
Pharmacoinformatics and Breed-Based De Novo Hybridization Studies to Develop New Neuraminidase Inhibitors as Potential Anti-Influenza Agents

 ,
,  , ,
, ,  and
and 
Abstract
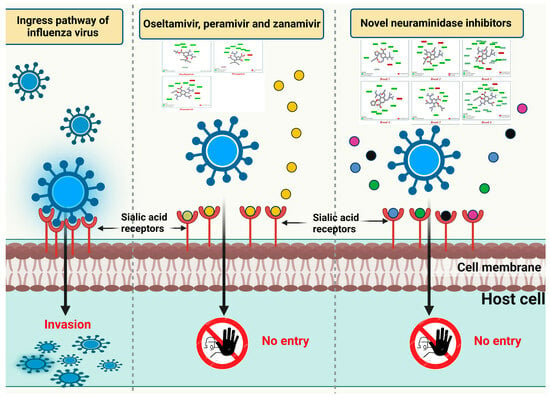
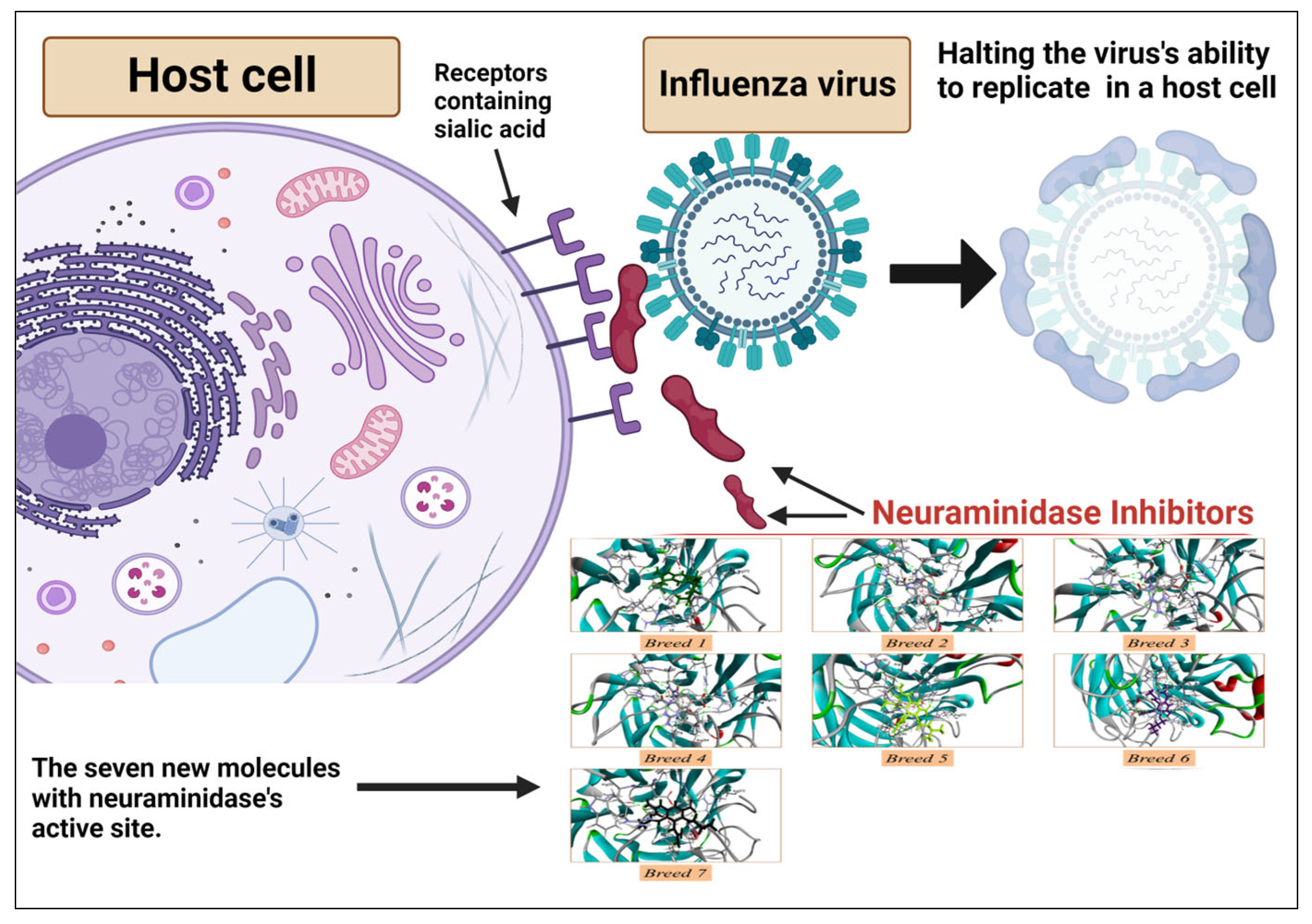
:
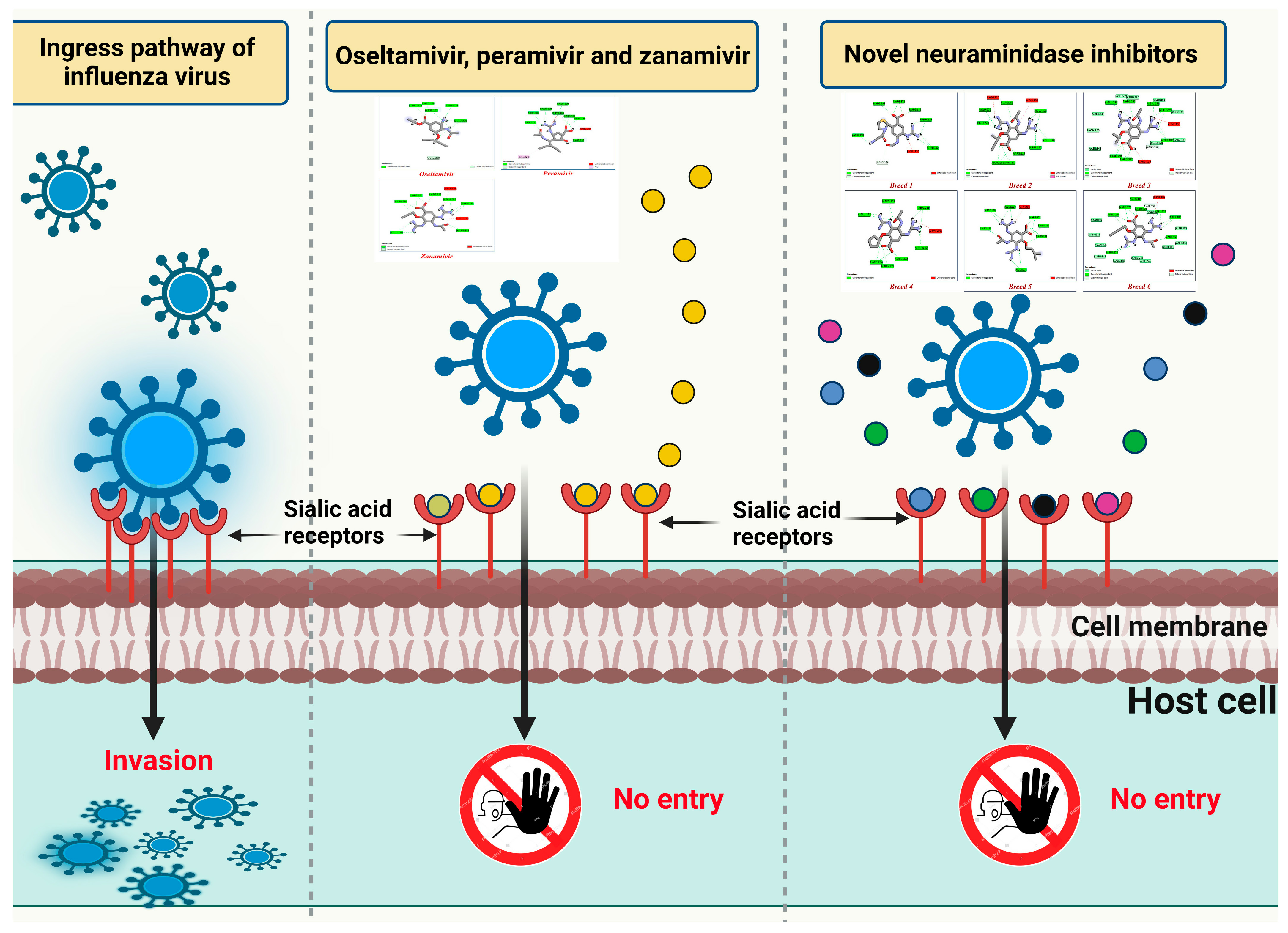
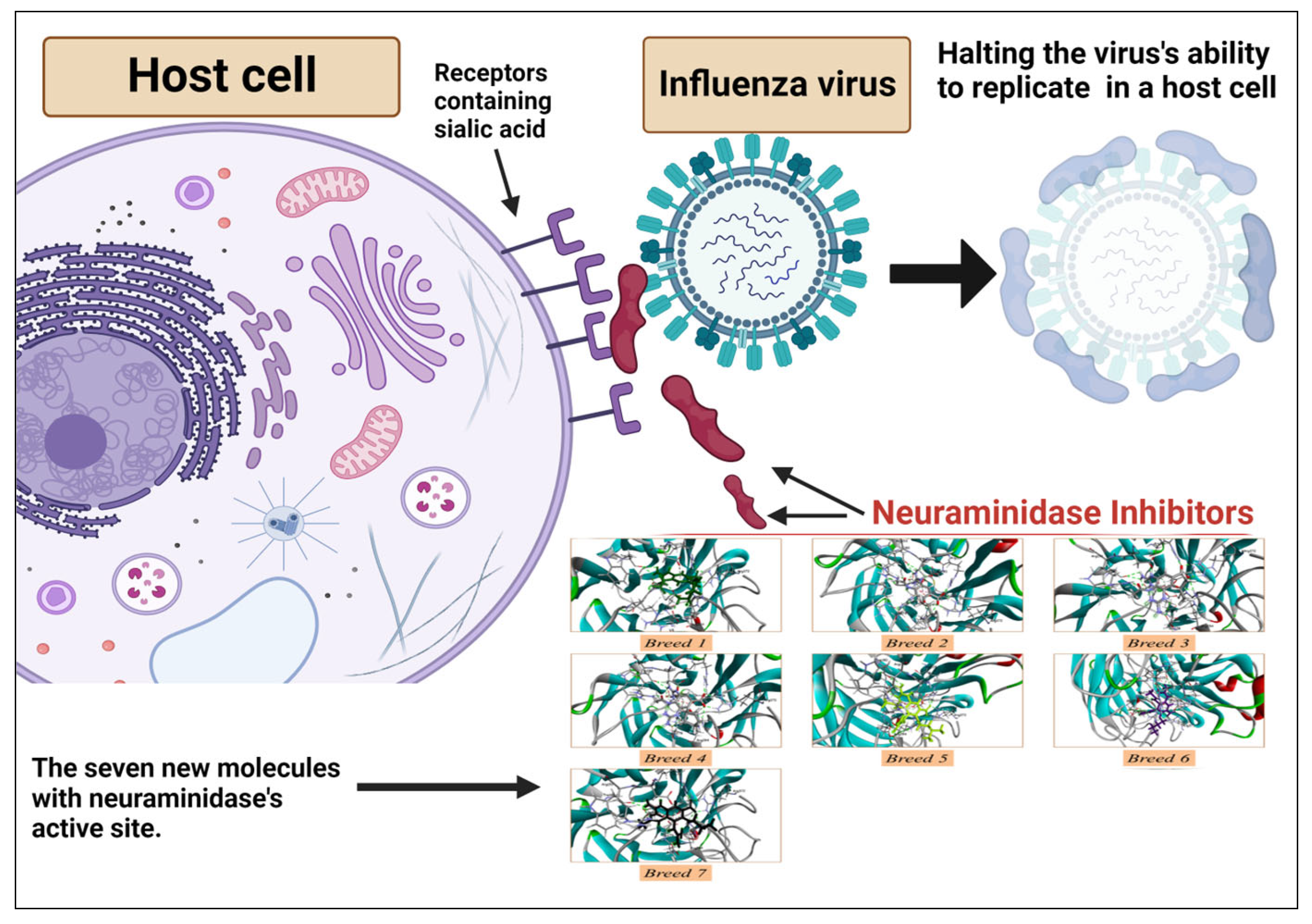
1. Introduction
2. Results
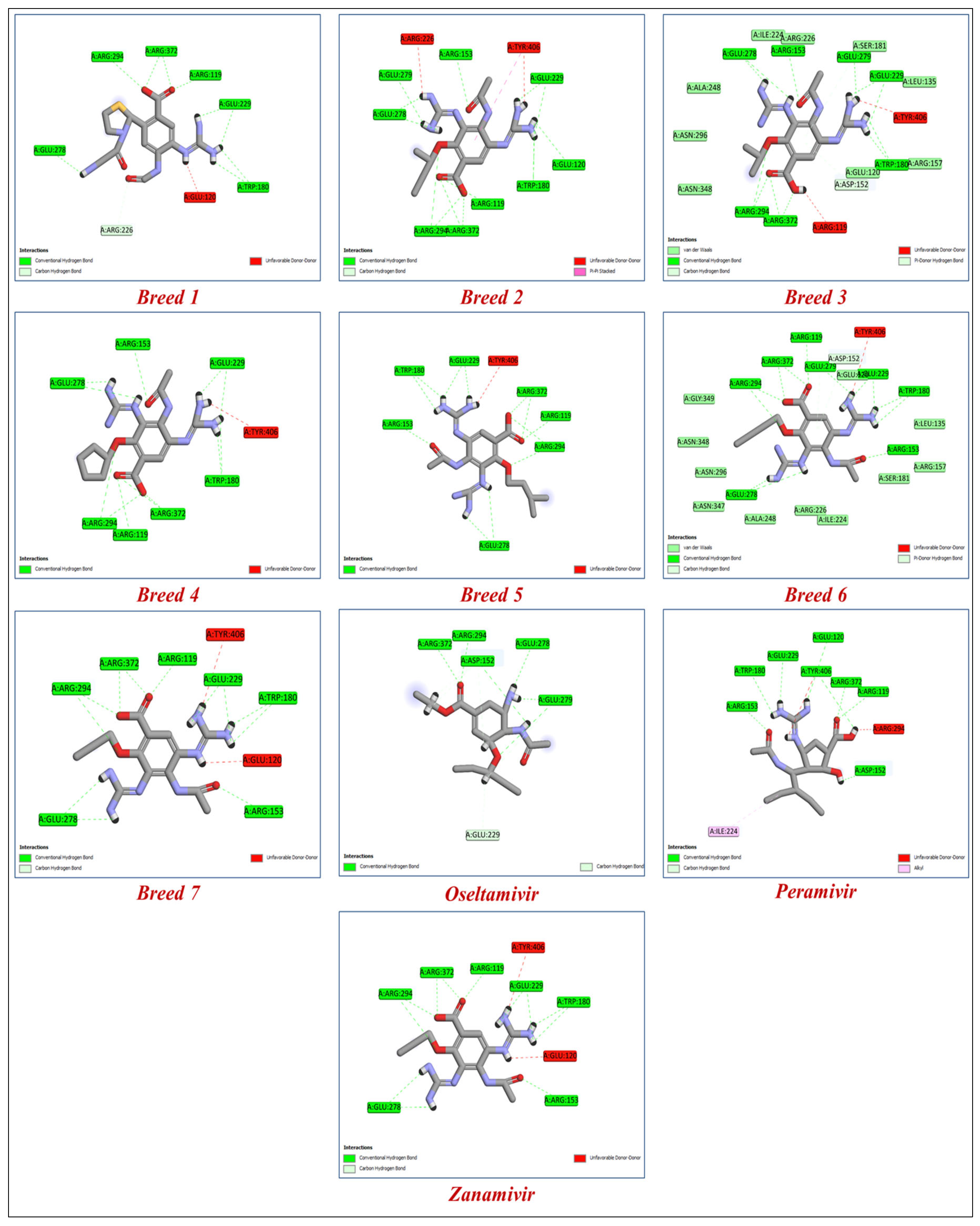
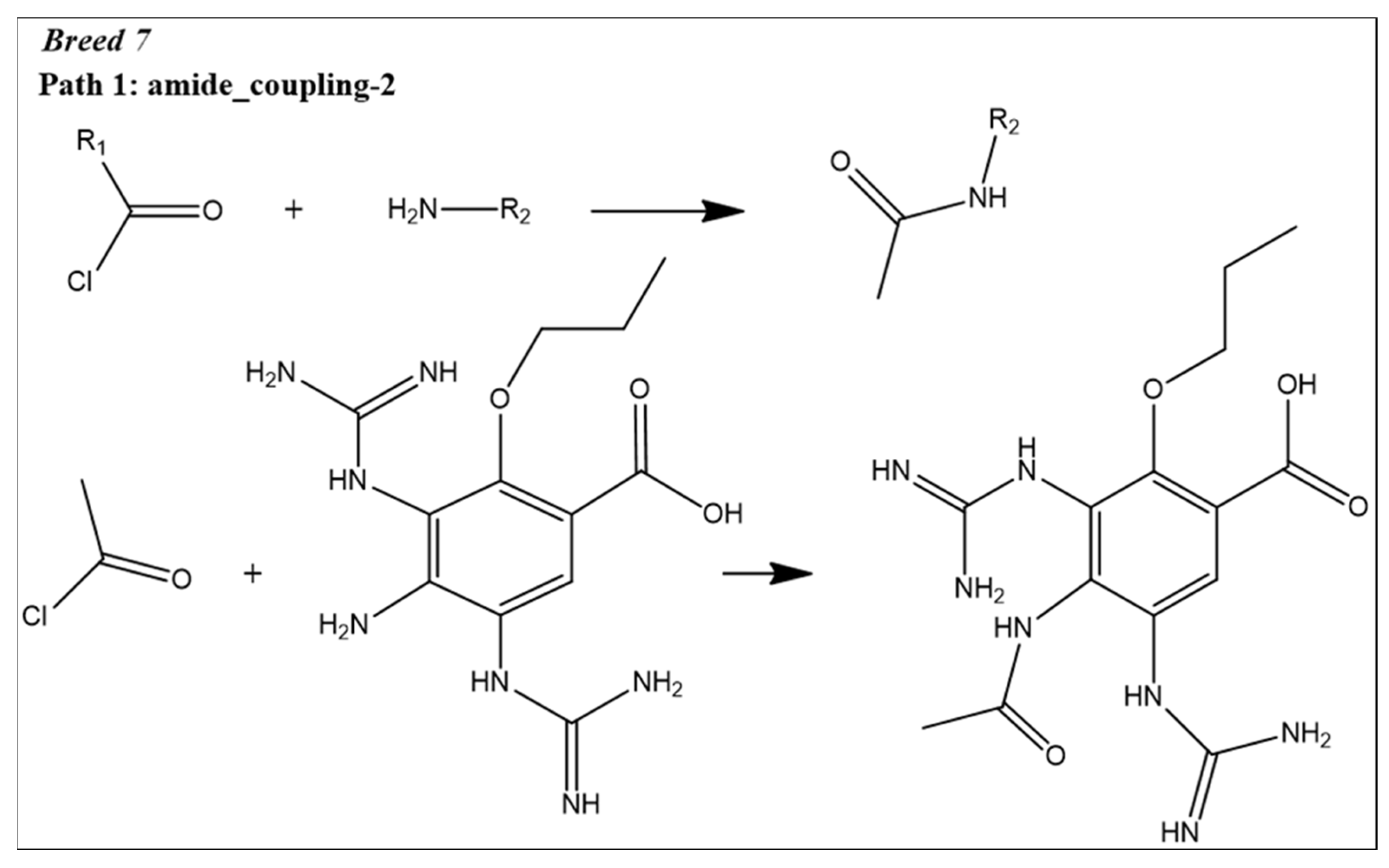
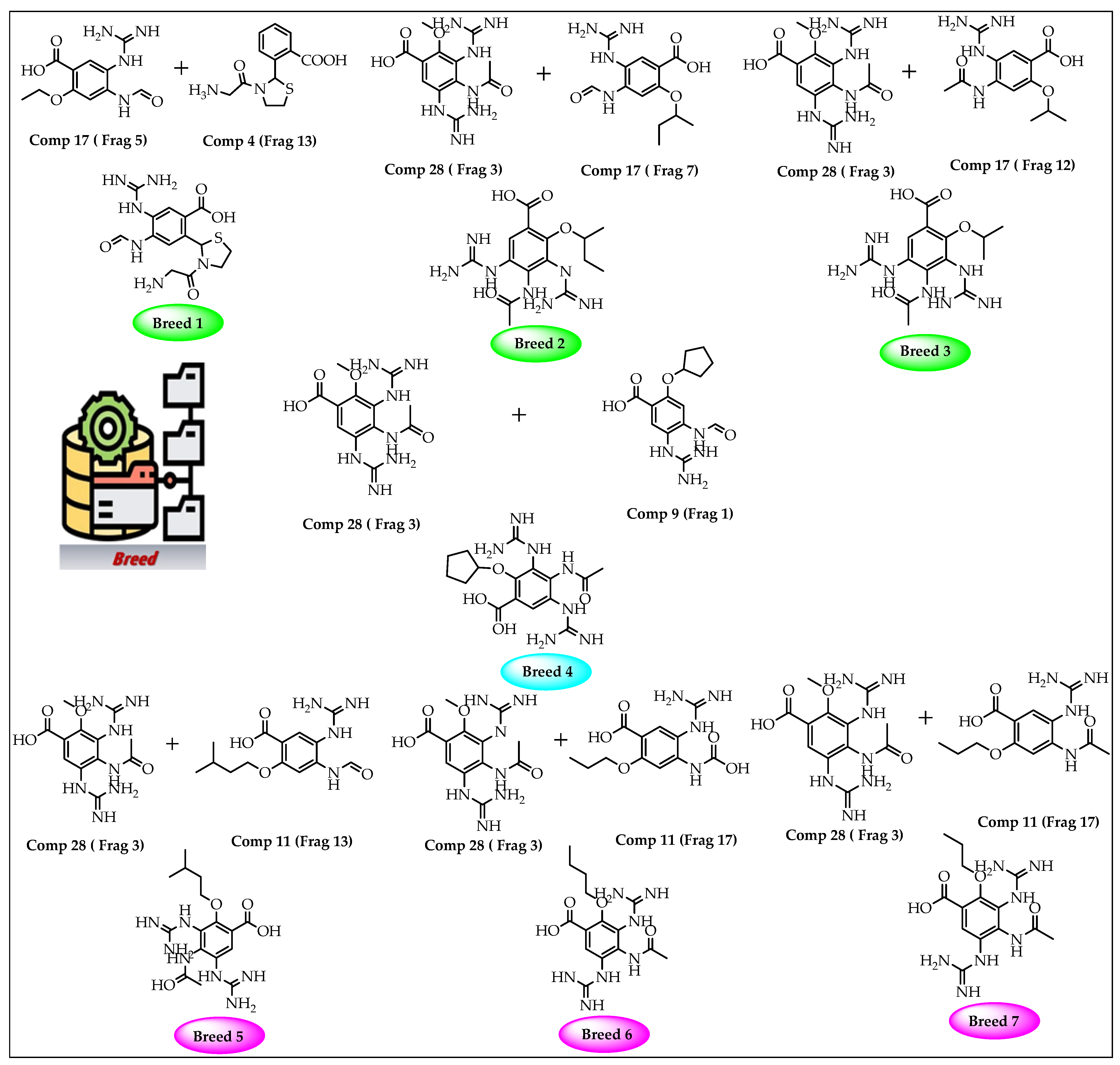
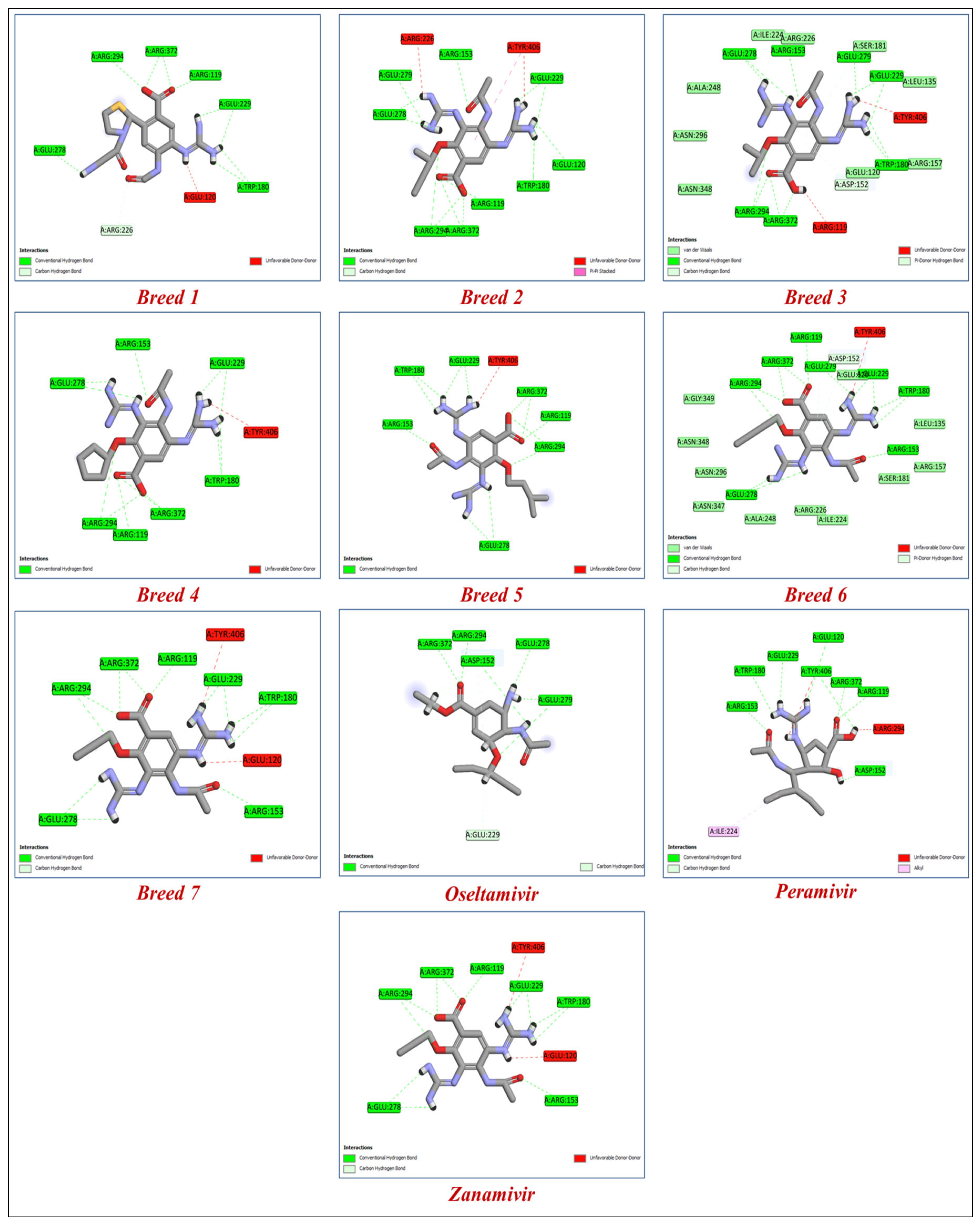
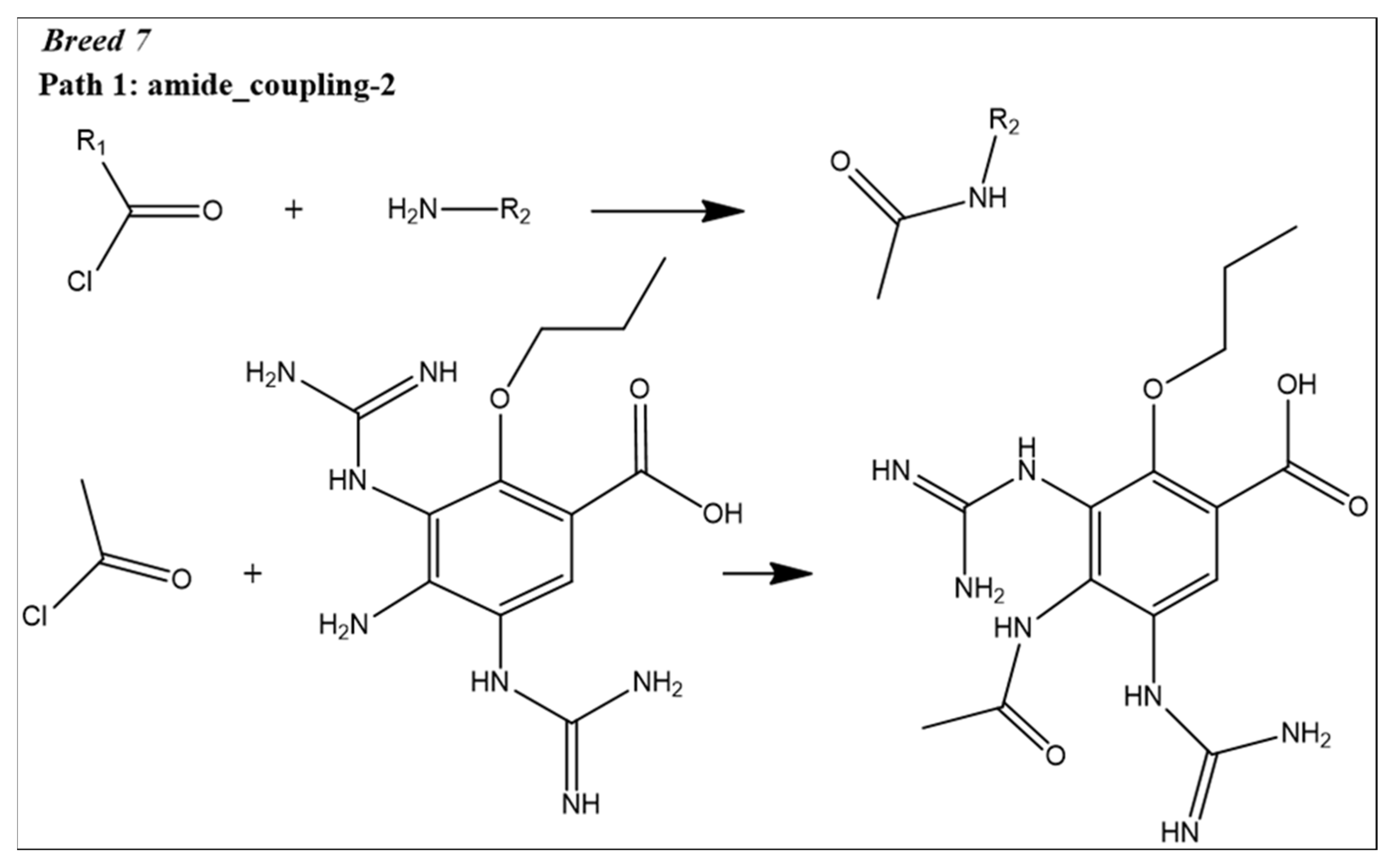
2.1. Breed-Based De Novo and Molecular Docking Approaches
2.2. ADME-Tox Prediction
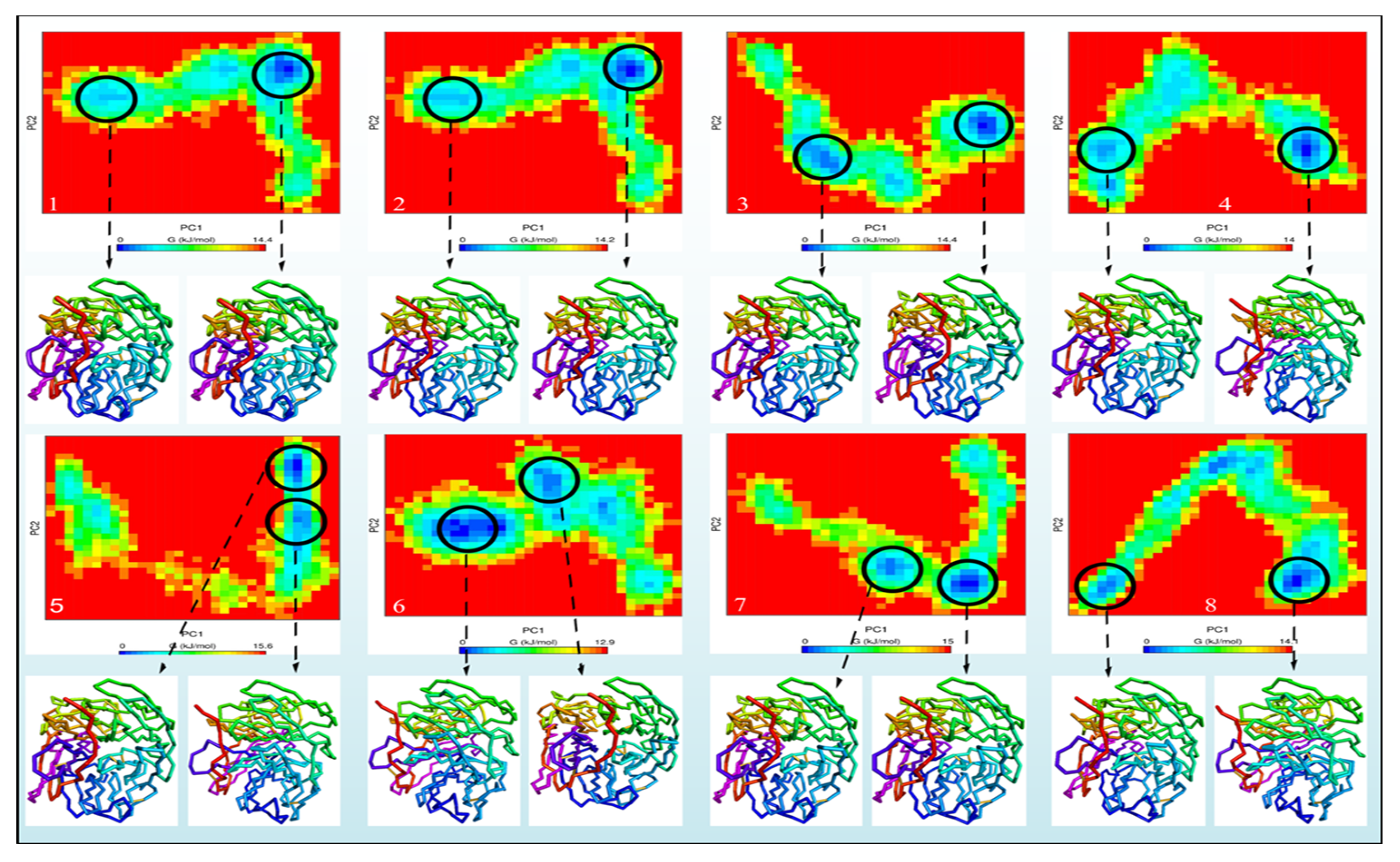
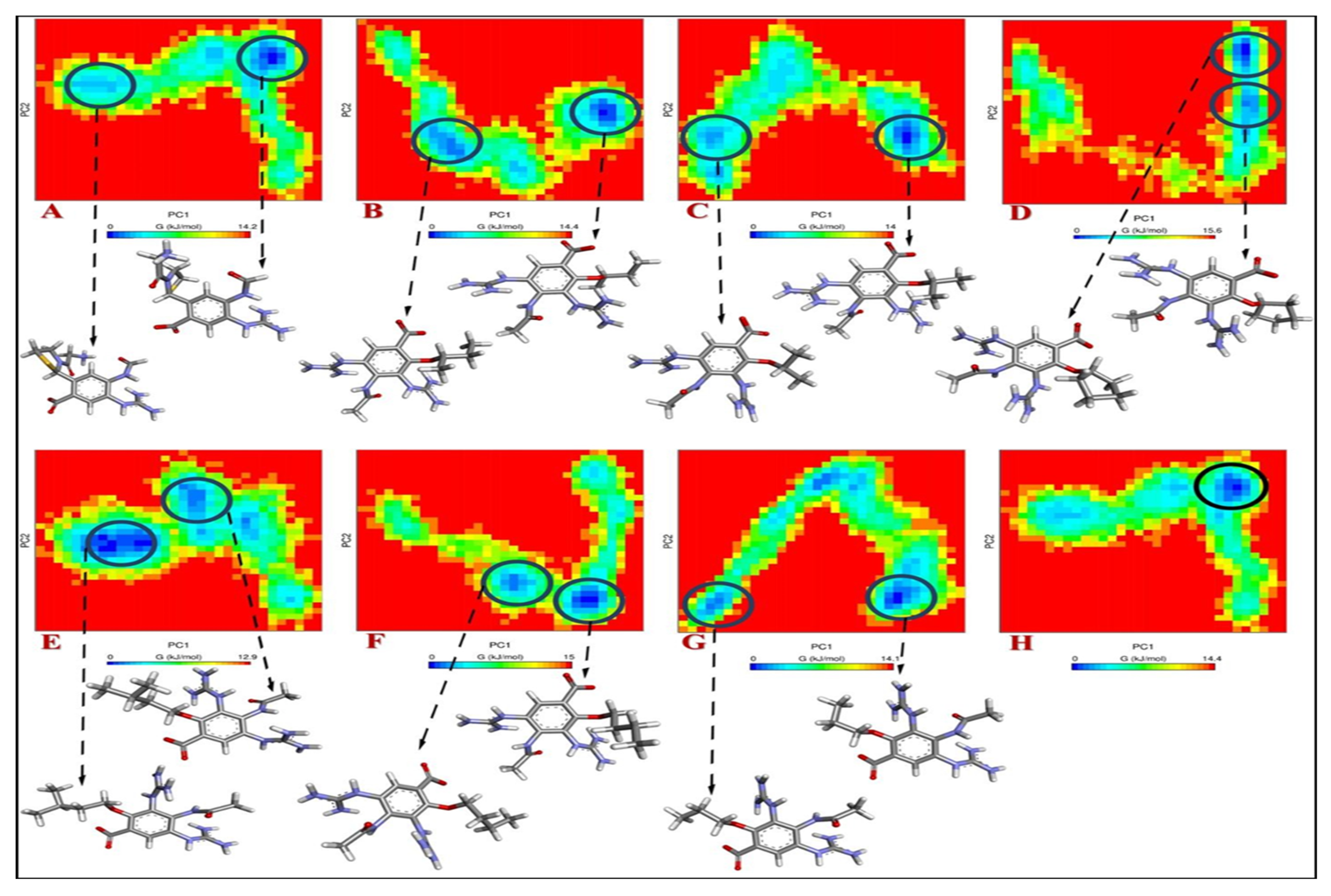
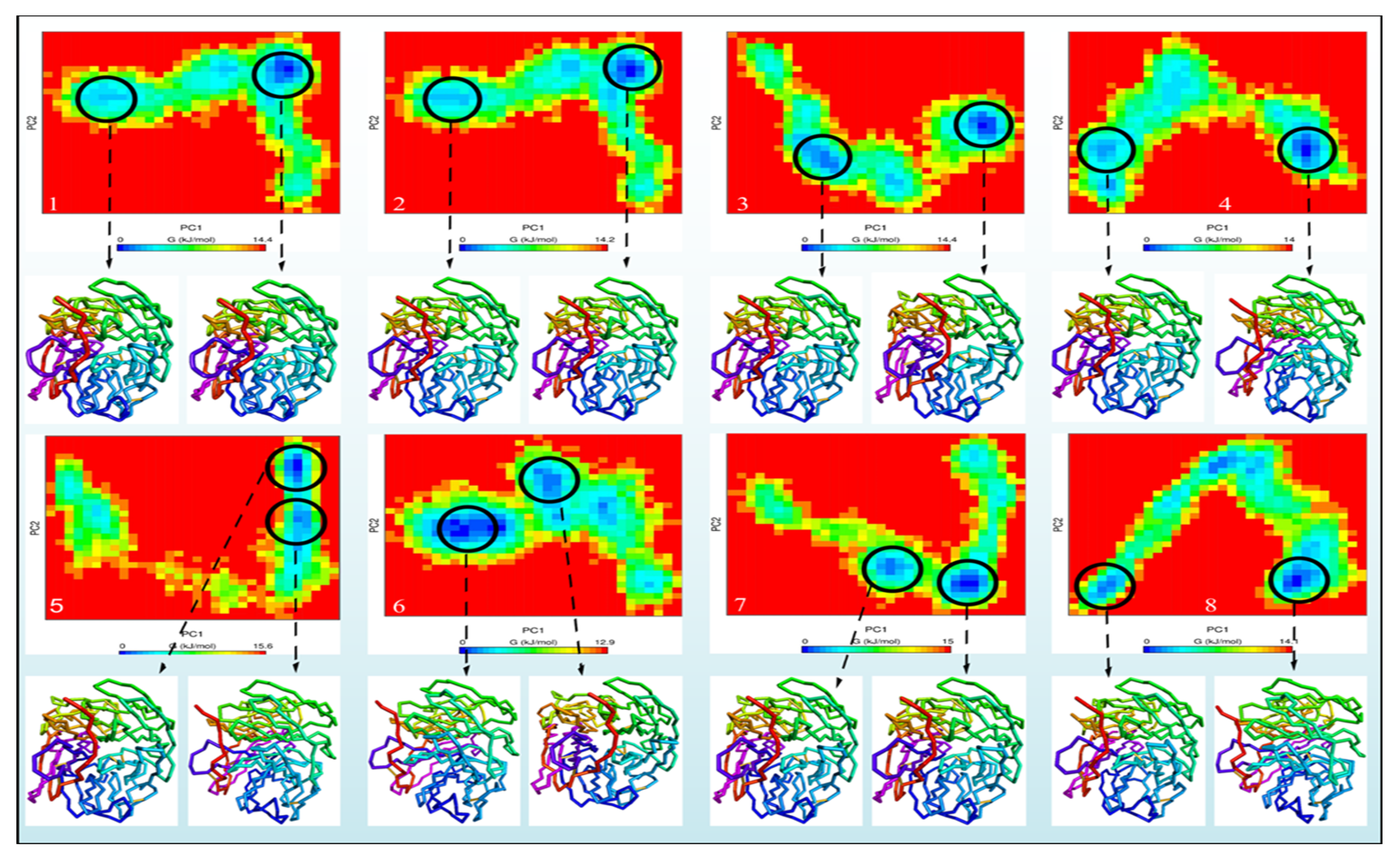
2.3. Study of Molecular Dynamics
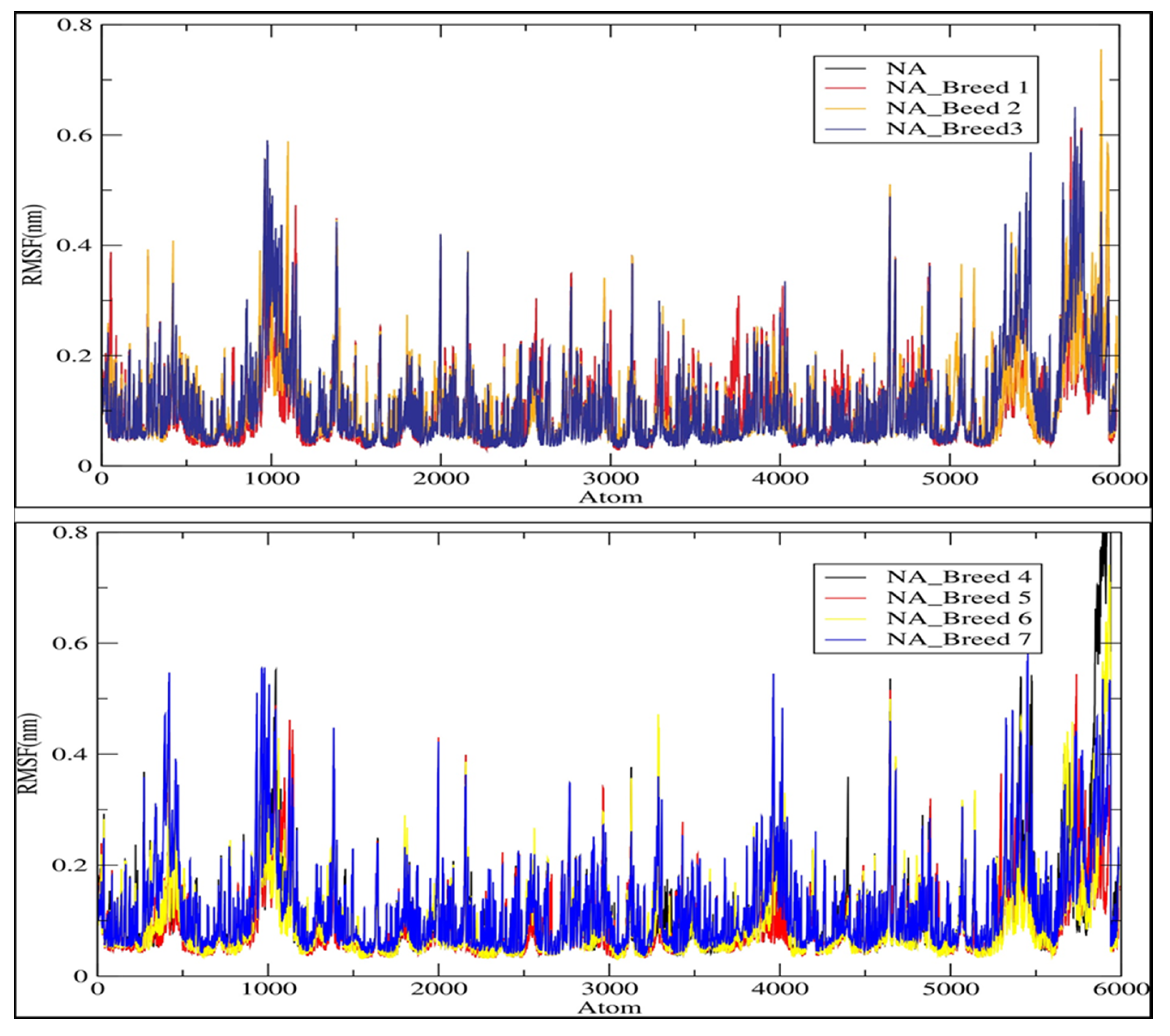
2.4. RMSD and RMSF Analysis
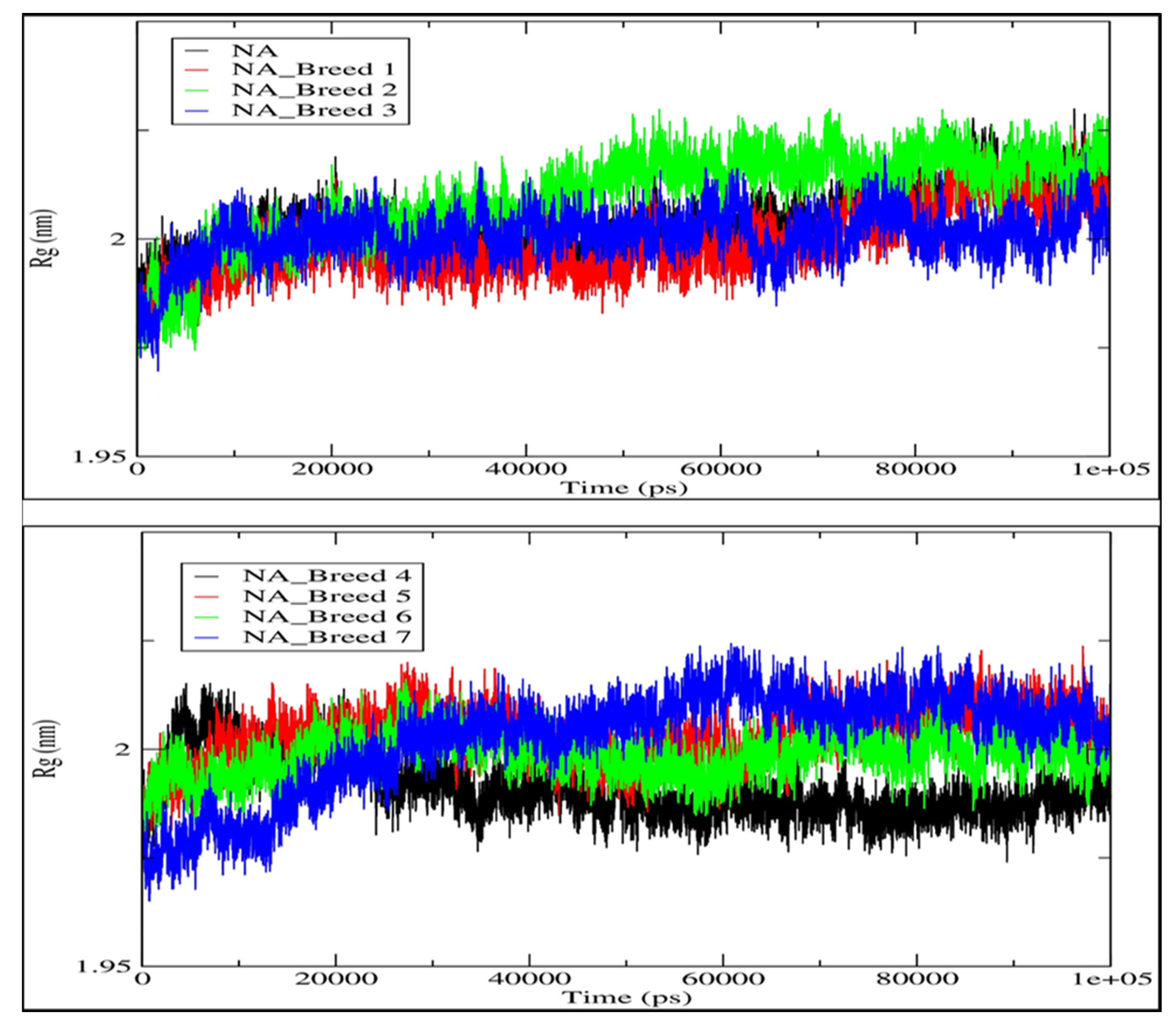
2.5. Radius of Gyration (Rg)
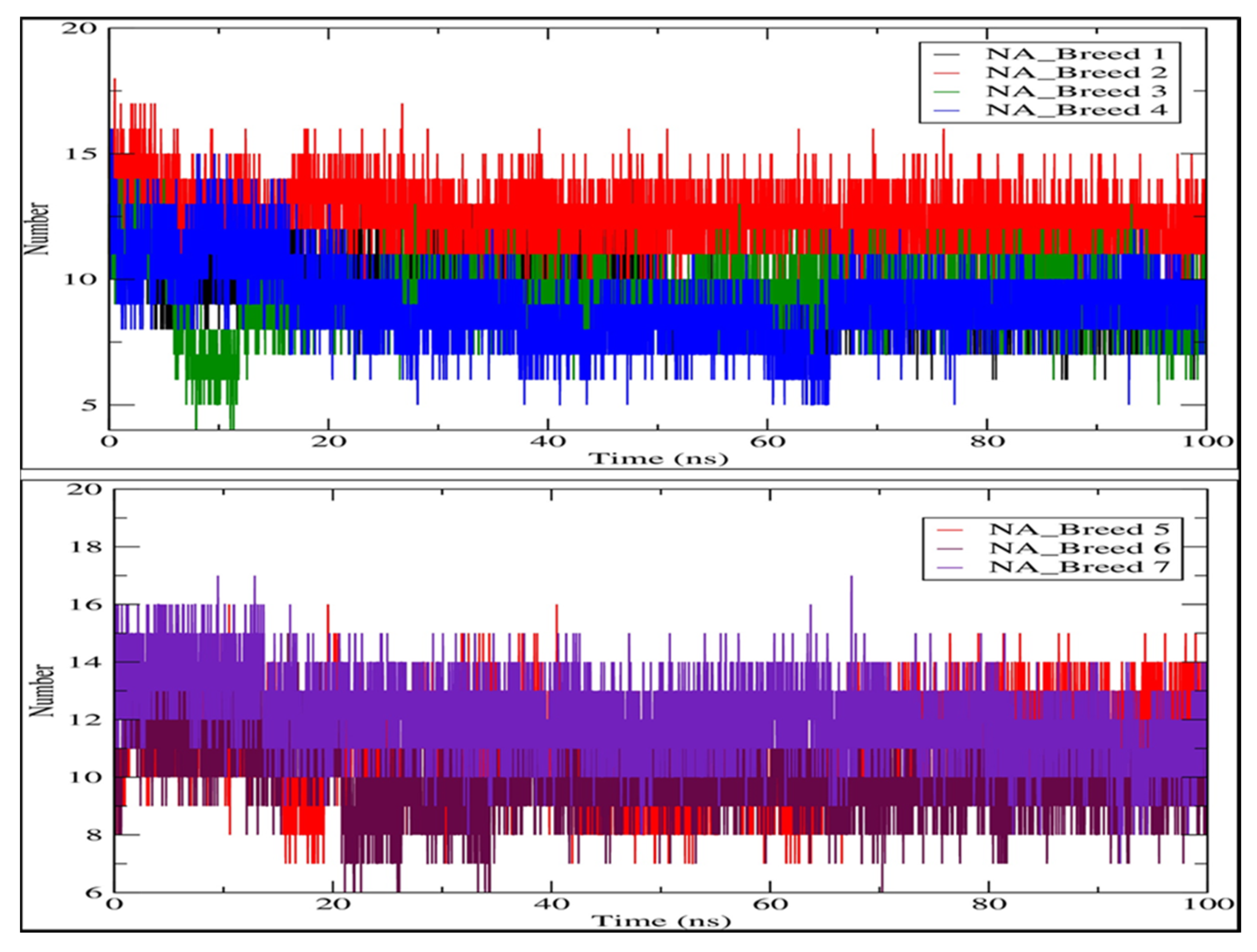
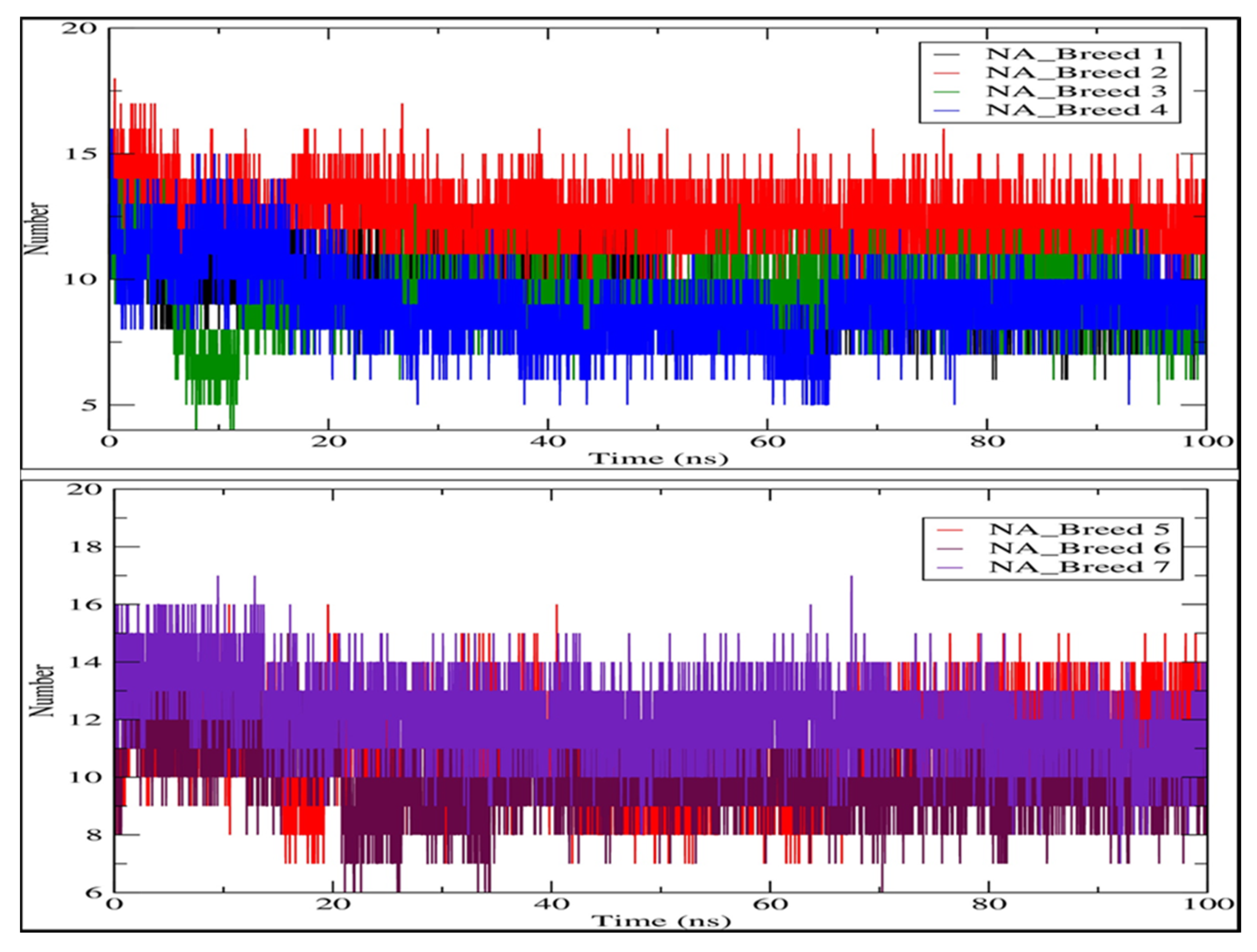
2.6. Hydrogen Bonding Analysis
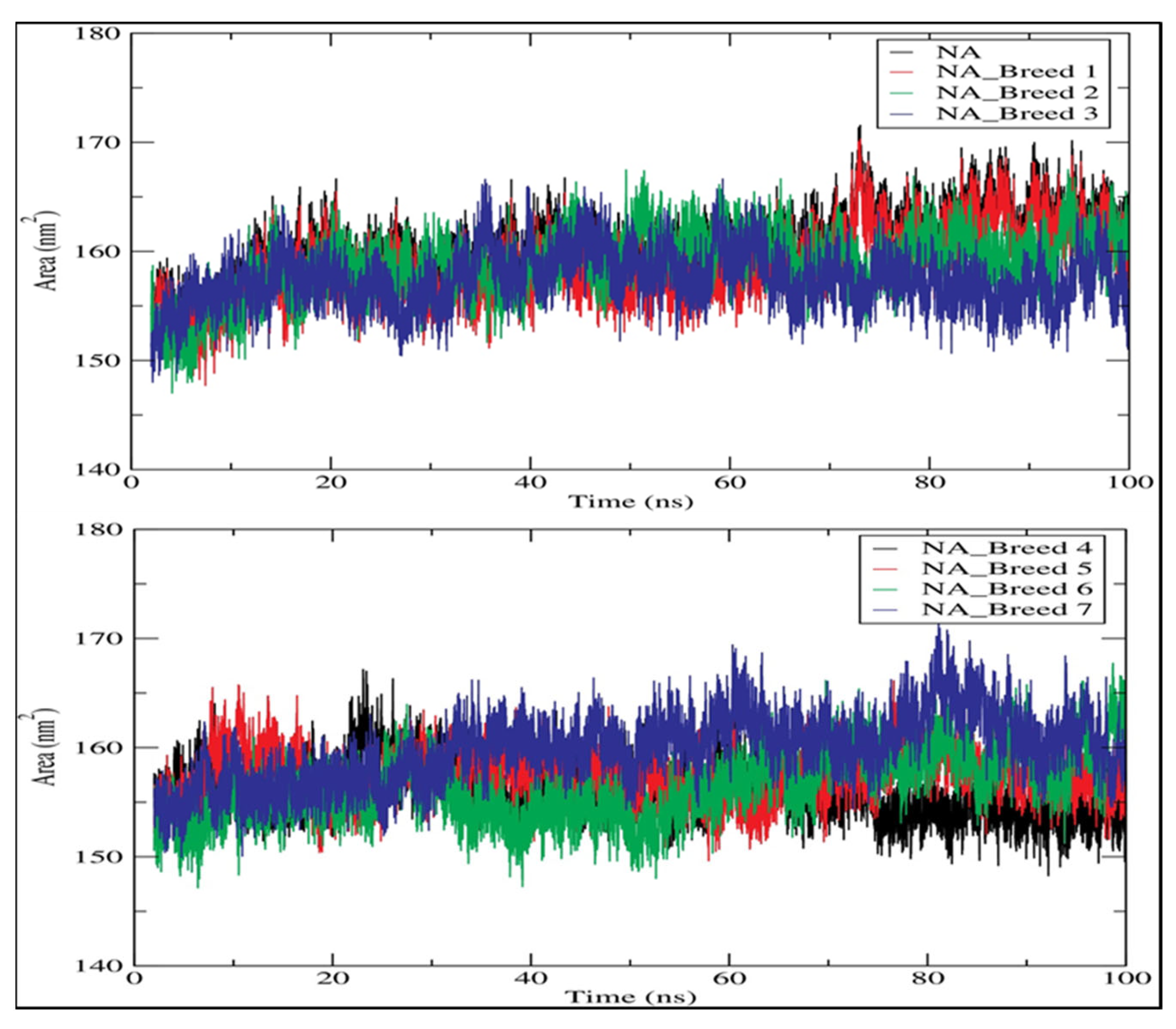
2.7. Solvent-Accessible Surface Area (SASA)
2.8. MM-PBSA Analysis
2.9. Reaction-Based Enumeration
3. Discussion
4. Materials and Methods
4.1. Breed De Novo Hybridization Approach
4.2. Molecular Docking Study
4.3. ADME-Tox Prediction
4.4. Molecular Dynamic Simulation
4.5. Binding Free Energy
4.6. Reaction-Based Enumeration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258. [Google Scholar] [CrossRef] [PubMed]
- Moghadami, M. A narrative review of influenza: A seasonal and pandemic disease. Iran. J. Med. Sci. 2017, 42, 2. [Google Scholar]
- Blut, A. Influenza virus. Transfus. Med. Hemotherapy 2009, 36, 32. [Google Scholar]
- Pei, S.; Shaman, J. Counteracting structural errors in ensemble forecast of influenza outbreaks. Nat. Commun. 2017, 8, 925. [Google Scholar] [CrossRef] [PubMed]
- Świerczyńska, M.; Mirowska-Guzel, D.M.; Pindelska, E. Antiviral drugs in influenza. Int. J. Environ. Res. Public Health 2022, 19, 3018. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Baloxavir marboxil: A review in acute uncomplicated influenza. Drugs 2020, 80, 1109–1118. [Google Scholar] [CrossRef]
