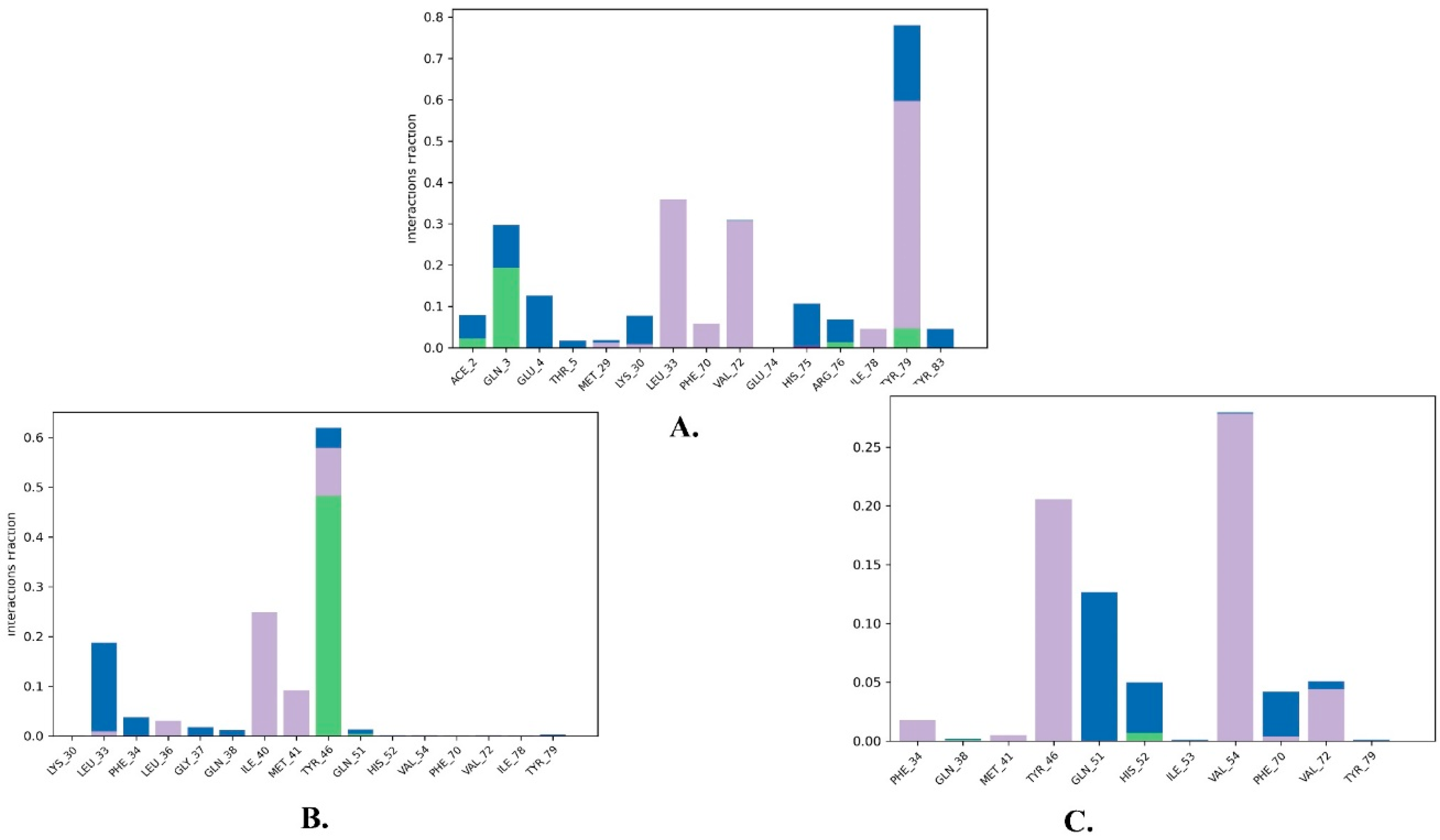
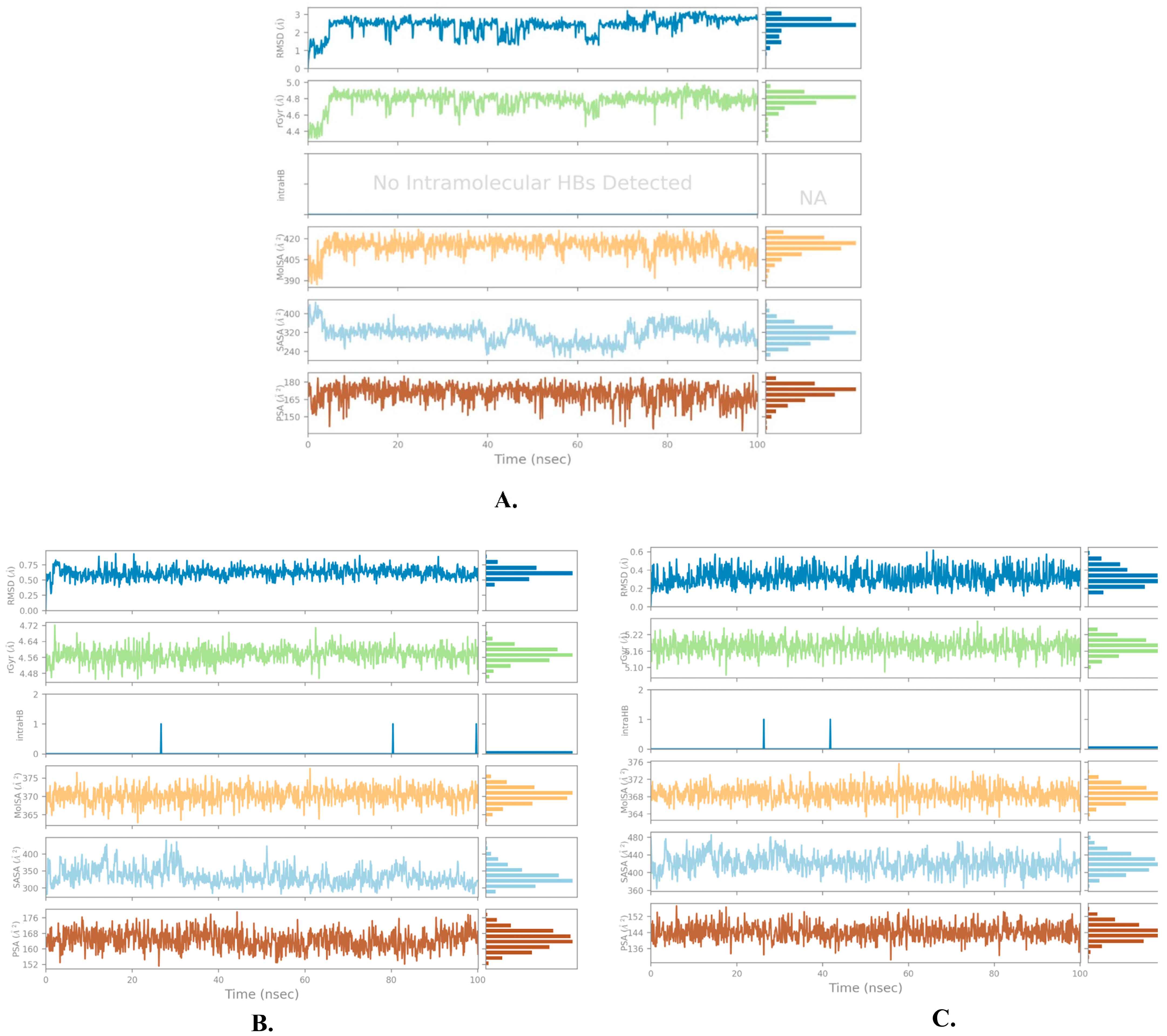
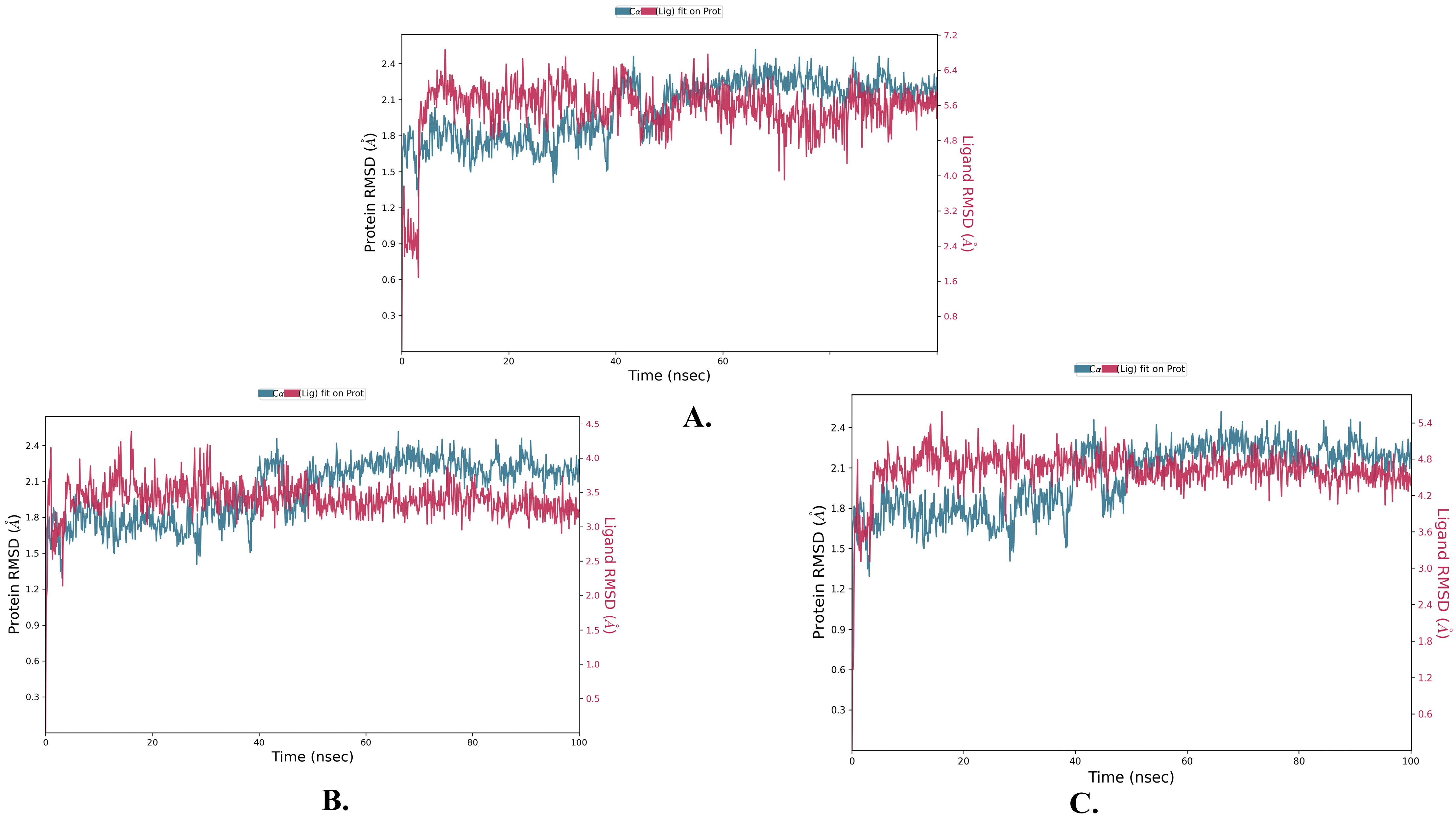
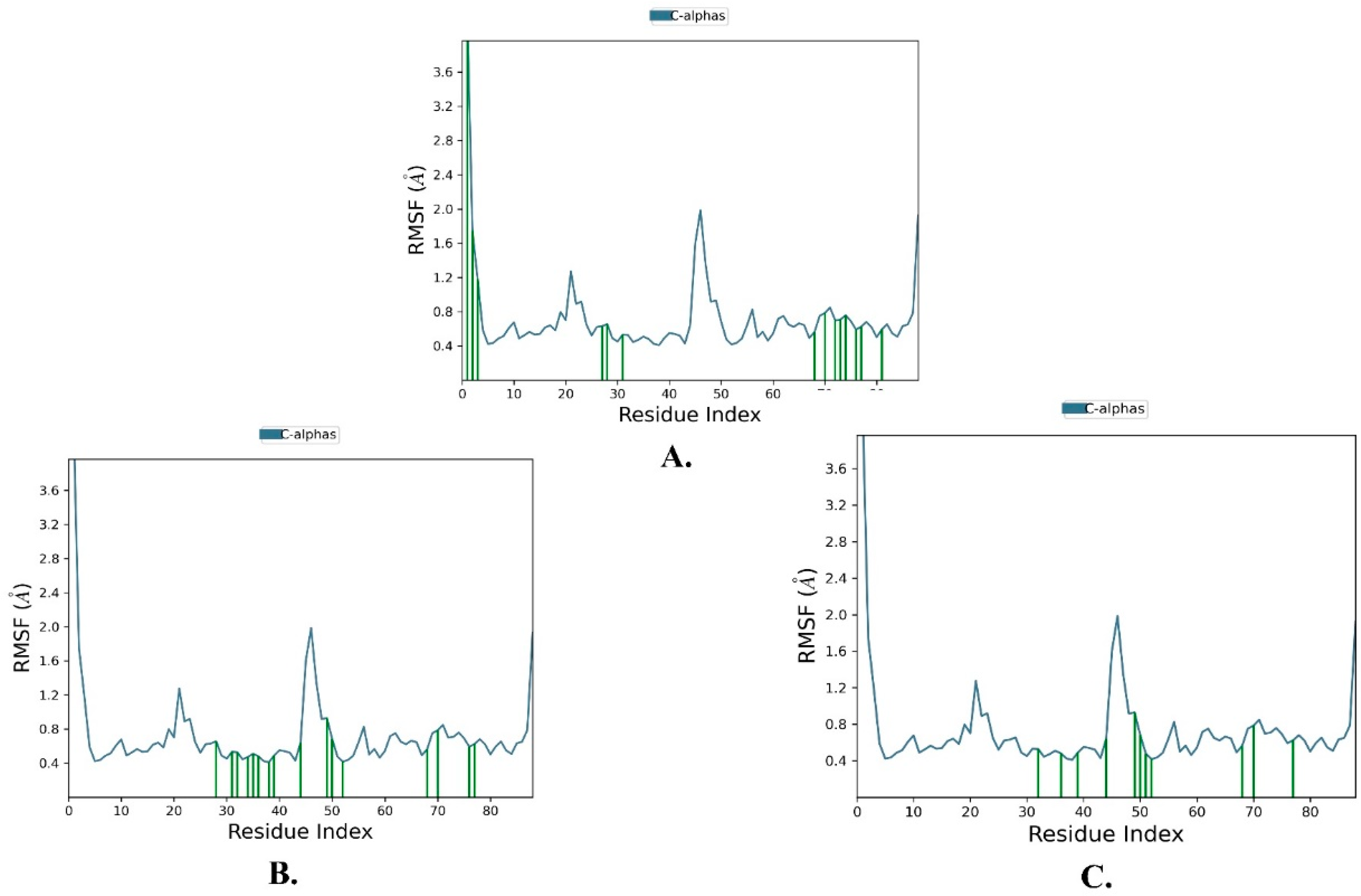
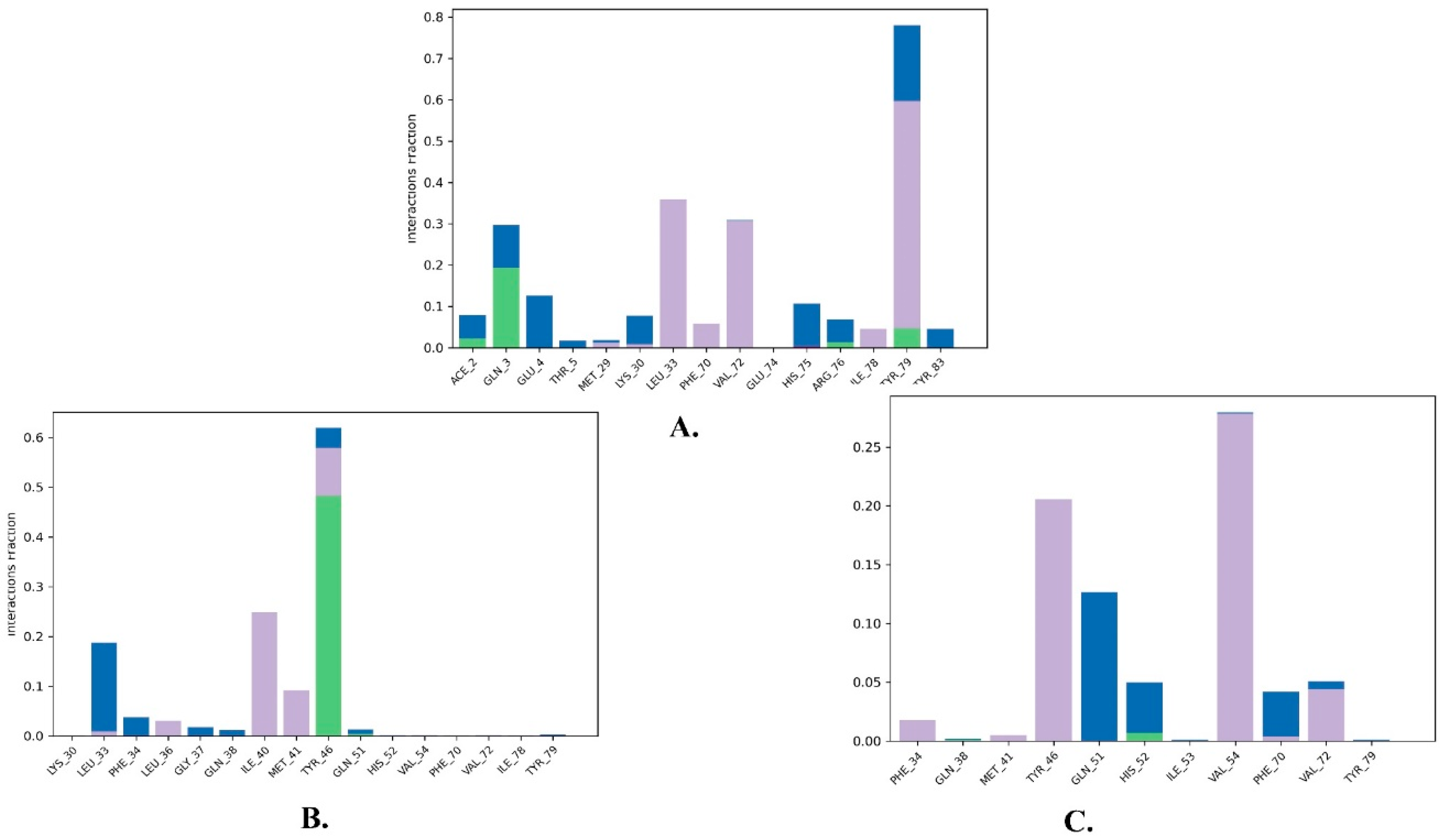
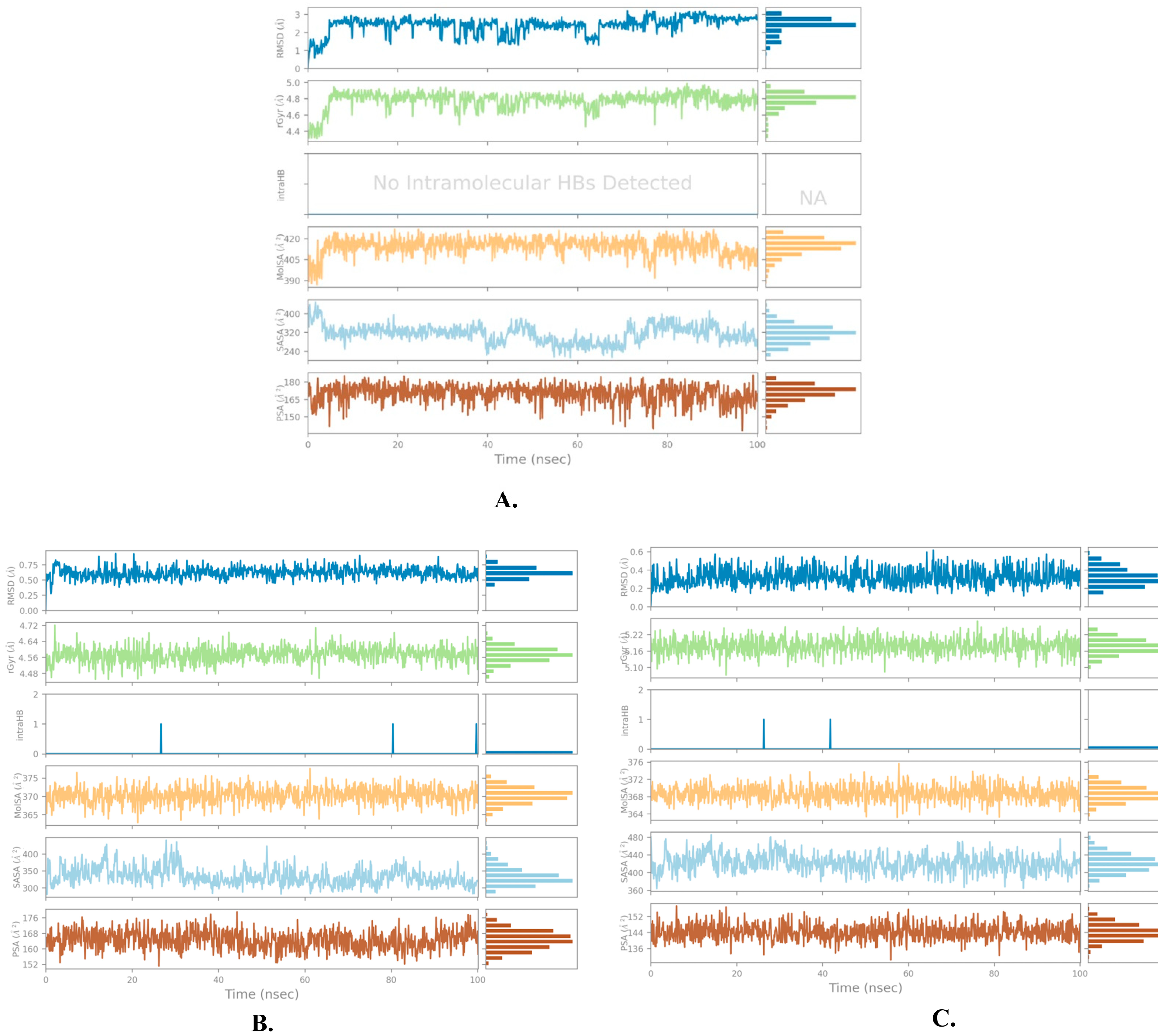
2.2. Molecular Dynamics Simulation
3. Materials and Methods
3.1. Protein and Ligand Preparation
3.2. Grid Generation and Molecular Docking
3.3. ADMET Prediction
3.4. MD Simulations and Post-MD MM-GBSA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Barakat, A.; Islam, M.S.; Ghawas, H.M.; Al-Majid, A.M.; El-Senduny, F.F.; Badria, F.A.; Elshaier, Y.A.; Ghabbour, H.A. Bioorganic Chemistry Design and synthesis of new substituted spirooxindoles as potential inhibitors of the MDM2–p53 interaction. Bioorg. Chem. 2019, 86, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53–MDM2 interaction by small-molecule inhibitors: Learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Matos, B.; Howl, J.; Jerónimo, C.; Fardilha, M. The disruption of protein-protein interactions as a therapeutic strategy for prostate cancer. Pharmacol. Res. 2020, 161, 105145. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Wynford-Thomas, D. Proliferative lifespan checkpoints: Cell-type specificity and influence on tumour biology. Eur. J. Cancer Part A 1997, 33, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Gonzalez, A.Z.; Li, Z.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Duquette, J.; Eksterowicz, J.; Fox, B.M.; Fu, J.; et al. Novel inhibitors of the MDM2-p53 interaction featuring hydrogen bond acceptors as carboxylic acid isosteres. J. Med. Chem. 2014, 57, 2963–2988. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-Associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Chène, P. Inhibiting the p53-MDM2 interaction: An important target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Bert, V.; David, L.; Arnold, L. