Abstract
In the Central Nervous System (CNS), Nitric Oxide (NO) is mainly biosynthesized by neuronal Nitric Oxide Synthase (nNOS). The dysregulated activation of nNOS in neurons is critical in the development of different conditions affecting the CNS. The excessive production of NO by nNOS is responsible for a number of proteins’ post-translational modifications (PTMs), which can lead to aberrant biochemical pathways, impairing CNS functions. In this review, we briefly revise the main implications of dysregulated nNOS in the progression of the most prevalent CNS neurodegenerative disorders, i.e., Alzheimer’s disease (AD) and Parkinson’s disease, as well as in the development of neuronal disorders. Moreover, a specific focus on compounds able to modulate nNOS activity as promising therapeutics to tackle different neuronal diseases is presented.
1. Introduction
The enzyme neuronal Nitric Oxide Synthase (nNOS) is an oxidoreductase primarily expressed in the nervous system and it is responsible for the conversion of L-Arg into L-Citr and Nitric Oxide (NO). This is a pleiotropic molecule, playing essential roles in mammalians [1]. In the nervous system, it mediates neuronal signaling as an unconventional neurotransmitter, since it is a gaseous molecule, not stored in synaptic vesicles but synthesized on demand by neurons [2,3]. Moreover, NO is a radical molecule, which can react with a variety of biological substrates both by nitrosylation, e.g., the direct reaction of NO with a reactant, or nitrosation, e.g., the indirect transfer of NO to biological nucleophiles via nitrosonium ions (NO+), which are preliminary generated from NO oxidation [4]. Under oxidative stress conditions, NO can react with the superoxide anion (O2−), thus forming Reactive Nitrogen Species (RNS), such as the peroxynitrite (ONOO−), which can generate potent oxidant intermediates responsible, for example, for proteins’ nitration. In any case, NO can regulate the neuronal physiologic and pathologic responses by finely tuning multiple pathways. Basically, they can be categorized as “canonical pathways” and “non-canonical pathways” (Figure 1). The first group are initiated by the nitrosylation of the metalloenzyme soluble guanylate cyclase (sGC), which catalyzes the production of cyclic guanosine monophosphate (cGMP) from triphosphate guanosine (GTP) [5,6]. Then, cGMP mediates downstream effects mainly through the interaction with protein kinase G (PKG), phosphodiesterase (PDE), and cGMP-gated cation channels [7,8]. NO can affect its downstream targets also through “non-canonical pathways”, i.e., inducing post-translational modifications (PTMs) of various proteins (Figure 1). In particular, S-nitrosylation, e.g., the reaction of NO with reactive cysteine thiols to form nitrosothiol groups, is a determining mechanism of NO-mediated signaling in the brain [9]. Being the primary source of NO in the Central Nervous System (CNS), nNOS is implicated in both physiological and pathological responses of neuronal cells [10,11]. Besides nNOS, there are also the endothelial NOS (eNOS) and the inducible NOS (iNOS), which are two other homolog NOS isoforms, playing a different role in the brain. In fact, eNOS is almost exclusively expressed in the vascular endothelial cells where it regulates vascular homeostasis [12], while iNOS is poorly found in some specific regions of the healthy brain, such as the hypothalamus, thalamus, and the amygdaloid nuclei, but is largely expressed mainly in the microglia after pro-inflammatory stimuli, as part of the innate immunity response [13,14]. The overexpression of iNOS is a major feature in different CNS diseases associated with neuroinflammation, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) [15], as well as in several psychiatric illnesses [16]. The consequent overproduction of NO is responsible not only for neurotoxicity due its conversion in the above-mentioned RNS, but it is also implicated in the loss of blood–brain barrier (BBB) integrity, inducing its increased permeability to different substances [17]. It is interesting to note that specific drugs for the therapy of AD, such as donezepil [18], or long-term treatment with the antidepressants fluoxetine or vortioxetine [19] can prevent the overexpression of iNOS connected to the oxidative stress observed in animal models of AD. The implications of the excessive production of NO by iNOS in the development of neuroinflammation and its specific role in CNS diseases have been extensively studied in recent years and they have been reported elsewhere [20,21].
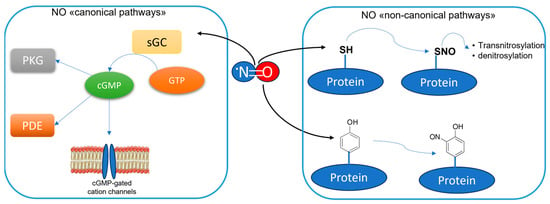
Figure 1.
NO signaling pathways in the brain. The “canonical pathways” of NO signaling are initiated through the stimulation of soluble guanylate cyclase (sGC). The produced cGMP mediates downstream signaling mainly by interacting with PKG, PDE, and cGMP-gated cation channels. The “non-canonical pathways” of NO signaling involve the proteins’ post-translational modifications. Mainly, they are protein nitrosylation, which can lead to the transnitrosylation of other proteins as well as to their denitrosylation by denitrosilases, and protein nitrotyrosination.
In this review, given the prominent role of nNOS-derived NO in the brain, we briefly revise the main implications of dysregulated nNOS in the initiation and progression of the most prevalent CNS neurodegenerative disorders, i.e., AD and PD, as well as in the development of neuronal disorders. Moreover, a specific focus on compounds able to modulate nNOS activity as promising therapeutics to tackle different neuronal diseases is presented.