- Ghaffari, H.; Tavakoli, A.; Moradi, A.; Tabarraei, A.; Bokharaei-Salim, F.; Zahmatkeshan, M.; Farahmand, M.; Javanmard, D.; Kiani, S.J.; Esghaei, M. Inhibition of H1N1 influenza virus infection by zinc oxide nanoparticles: Another emerging application of nanomedicine. J. Biomed. Sci. 2019, 26, 70. [Google Scholar] [CrossRef]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.-D. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 2004, 78, 12665–12667. [Google Scholar] [CrossRef]
- Ding, Y.; Cao, Z.; Cao, L.; Ding, G.; Wang, Z.; Xiao, W. Antiviral activity of chlorogenic acid against influenza A (H1N1/H3N2) virus and its inhibition of neuraminidase. Sci. Rep. 2017, 7, 45723. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Smee, D.F.; Barnard, D.L.; Julander, J.G.; Gross, M.; de Jong, M.D.; Went, G.T. Efficacy of combined therapy with amantadine, oseltamivir, and ribavirin in vivo against susceptible and amantadine-resistant influenza A viruses. PLoS ONE 2012, 7, e31006. [Google Scholar] [CrossRef]
- Regoes, R.R.; Bonhoeffer, S. Emergence of drug-resistant influenza virus: Population dynamical considerations. Science 2006, 312, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Lee, Y.-T.; Park, S.; Jung, Y.-J.; Lee, Y.; Ko, E.-J.; Kim, Y.-J.; Li, X.; Kang, S.-M. Neuraminidase expressing virus-like particle vaccine provides effective cross protection against influenza virus. Virology 2019, 535, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.; Whitley, R.J.; Pocock, S.; Monto, A.S. Oseltamivir treatment for influenza in adults: A meta-analysis of randomised controlled trials. Lancet 2015, 385, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, T.; Demicheli, V.; Di Pietrantonj, C.; Rivetti, D.; Cochrane Acute Respiratory Infections Group. Amantadine and rimantadine for influenza A in adults. Cochrane Database Syst. Rev. 1996, 2012. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, L.; Wat, C.; Mills, T.; Mahoney, P.; Ward, P.; Hayden, F. Impact of oseltamivir treatment on influenza-related lower respiratory tract complications and hospitalizations. Arch. Intern. Med. 2003, 163, 1667–1672. [Google Scholar] [CrossRef] [PubMed]