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Lane, D.P. What the papers say: The p53-mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth control by p53? BioEssays 1993, 15, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Munisamy, M.; Mukherjee, N.; Thomas, L.; Pham, A.T.; Shakeri, A.; Zhao, Y.; Kolesar, J.; Rao, P.P.N.; Rangnekar, V.M.; Rao, M. Therapeutic opportunities in cancer therapy: Targeting the p53-MDM2/MDMX interactions. Am. J. Cancer Res. 2021, 11, 5762–5781. [Google Scholar] [PubMed]
- Reutershan, M.H.; MacHacek, M.R.; Altman, M.D.; Bogen, S.; Cai, M.; Cammarano, C.; Chen, D.; Christopher, M.; Cryan, J.; Daublain, P.; et al. Discovery of MK-4688: An Efficient Inhibitor of the HDM2-p53 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 16213–16241. [Google Scholar] [CrossRef]
- Wang, S.; Chen, F.E. Small-molecule MDM2 inhibitors in clinical trials for cancer therapy. Eur. J. Med. Chem. 2022, 236, 114334. [Google Scholar] [CrossRef]
- Gezici, S.; Şekeroğlu, N. Current Perspectives in the Application of Medicinal Plants Against Cancer: Novel Therapeutic Agents. Anti-Cancer Agents Med. Chem. 2019, 19, 101–111. [Google Scholar] [CrossRef]
- Majolo, F.; de Oliveira Becker Delwing, L.K.; Marmitt, D.J.; Bustamante-Filho, I.C.; Goettert, M.I. Medicinal plants and bioactive natural compounds for cancer treatment: Important advances for drug discovery. Phytochem. Lett. 2019, 31, 196–207. [Google Scholar] [CrossRef]
- Khazir, J.; Riley, D.L.; Pilcher, L.A.; De-Maayer, P.; Mir, B.A. Anticancer agents from diverse natural sources. Nat. Prod. Commun. 2014, 9, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Shaik, B.B.; Katari, N.K.; Jonnalagadda, S.B. Role of Natural Products in Developing Novel Anticancer Agents: A Perspective. Chem. Biodivers. 2022, 19, e202200535. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Pianjing, P.; Thiantanawat, A.; Rangkadilok, N.; Watcharasit, P.; Mahidol, C.; Satayavivad, J. Estrogenic activities of sesame lignans and their metabolites on human breast cancer cells. J. Agric. Food Chem. 2011, 59, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.Y.; Kim, M.Y.; Cho, J.Y. Antioxidant, Anti-Inflammatory, Anti-Menopausal, and Anti-Cancer Effects of Lignans and Their Metabolites. Int. J. Mol. Sci. 2022, 23, 15482. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, P.; Ozimek, L.; Kozłowska, J. The application of flax and hemp seeds in food, animal feed and cosmetics production. In Handbook of Natural Fibres; Woodhead Publishing Limited: Sawston, UK, 2012. [Google Scholar] [CrossRef]
- Mukherjee, P.K. High-Performance Liquid Chromatography for Analysis of Herbal Drugs; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780128133743. [Google Scholar] [CrossRef]
- Mukhija, M.; Joshi, B.C.; Bairy, P.S.; Bhargava, A.; Sah, A.N. Lignans: A versatile source of anticancer drugs. Beni-Suef. Univ. J. Basic Appl. Sci. 2022, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Teodor, E.D.; Moroeanu, V.; Radu, G.L. Lignans from Medicinal Plants and their Anticancer Effect. Mini Rev. Med. Chem. 2020, 20, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Khayat, M.T.; Alharbi, M.; Ghazawi, K.F.; Mohamed, G.A.; Ibrahim, S.R.M. Ferula sinkiangensis (Chou-AWei, Chinese Ferula): Traditional Uses, Phytoconstituents, Biosynthesis, and Pharmacological Activities. Plants 2023, 12, 902. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, S.G.A.; Abdallah, H.M.; Mohamed, G.A. Ethnomedicinal uses, phytochemistry, and pharmacological relevance of Justicia procumbens (Oriental Water Willow)—A promising traditional plant. J. Ethnopharmacol. 2023, 317, 116819. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, J.; Yan, H. Therapeutic Potential of Naturally Occurring Lignans as Anticancer Agents. Curr. Top. Med. Chem. 2022, 22, 1393–1405. [Google Scholar] [CrossRef]