- Kohno, S.; Yen, M.-Y.; Cheong, H.-J.; Hirotsu, N.; Ishida, T.; Kadota, J.-i.; Mizuguchi, M.; Kida, H.; Shimada, J. Phase III randomized, double-blind study comparing single-dose intravenous peramivir with oral oseltamivir in patients with seasonal influenza virus infection. Antimicrob. Agents Chemother. 2011, 55, 5267–5276. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.; Laughlin, A.; Carson, S.; Mitha, E.; Tellier, G.; Stich, M.; Elder, J.; Alexander, W.J.; Dobo, S.; Collis, P. Single dose peramivir for the treatment of acute seasonal influenza: Integrated analysis of efficacy and safety from two placebo-controlled trials. Antivir. Ther. 2015, 20, 709–719. [Google Scholar] [CrossRef]
- Pierce, A.C.; Rao, G.; Bemis, G.W. BREED: Generating novel inhibitors through hybridization of known ligands. Application to CDK2, p38, and HIV protease. J. Med. Chem. 2004, 47, 2768–2775. [Google Scholar] [CrossRef]
- Ho, C.M.; Marshall, G.R. SPLICE: A program to assemble partial query solutions from three-dimensional database searches into novel ligands. J. Comput.-Aided Mol. Des. 1993, 7, 623–647. [Google Scholar] [CrossRef]
- Patel, H.M.; Shaikh, M.; Ahmad, I.; Lokwani, D.; Surana, S.J. BREED based de novo hybridization approach: Generating novel T790M/C797S-EGFR tyrosine kinase inhibitors to overcome the problem of mutation and resistance in non small cell lung cancer (NSCLC). J. Biomol. Struct. Dyn. 2021, 39, 2838–2856. [Google Scholar] [CrossRef]
- Schmitt, S.; Kuhn, D.; Klebe, G. A new method to detect related function among proteins independent of sequence and fold homology. J. Mol. Biol. 2002, 323, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L. OPLS3e: Extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, S. In silico ADME-Tox modeling: Progress and prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Maowa, J.; Hosen, M.A.; Alam, A.; Rana, K.M.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. Pharmacokinetics and Molecular Docking Studies of Uridine Derivatives as SARS-CoV-2 Mpro Inhibitors. Phys. Chem. Res. 2021, 9, 385–412. [Google Scholar]
- Rana, K.M.; Maowa, J.; Alam, A.; Hosen, A.; Dey, S.; Hasan, I.; Fujii, Y.; Ozeki, Y.; Kawsar, S.M.A. In Silico DFT Study, Molecular Docking, and ADMET Predictions of Cytidine Analogs with Antimicrobial and Anticancer Properties. In Silico Pharmacol. 2021, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, F.; Zomorodipour, A.; Karkhane, A.A.; Khorramizadeh, M.R. In silico designing of hyperglycosylated analogs for the human coagulation factor IX. J. Mol. Graph. Model. 2016, 68, 39–47. [Google Scholar] [CrossRef]
- Kawsar, S.M.A.; Kumer, A.; Munia, N.S.; Hosen, M.A.; Chakma, U.; Akash, S. Chemical descriptors, PASS, molecular docking, molecular dynamics and ADMET predictions of glucopyranoside derivatives as inhibitors to bacteria and fungi growth. Org. Commun. 2022, 15, 184–203. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef]
- Islam, R.; Parves, M.R.; Paul, A.S.; Uddin, N.; Rahman, M.S.; Mamun, A.A.; Hossain, M.N.; Ali, M.A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3213–3224. [Google Scholar] [CrossRef]
- Zhang, J.; Shan, Y.; Pan, X.; Wang, C.; Xu, W.; He, L. Molecular docking, 3D-QSAR Studies, and in silico ADME prediction of p-aminosalicylic acid derivatives as neuraminidase inhibitors. Chem. Biol. Drug Des. 2011, 78, 709–717. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Targowska-Duda, K.M.; Patel, J.Z.; Laitinen, T.; Parkkari, T.; Adams, Y.; Nevalainen, T.J.; Poso, A. Comparative molecular field analysis and molecular dynamics studies of α/β hydrolase domain containing 6 (ABHD6) inhibitors. J. Mol. Model. 2015, 21, 250. [Google Scholar] [CrossRef] [PubMed]
- Opoku, F.; Govender, P.P.; Pooe, O.J.; Simelane, M.B. Evaluating iso-mukaadial acetate and ursolic acid acetate as plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase inhibitors. Biomolecules 2019, 9, 861. [Google Scholar] [CrossRef] [PubMed]
- Usha, T.; Shanmugarajan, D.; Goyal, A.K.; Kumar, C.S.; Middha, S.K. Recent updates on computer-aided drug discovery: Time for a paradigm shift. Curr. Top. Med. Chem. 2017, 17, 3296–3307. [Google Scholar] [CrossRef] [PubMed]