- Ai, Z.; Wang, M.; Zhou, Y.; Yuan, D.; Jian, Q.; Wu, S.; Liu, B.; Yang, Y. Deciphering the pharmacological mechanisms of Rostellularia procumbens (L) Nees. Extract alleviates adriamycin-induced nephropathy in vivo and in vitro. Phytomedicine 2023, 113, 154736. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, T.; Xie, Z.-T.; Hong, Z.-C.; Lu, Y.; Long, Y.-M.; Ji, C.-Z.; Liu, Y.-T.; Yang, Y.-F.; Wu, H.-Z. Effective components and mechanism analysis of anti-platelet aggregation effect of Justicia procumbens L. J. Ethnopharmacol. 2022, 294, 115392. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Gong, Y. Chapter 20-Herbs That Counteract Toxins, Kill Parasites, and Relieve Itching. In Essentials of Chinese Materia Medica and Medical Formulas; Xi, S., Gong, Y.B.T.-E., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 401–410. ISBN 978-0-12-812722-3. [Google Scholar] [CrossRef]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef] [PubMed]
- Abd Emoniem, N.; Mukhtar, R.M.; Ghaboosh, H.; Elshamly, E.M.; Mohamed, M.A.; Elsaman, T.; Alzain, A.A. Turning down PI3K/AKT/mTOR signalling pathway by natural products: An in silico multi-target approach. SAR QSAR Environ. Res. 2023, 34, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Shoaib, T.H.; Ibraheem, W.; Abdelrahman, M.; Osman, W.; Sherif, A.E.; Ashour, A.; Ibrahim, S.R.M.; Ghazawi, K.F.; Miski, S.F.; Almadani, S.A.; et al. Exploring the potential of approved drugs for triple-negative breast cancer treatment by targeting casein kinase 2: Insights from computational studies. PLoS ONE 2023, 18, e0289887. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, R. Classification of current scoring functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Iancu-Rubin, C.; Mosoyan, G.; Glenn, K.; Gordon, R.E.; Nichols, G.L.; Hoffman, R. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Exp. Hematol. 2014, 42, 137–145.e5. [Google Scholar] [CrossRef] [PubMed]