- Gubareva, L.V.; Sleeman, K.; Guo, Z.; Yang, H.; Hodges, E.; Davis, C.T.; Baranovich, T.; Stevens, J. Drug susceptibility evaluation of an influenza A (H7N9) virus by analyzing recombinant neuraminidase proteins. J. Infect. Dis. 2017, 216 (Suppl. S4), S566–S574. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.-Y.; Li, J.-Y.; Hao, G.-F.; Yang, G.-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today 2020, 25, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Raies, A.B.; Bajic, V.B. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Ahmad, S.S.; Sinha, M.; Ahmad, K.; Khalid, M.; Choi, I. Study of Caspase 8 inhibition for the management of Alzheimer’s disease: A molecular docking and dynamics simulation. Molecules 2020, 25, 2071. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Kant, K.; Kumar, A.; Bhati, K.; Verma, S.M. HeroMDAnalysis: An automagical tool for GROMACS-based molecular dynamics simulation analysis. Future Med. Chem. 2021, 13, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Singh, R.; Subbarao, N. Exploring the interaction mechanism between potential inhibitor and multitarget Mur enzymes of mycobacterium tuberculosis using molecular docking, molecular dynamics simulation, principal component analysis, free energy landscape, dynamic cross-correlation matrices, vector movements, and binding free energy calculation. J. Biomol. Struct. Dyn. 2022, 40, 13497–13526. [Google Scholar] [PubMed]
- Singh, N.; Siddiqi, M.I. Computational evaluation of glutamine synthetase as drug target against infectious diseases: Molecular modeling, substrate-binding analysis, and molecular dynamics simulation studies. Med. Chem. Res. 2017, 26, 450–460. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Konze, K.D.; Bos, P.H.; Dahlgren, M.K.; Leswing, K.; Tubert-Brohman, I.; Bortolato, A.; Robbason, B.; Abel, R.; Bhat, S. Reaction-based enumeration, active learning, and free energy calculations to rapidly explore synthetically tractable chemical space and optimize potency of cyclin-dependent kinase 2 inhibitors. J. Chem. Inf. Model. 2019, 59, 3782–3793. [Google Scholar] [CrossRef]
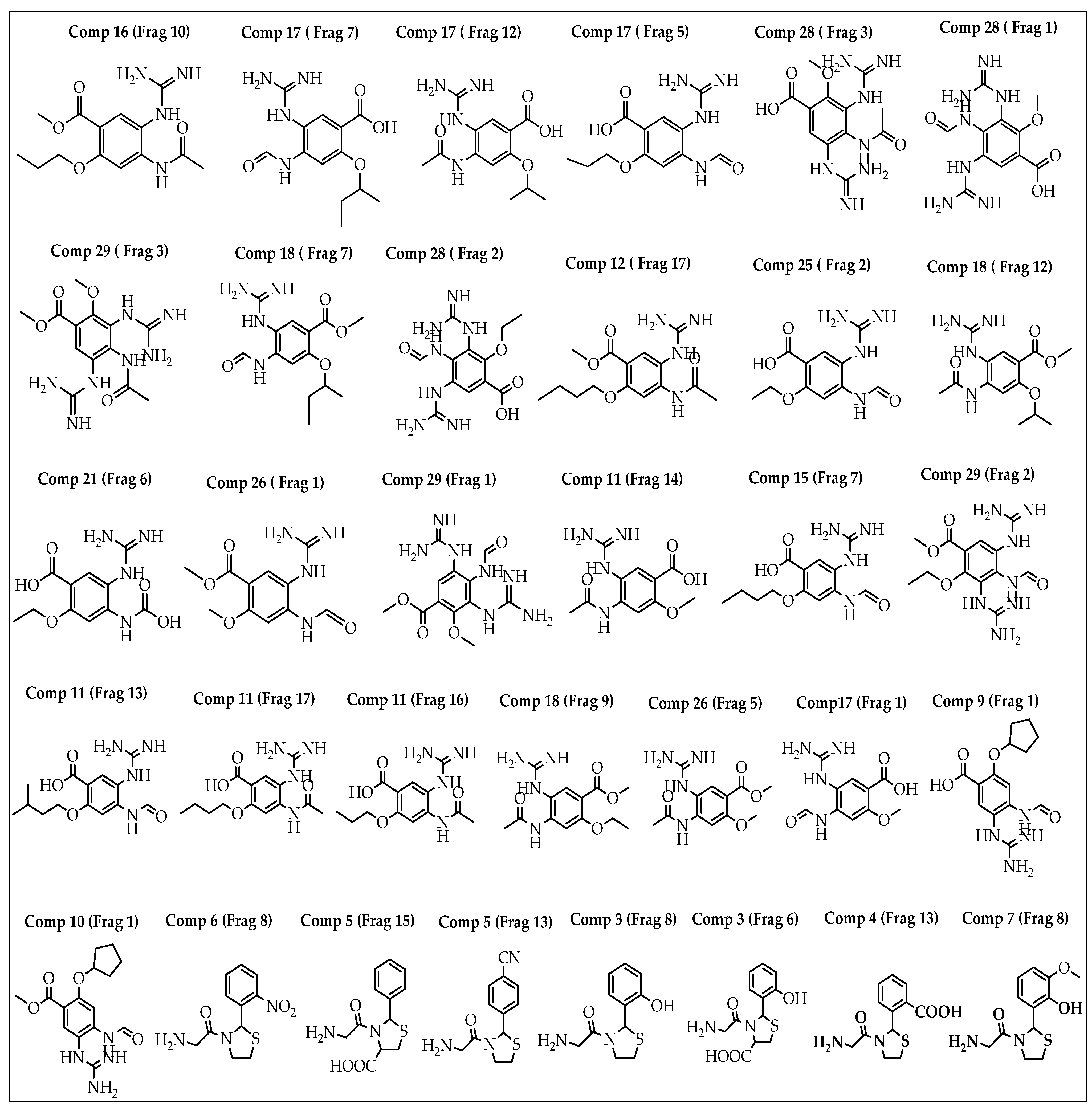







{kind=link}