- Higgins, B.; Glenn, K.; Walz, A.; Tovar, C.; Filipovic, Z.; Hussain, S.; Lee, E.; Kolinsky, K.; Tannu, S.; Adames, V.; et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin. Cancer Res. 2014, 20, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Takaba, H.; Del Carpio, C.A.; Miyamoto, A. How Nutlin-3 disrupts the MDM2-p53 interaction: A theoretical investigation. Med. Chem. Res. 2014, 23, 1998–2006. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Rutz, A.; Sorokina, M.; Galgonek, J.; Mietchen, D.; Willighagen, E.; Gaudry, A.; Graham, J.G.; Stephan, R.; Page, R.; Vondrášek, J.; et al. The LOTUS initiative for open knowledge management in natural products research. Elife 2022, 11, e70780. [Google Scholar] [CrossRef]
- Qiu, F.; Zhou, S.; Fu, S.; Kong, W.; Yang, S.; Yang, M. LC-ESI-MS/MS analysis and pharmacokinetics of 6′-hydroxy justicidin A, a potential antitumor active component isolated from Justicia procumbens, in rats. J. Pharm. Biomed. Anal. 2012, 70, 539–543. [Google Scholar] [CrossRef]
- He, X.L.; Zhang, P.; Dong, X.Z.; Yang, M.H.; Chen, S.L.; Bi, M.G. JR6, a new compound isolated from Justicia procumbens, induces apoptosis in human bladder cancer EJ cells through caspase-dependent pathway. J. Ethnopharmacol. 2012, 144, 284–292. [Google Scholar] [CrossRef]
- Pharmaceutical Composition for Prevention or Treatment of Diseases of Coronavirus Infection-Patent KR-20220051628-A-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/patent/KR-20220051628-A (accessed on 17 June 2023).
- Novel Dialkoxynaphtho[2,3-c]furan-1(3H)-one Derivatives and Pharmaceutical Composition for Preventing or Treating Respiratory Disease or SARS-CoV-2 Infection Disease Comprising the Same-Patent KR-20220136932-A-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/patent/KR-20220136932-A (accessed on 17 June 2023).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Schrödinger is the Scientific Leader in Developing State-of-The-Art Chemical Simulation Software for Use in Pharmaceutical, Biotechnology, and Materials Research. 2021. Available online: https://www.schrodinger.com/ (accessed on 7 March 2022).
- Schrödinger, Schrödinger Release 2018-1. 2021. Available online: https://www.schrodinger.com/products/ligprep (accessed on 7 March 2022).
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Schrödinger Release; 2021-3: MacroModel, Schrödinger, LLC: New York, NY, USA, 2021.
- Schrödinger Release; 2021-3: Receptor Grid Generation, Schrödinger, LLC: New York, NY, USA, 2021.
- Eltaib, L.; Alzain, A.A. Targeting the omicron variant of SARS-CoV-2 with phytochemicals from Saudi medicinal plants: Molecular docking combined with molecular dynamics investigations. J. Biomol. Struct. Dyn. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alzain, A.A.; Elbadwi, F.A. Identification of novel TMPRSS2 inhibitors for COVID-19 using e-pharmacophore modelling, molecular docking, molecular dynamics and quantum mechanics studies. Inform. Med. Unlocked 2021, 26, 100758. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release; 2021-3: QikProp, Schrödinger, LLC: New York, NY, USA, 2021.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Obubeid, F.O.; Eltigani, M.M.; Mukhtar, R.M.; Ibrahim, R.A.; Alzain, M.A.; Elbadawi, F.A.; Ghaboosh, H.; Alzain, A.A. Dual targeting inhibitors for HIV-1 capsid and cyclophilin A: Molecular docking, molecular dynamics, and quantum mechanics. Mol. Simul. 2022, 48, 1476–1489. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
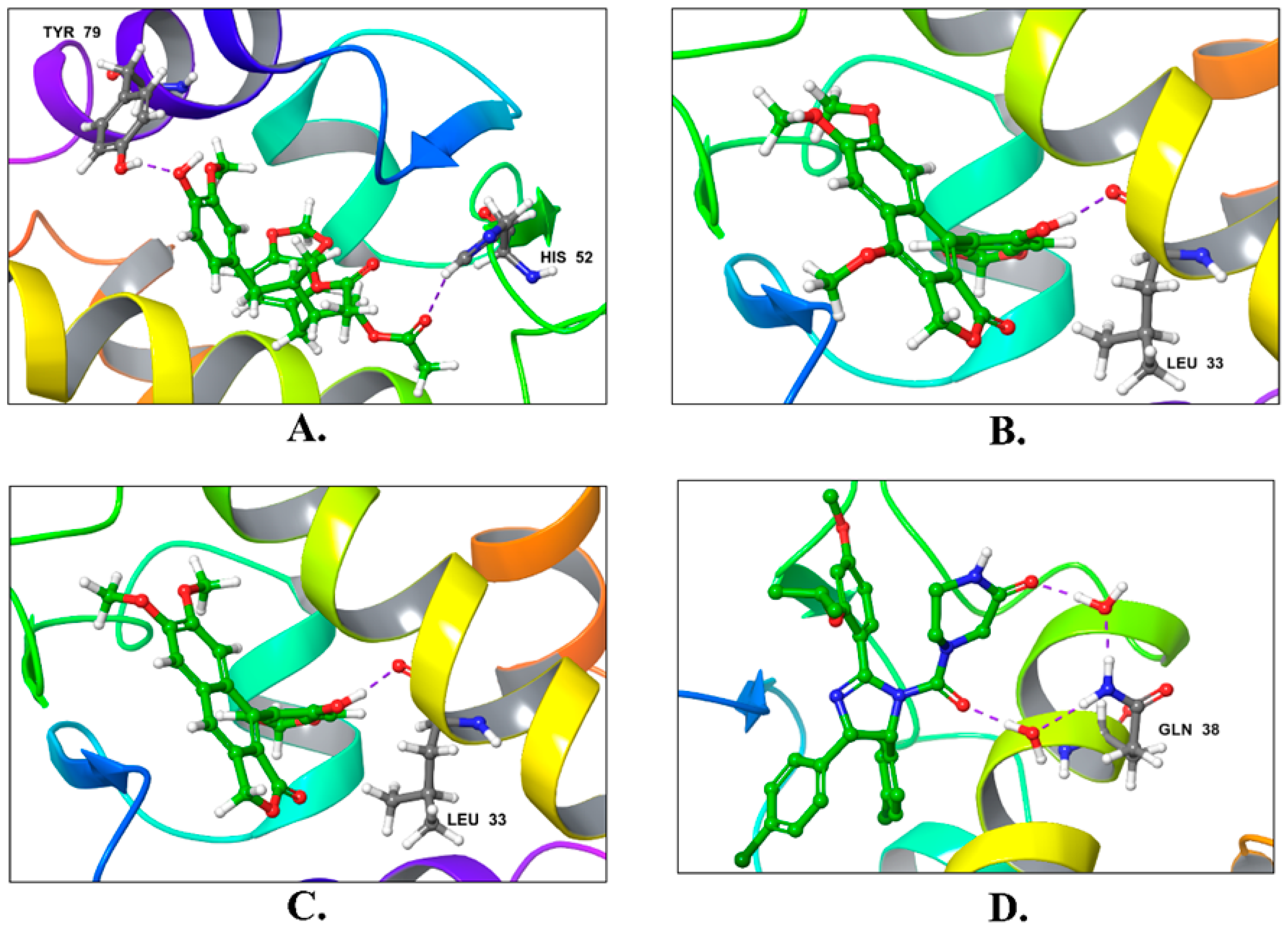
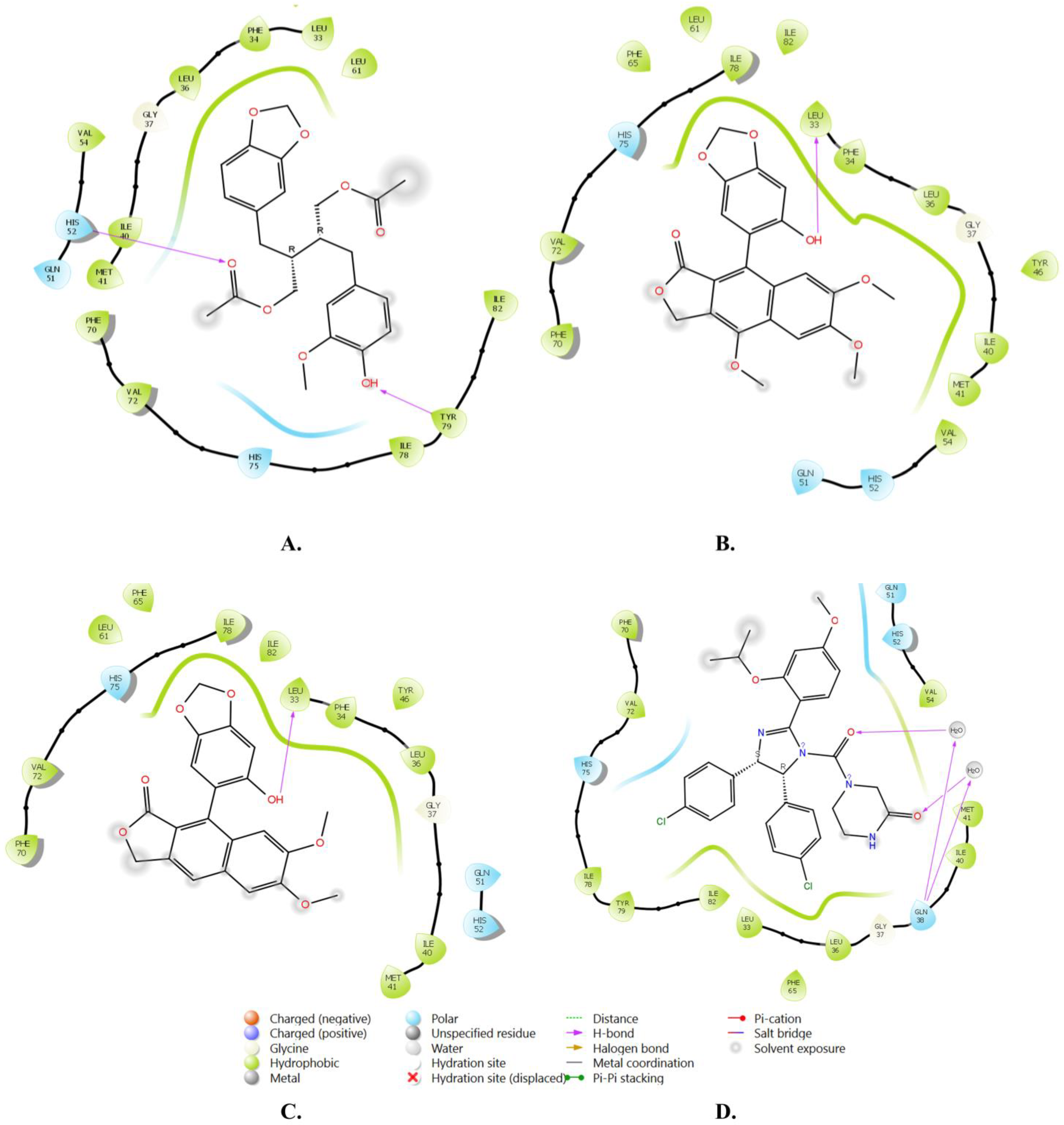
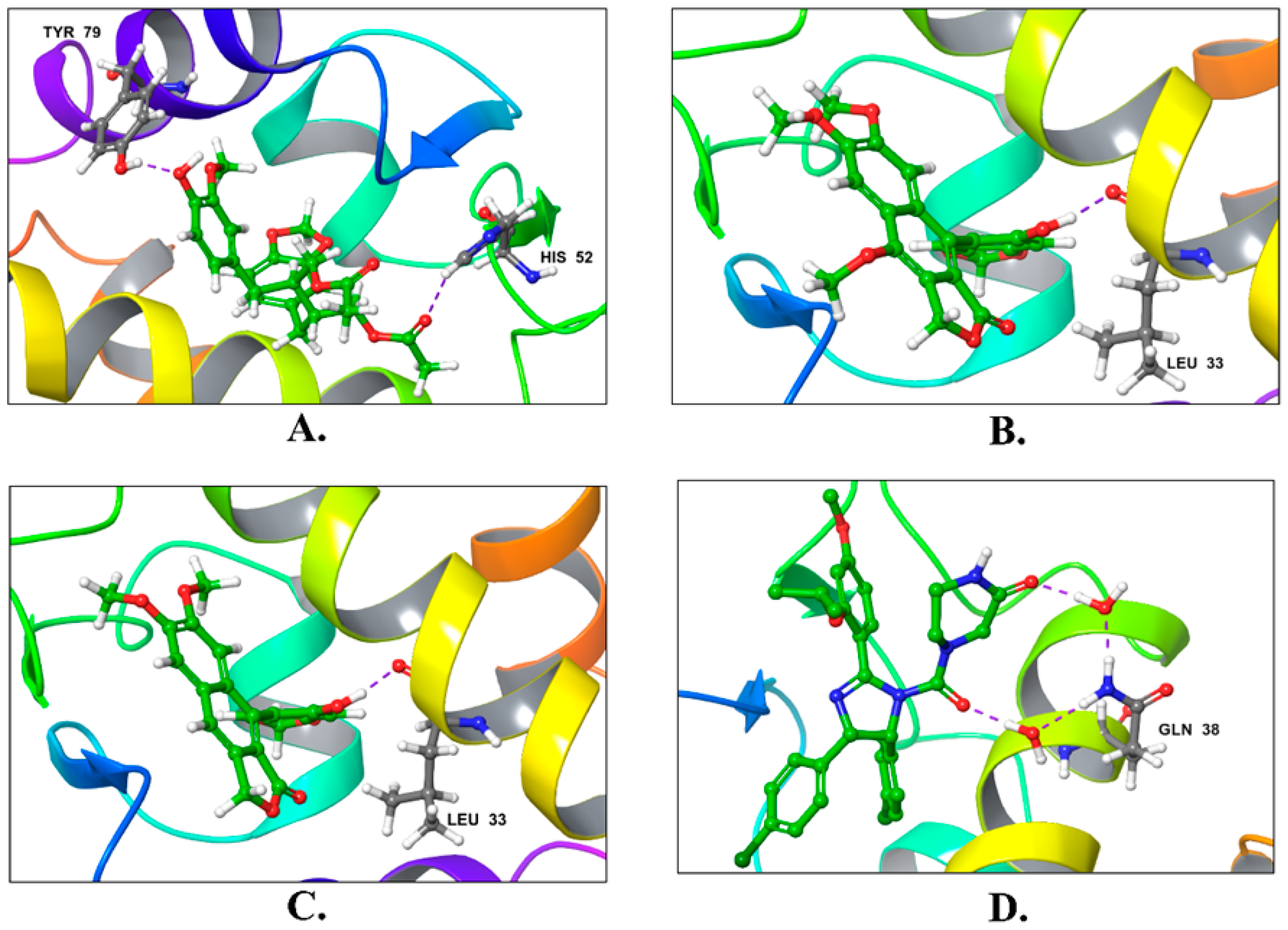
| Compound | Title | Docking Score Kcal/mol | H-Bonding | Hydrophobic Interactions |
|---|---|---|---|---|
| 1 | Justin A | −7.526 | HIS-52 (2.73 Å) TYR-79 (1.97 Å) | LEU-33, ILE-40, MET-41, PHE-65, PHE-70, VAL-72, ILE-78, TYR-79 |
| 2 | 6′-Hydroxy justicidin A | −7.438 | LEU-33 (1.87 Å) | LEU-33, LEU-36, ILE-40, MET-41, PHE-65, PHE-70, VAL-72, ILE-78 |
| 3 | 6′-Hydroxy justicidin B | −7.240 | LEU-33 (1.94 Å) | LEU-33, LEU-36, ILE-40, MET-41, PHE-65, PHE-70, VAL-72, ILE-78 |
| 4 | Lariciresinol | −7.067 | Bridged H-bond with PHE-34 GLN-51 (1.88 Å) HIS-52 (2.44 Å) TYR-79 (2.36 Å) | LEU-33, VAL-72, TYR-79 |