{kind=link}
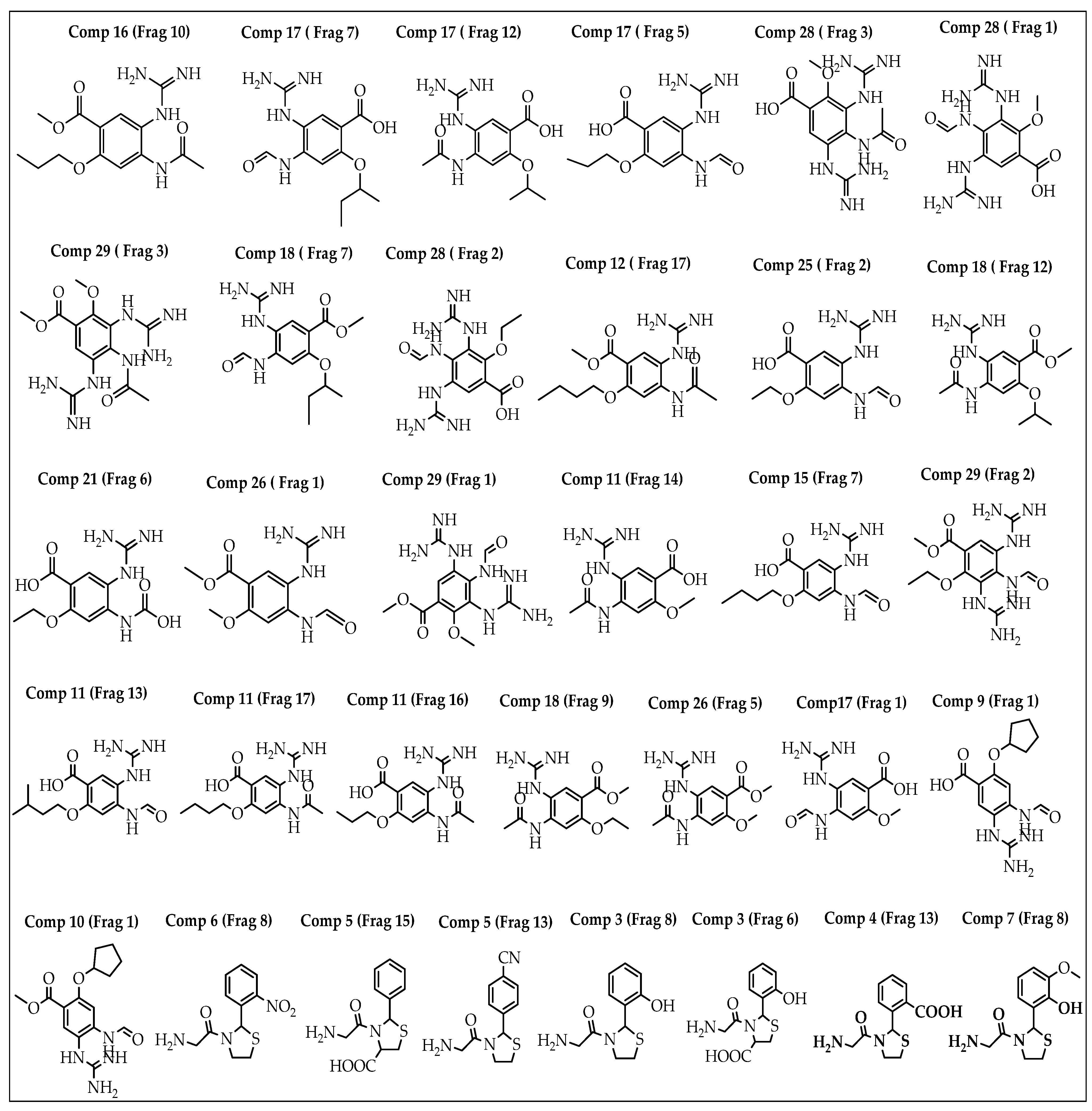{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
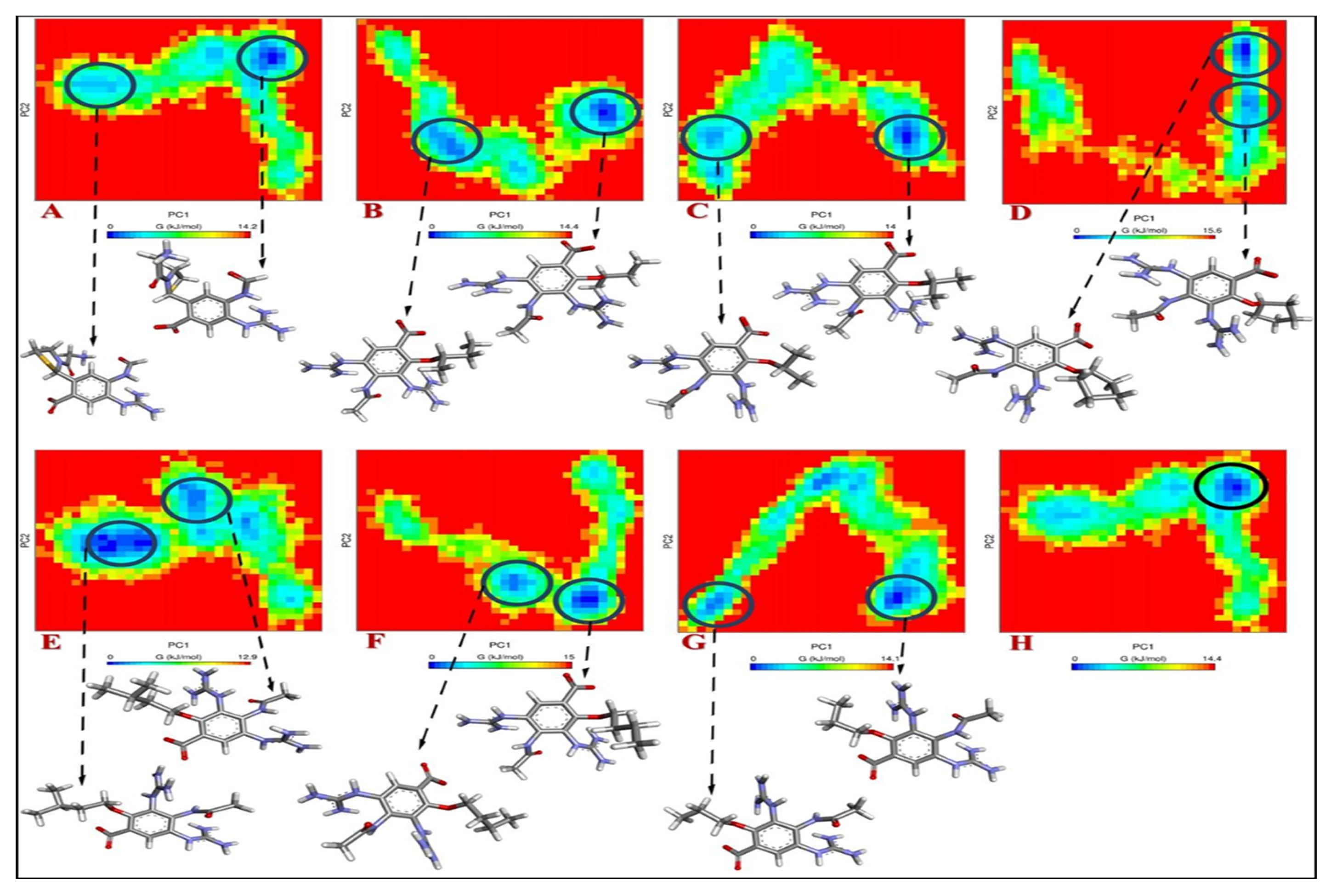{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
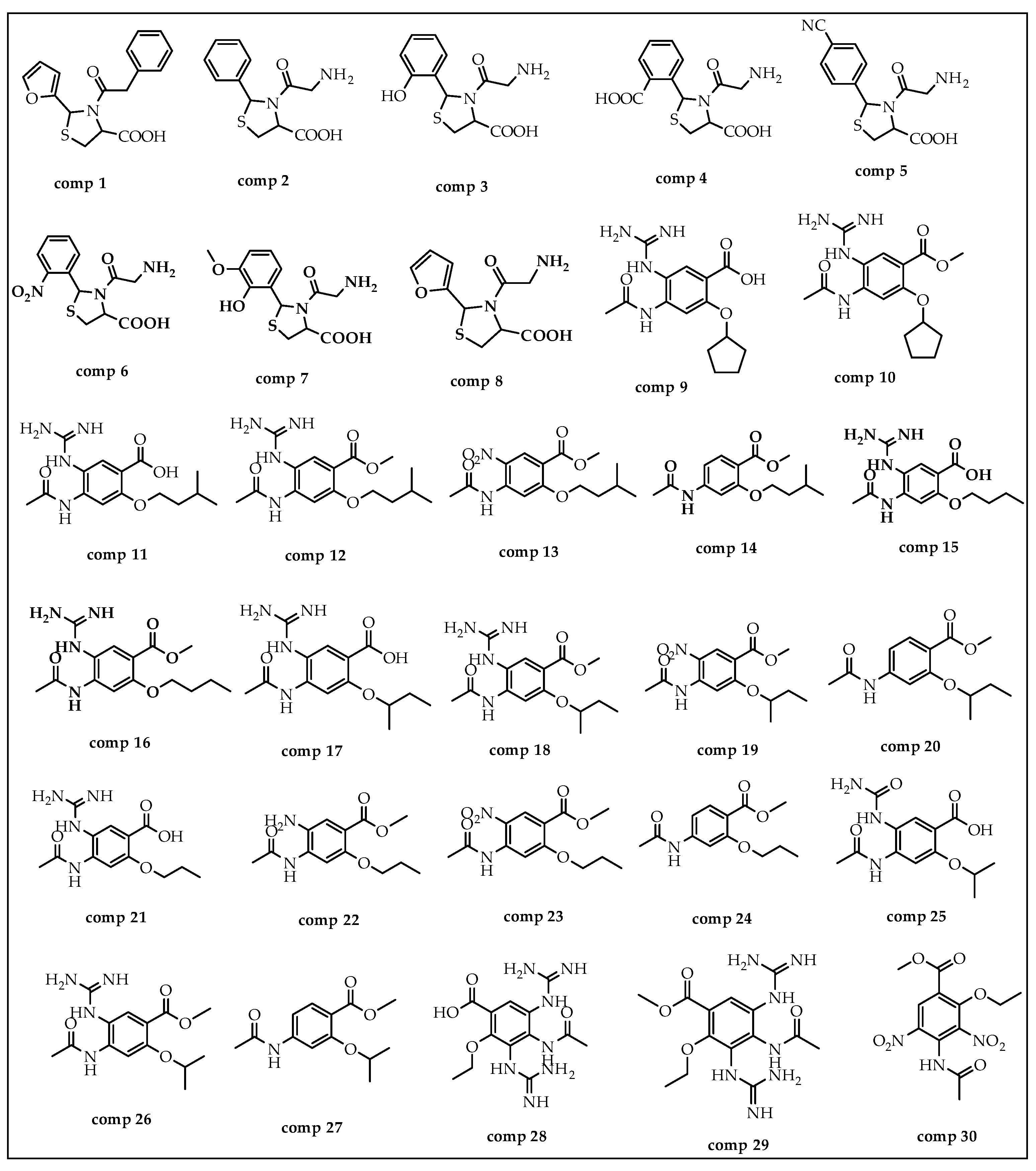{kind=link}
| Fragment | SP-Score (kcal/mol) | Fragment | SP-Score (kcal/mol) | Fragment | SP-Score (kcal/mol) |
|---|---|---|---|---|---|
| Comp28 (Frag3) | −8.700 | Comp5 (Frag15) | −7.085 | Comp26 (Frag5) | −6.425 |
| Comp28 (Frag1) | −8.425 | Comp17 (Frag1) | −7.055 | Comp6 (Frag8) | −6.370 |
| Comp17 (Frag12) | −8.030 | Comp17 (Frag7) | −7.048 | Comp18 (Frag9) | −6.264 |
| Comp11 (Frag16) | −7.897 | Comp3 (Frag8) | −7.030 | Comp16 (Frag10) | −6.117 |
| Comp9 (Frag1) | −7.679 | Comp29 (Frag1) | −7.002 | Comp18 (Frag12) | −6.166 |
| Comp17 (Frag5) | −7.661 | Comp25 (Frag2) | −6.960 | Comp12 (Frag17) | −6.144 |
| Comp28 (Frag2) | −7.654 | Comp29 (Frag3) | −6.883 | Comp26 (Frag1) | −6.135 |
| Comp11 (Frag14) | −7.409 | Comp11 (Frag13) | −6.883 | Comp5 (Frag13) | −6.073 |
| Comp11 (Frag17) | −7.296 | Comp29 (Frag2) | −6.743 | Comp15 (Frag7) | −6.042 |
| Comp21 (Frag6) | −7.287 | Comp7 (Frag8) | −6.518 | Comp10 (Frag1) | −6.031 |
| Comp4 (Frag13) | −7.263 | Comp18 (Frag7) | −6.464 | Comp3 (Frag6) | −6.012 |
| Molecules | Breed Score | SP-Score | XP-Score | H-Bond Interactions | Distance |
|---|---|---|---|---|---|
| Breed 1 | 15.623 | −8.799 | −11.867 | Arg119, Asp152, Trp180, Arg226, Glu226, Glu278, Arg294, Arg372. | 1.61–3.33 |
| Breed 2 | 11.080 | −8.372 | −10.804 | Arg119, Glu120, Arg153, Trp180, Glu229, Glu278, Glu279, Arg294, Arg372. | 1.57–5.98 |
| Breed 3 | 9.952 | −8.316 | −10.791 | Arg119, Asp152, Arg153, Trp180, Glu229, Glu278, Glu279, Arg294, Arg372. | 1.48–3.99 |
| Breed 4 | 13.604 | −8.278 | −10.765 | Arg119, Asp152, Arg153, Trp180, Glu229, Glu278, Glu279, Arg294, Arg372. | 1.50–4.91 |
| Breed 5 | 7.886 | −8.457 | −10.706 | Arg119, Asp152, Arg153, Trp180, Arg194, Glu229, Glu278, Glu279, Arg372. | 1.22–3.81 |
| Breed 6 | 8.298 | −7.949 | −10.628 | Arg119, Asp152, Arg153, Trp180, Glu229, Glu278, Glu279, Arg294, Arg372. | 1.48–3.99 |