| 5 | Procumbiene | −7.027 | Bridged H-bond with GLN-38 | LEU-33, ILE-40, PHE-65, PHE-70, VAL-72, ILE-78, TYR-79 |
| 6 | Diphyllin | −6.985 | - | LEU-33, LEU-36, ILE-40, MET-41, PHE-65, PHE-70, VAL-72, ILE-78 |
| 7 | 6′-Hydroxy justicidin C | −6.966 | LEU-33 (1.90 Å) | LEU-33, LEU-36, ILE-40, MET-41, PHE-65, PHE-70, VAL-72, ILE-78 |
| 8 | (+)-Sinkianlignan E | −6.877 | TYR-79 (2 Å) GLN-51 (1.69 Å) | LEU-33, ILE-40, PHE-65, PHE-70, VAL-72, ILE-78, TYR-79 |
| 9 | Pinoresinol | −6.831 | TYR-79 (2.18 Å) GLN-51 (1.87 Å) HIS-52 (2.5, 2.8 Å) | LEU-33, ILE-40, VAL-72, ILE-78, TYR-79 |
| Reference | Nutlin-3a | −6.830 | 2 bridged H-bonds with GLN-38 | LEU-33, LEU-36, ILE-40, MET-41, PHE-65, VAL-72, ILE-78, TYR-79 |
| Compound | Donor HB a | Accpt HB b | QPlog Po/w c | QPlog S d | QPlog HERG e | QPP Caco f | QPlog BB g | Mwt h | % HOR i | ROF j |
|---|---|---|---|---|---|---|---|---|---|---|
| Justin A | 1 | 7 | 4.074 | −4.924 | −5.086 | 380.953 | −1.546 | 444.480 | 96.9 | 0 |
| 6′−Hydroxy justicidin A | 1 | 7 | 2.617 | −3.414 | −4.082 | 1497.331 | −0.481 | 410.379 | 100 | 0 |
| 6′−Hydroxy justicidin B | 1 | 7 | 2.561 | −3.384 | −4.160 | 1471.994 | −0.417 | 380.353 | 100 | 0 |
| Lariciresinol | 3 | 6 | 2.651 | −3.815 | −4.919 | 528.187 | −1.169 | 360.406 | 91 | 0 |
| Procumbiene | 1 | 8 | 1.693 | −2.004 | −3.387 | 1121.839 | −0.494 | 368.342 | 91 | 0 |
| Diphyllin | 1 | 7 | 2.528 | −3.383 | −4.122 | 1332.149 | −0.455 | 380.353 | 100 | 0 |
| 6′−Hydroxy justicidin C | 1 | 7 | 2.532 | −3.339 | −4.005 | 1275.549 | −0.537 | 410.379 | 100 | 0 |
| (+)−Sinkianlignan E | 2 | 7 | 3.791 | −4.375 | −5.514 | 984.675 | −1.139 | 358.433 | 100 | 0 |
| Pinoresinol | 2 | 6 | 2.849 | −4.393 | −4.917 | 969.614 | −0.669 | 358.390 | 100 | 0 |
| Nutlin−3a (reference) | 1 | 7 | 5.098 | −5.968 | −2.904 | 296.106 | −0.288 | 581.497 | 75 | 2 |
| Standard values | ≤5 | ≤10 | −2.0–6.5 | −6.5–0.5 | Below −5 | >25 poor <500 great | −3–1.2 | >500 | >25% poor <80% great | 0–4 |