| Breed 7 | 10.740 | −7.890 | −10.529 | Arg119, Arg152, Arg153, Trp180, Glu229, Glu278, Arg294, Arg372. | 1.57–4.15 |
| Zanamivir | - | −7.610 | −9.848 | Arg119, Arg152, Arg153, Trp180, Glu229, Glu278, Glu279, Arg294, Arg372. | 1.50–4.15 |
| Peramivir | - | −7.370 | −8.844 | Arg119, Glu120, Asp152, Arg153, Trp180, Glu229, Glu279, Arg294, Arg372. | 1.55–4.39 |
| Oseltamivir | - | −5.588 | −6.326 | Asp152, Glu229, Glu278, Glu279, Arg294, Arg372. | 1.50–4.15 |
| Molecules | MW (g/mol) | Log S (ESOL) | Consensus Log P | Cytochrome P450 Inhibitors | Bioavailability Score | Log Kp cm/s | Synthetic Accessibility |
|---|---|---|---|---|---|---|---|
| Breed 1 | 366.40 | 0.77 | −1.55 | No | 0.55 | −11.38 | 3.60 |
| Breed 2 | 365.39 | −1.18 | −0.40 | No | 0.17 | −9.02 | 3.79 |
| Breed 3 | 351.36 | −0.83 | −0.74 | No | 0.17 | −9.31 | 3.19 |
| Breed 4 | 409.48 | −2.28 | 0.24 | No | 0.17 | −8.41 | 3.78 |
| Breed 5 | 379.41 | −1.42 | −0.13 | No | 0.17 | −8.85 | 3.46 |
| Breed 6 | 365.39 | −1.07 | −0.43 | No | 0.17 | −9.08 | 3.35 |
| Breed 7 | 351.36 | −0.83 | −0.59 | No | 0.17 | −9.24 | 3.24 |
| Molecules | Cytotoxicity | Carcinogenicity | Mutagenicity | Immunotoxicity | Toxicity Class | LD50 (mg/kg) |
|---|---|---|---|---|---|---|
| Breed 1 | Inactive | Inactive | Inactive | Inactive | 4 | 1098 |
| Breed 2 | Inactive | Inactive | Inactive | Inactive | 5 | 3200 |
| Breed 3 | Inactive | Inactive | Inactive | Inactive | 5 | 3200 |
| Breed 4 | Inactive | Inactive | Inactive | Inactive | 5 | 4000 |
| Breed 5 | Inactive | Inactive | Inactive | Inactive | 5 | 3200 |
| Breed 6 | Inactive | Inactive | Inactive | Inactive | 5 | 3200 |
| Breed 7 | Inactive | Inactive | Inactive | Inactive | 5 | 3200 |
| Complex | Average RMSD (nm) | Average RMSF (nm) | Average Rg (nm) | Average H-Bonds (nm) | SASA (nm2) |
|---|---|---|---|---|---|
| NA_Breed 1 | 0.149 | 0.105 | 1.998 | 9.428 | 158.812 |
| NA_Breed 2 | 0.131 | 0.109 | 2.009 | 12.444 | 158.835 |
| NA_Breed 3 | 0.135 | 0.110 | 2.000 | 9.196 | 157.379 |
| NA_Breed 4 | 0.166 | 0.115 | 1.991 | 9.018 | 156.121 |
| NA_Breed 5 | 0.172 | 0.100 | 2.003 | 11.057 | 157.397 |
| NA_Breed 6 | 0.172 | 0.106 | 1.998 | 9.964 | 156.429 |
| NA_Breed 7 | 0.148 | 0.123 | 2.002 | 12.064 | 159.806 |
| NA | 0.148 | 0.106 | 2.003 | - | 160.245 |
| Protein-Ligand Complexes | ΔEVDW (KJ/mol) | ΔEEEL (KJ/mol) | ΔEPB (KJ/mol) | ΔENPOLAR (KJ/mol) | ΔEDISPER (KJ/mol) | ΔGGAS (KJ/mol) | ΔGSOLV (KJ/mol) | ΔTOTAL (KJ/mol) |
|---|---|---|---|---|---|---|---|---|
| NA_Breed 1 | −11.61 | −385.46 | 319.53 | −25.45 | 47.99 | −397.07 | 342.07 | −55.00 |
| NA_Breed 2 | −15.34 | −400.88 | 320.91 | −29.39 | 50.15 | −416.22 | 341.67 | −74.55 |
| NA_Breed 3 | −12.39 | −363.90 | 306.55 | −27.48 | 49.61 | −376.28 | 328.67 | −47.61 |
| NA_Breed 4 | −17.29 | −347.68 | 297.99 | −29.23 | 51.32 | −364.97 | 320.08 | −44.89 |
| NA_Breed 5 | −19.90 | −328.15 | 290.05 | −29.25 | 52.29 | −348.05 | 313.10 | −34.96 |
| NA_Breed 6 | −24.00 | −338.25 | 297.31 | −28.89 | 51.35 | −362.25 | 319.77 | −42.48 |
| NA_Breed 7 | −10.87 | −407.23 | 321.76 | −28.41 | 48.69 | −418.10 | 342.04 | −76.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lotfi, B.; Mebarka, O.; Alhatlani, B.Y.; Abdallah, E.M.; Kawsar, S.M.A. Pharmacoinformatics and Breed-Based De Novo Hybridization Studies to Develop New Neuraminidase Inhibitors as Potential Anti-Influenza Agents. Molecules 2023, 28, 6678. https://doi.org/10.3390/molecules28186678
Lotfi B, Mebarka O, Alhatlani BY, Abdallah EM, Kawsar SMA. Pharmacoinformatics and Breed-Based De Novo Hybridization Studies to Develop New Neuraminidase Inhibitors as Potential Anti-Influenza Agents. Molecules. 2023; 28(18):6678. https://doi.org/10.3390/molecules28186678
Chicago/Turabian StyleLotfi, Bourougaa, Ouassaf Mebarka, Bader Y. Alhatlani, Emad M. Abdallah, and Sarkar M. A. Kawsar. 2023. "Pharmacoinformatics and Breed-Based De Novo Hybridization Studies to Develop New Neuraminidase Inhibitors as Potential Anti-Influenza Agents" Molecules 28, no. 18: 6678. https://doi.org/10.3390/molecules28186678
APA StyleLotfi, B., Mebarka, O., Alhatlani, B. Y., Abdallah, E. M., & Kawsar, S. M. A. (2023). Pharmacoinformatics and Breed-Based De Novo Hybridization Studies to Develop New Neuraminidase Inhibitors as Potential Anti-Influenza Agents. Molecules, 28(18), 6678. https://doi.org/10.3390/molecules28186678