| Name | RMSD | RMSF of Cα | |
|---|---|---|---|
| Cα | Ligand with Protein | ||
| Justin A | 2.025 ± 0.252 | 2.066 ± 0.256 | 0.721 ± 0.489 |
| 6′-Hydroxy justicidin A | 2.025 ± 0.252 | 2.042 ± 0.154 | 0.721 ± 0.489 |
| 6′-Hydroxy justicidin B | 2.025 ± 0.252 | 2.306 ± 0.178 | 0.721 ± 0.489 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoaib, T.H.; Abdelmoniem, N.; Mukhtar, R.M.; Alqhtani, A.T.; Alalawi, A.L.; Alawaji, R.; Althubyani, M.S.; Mohamed, S.G.A.; Mohamed, G.A.; Ibrahim, S.R.M.; et al. Molecular Docking and Molecular Dynamics Studies Reveal the Anticancer Potential of Medicinal-Plant-Derived Lignans as MDM2-P53 Interaction Inhibitors. Molecules 2023, 28, 6665. https://doi.org/10.3390/molecules28186665
Shoaib TH, Abdelmoniem N, Mukhtar RM, Alqhtani AT, Alalawi AL, Alawaji R, Althubyani MS, Mohamed SGA, Mohamed GA, Ibrahim SRM, et al. Molecular Docking and Molecular Dynamics Studies Reveal the Anticancer Potential of Medicinal-Plant-Derived Lignans as MDM2-P53 Interaction Inhibitors. Molecules. 2023; 28(18):6665. https://doi.org/10.3390/molecules28186665
Chicago/Turabian StyleShoaib, Tagyedeen H., Nihal Abdelmoniem, Rua M. Mukhtar, Amal Th. Alqhtani, Abdullah L. Alalawi, Razan Alawaji, Mashael S. Althubyani, Shaimaa G. A. Mohamed, Gamal A. Mohamed, Sabrin R. M. Ibrahim, and et al. 2023. "Molecular Docking and Molecular Dynamics Studies Reveal the Anticancer Potential of Medicinal-Plant-Derived Lignans as MDM2-P53 Interaction Inhibitors" Molecules 28, no. 18: 6665. https://doi.org/10.3390/molecules28186665
APA StyleShoaib, T. H., Abdelmoniem, N., Mukhtar, R. M., Alqhtani, A. T., Alalawi, A. L., Alawaji, R., Althubyani, M. S., Mohamed, S. G. A., Mohamed, G. A., Ibrahim, S. R. M., Hussein, H. G. A., & Alzain, A. A. (2023). Molecular Docking and Molecular Dynamics Studies Reveal the Anticancer Potential of Medicinal-Plant-Derived Lignans as MDM2-P53 Interaction Inhibitors. Molecules, 28(18), 6665. https://doi.org/10.3390/molecules28186665