2. The Overstimulation of nNOS in CNS Disease Development
nNOS shares a similar architecture to eNOS and iNOS, being a homodimer in which each monomer contains an N-terminal oxygenase domain hosting the catalytic site, and a reductase domain containing the cofactors binding sites (Figure 2). The two domains are connected by a Ca2+-calmodulin (CaM) binding sequence and, specifically for nNOS, the oxygenase domain is also connected to the postsynaptic density protein, discs-large, ZO-1 (PDZ) domain, which is characterized by a β-hairpin sequence. Through the PDZ domain, nNOS can interact with other proteins, triggering different cascades of protein–protein interactions [22]. This also determines the specific subcellular localization and functions of nNOS. Indeed, nNOS can be found both in the neurons’ cytosol as well as bound to the postsynaptic membrane through the interaction with the postsynaptic density protein-95 (PSD-95)-NMDAR complex, or with other different proteins, such as Capon, syntrophin, or postsynaptic density-93 (PSD-93) [23,24,25]. These proteins regulate the activation of nNOS and its physiological functions.
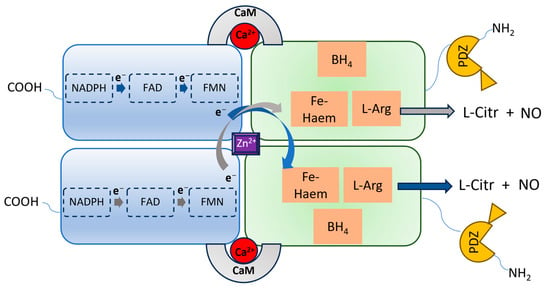
Figure 2.
Representation of the nNOS homodimeric architecture. Each monomer has a carboxy-terminal reductase domain and an amino-terminal oxygenase domain, containing the L-Arg binding site and linked to the PDZ domain. In the reductase domain, electrons via the NADPH-FAD-FMN transport chain reach the heme iron of the opposite monomer oxygenase domain, facilitated by CaM, enabling the oxidation of L-arginine to L-citrulline, accompanied by the release of NO.
In physiological conditions, the stimulated N-methyl-D-aspartate receptors (NMDARs) mediate the intracellular influx of Ca2+, which can bind the CaM sequence of nNOS, initiating its activity. The produced NO can behave as an anterograde or retrograde neurotransmitter, regulating memory, learning, and synaptic plasticity [26,27,28]. Indeed, nNOS-derived NO induces the full expression of c-Fos, Egr-1, Arc, and brain-derived neurotrophic factor (BDNF), which are key proteins associated with neuroplasticity. This was observed both in cortical cultures after bicuculline-evoked synaptic activity, and in in vivo mice models of experience-dependent plasticity in the whisker barrel cortex [29,30]. In particular, the signaling pathways involved in this effect include GMP, PKG, extracellular-signal-regulated kinase (ERK), and calcium–calmodulin (CaM)-dependent protein kinase II (CaMKII) [29].
However, in the early phases of pathological conditions such as stroke, AD, and PD, as well as in other neuropsychiatric conditions such as epilepsy and autism, an overactivation of NMDARs can be observed, followed by an excessive Ca2+ influx, with a loss of its homeostasis (Figure 3). These events lead to the prolonged overstimulation of nNOS, due to excessive intracellular Ca2+ levels [31,32]. The overproduced NO contributes to the development of such diseases by mediating proteins’ PTM, such as nitrotyrosination (Tyr-NO2), i.e., the reaction of tyrosine residues with ONOO−, and cysteine nitrosylation (SNO) (Figure 1). In the specific diseases’ context, these PTMs are mostly pathological, leading to proteins’ loss- or gain-of-function.
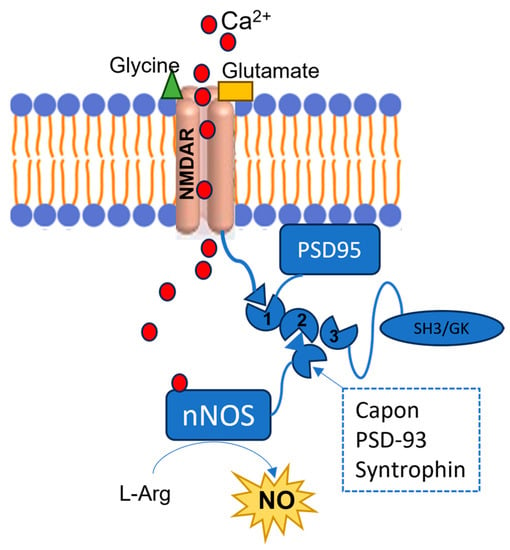
Figure 3.
Representation of the NMDARs-PSD-95-nNOS complex. The red circles represent the Ca2+ ions, the green triangle represents a glycine molecule, and the yellow rectangle represents a glutamate molecule. NMDARs are bound to the PDZ1 domain of the PSD95, and when they are stimulated by the interaction with glycine and glutamate, they allow Ca2+ intracellular influx. This cation binds the nNOS CaM, and the enzyme interacts with the PSD95’s PDZ2 domain by its PDZ β-hairpin motif or with other proteins such as Capon, synthropin, or PSD-93. As a consequence, the enzyme’s activity is stimulated, although an excessive nNOS activation is observed in different SNC diseases, due to the NMDARs’ overstimulation.
2.1. NO-Mediated PTM and Alzheimer Disease
AD is a progressive type of dementia characterized by the aggregation into β-sheets of the amyloid β peptide (Aβ), which is derived from the cleavage of the amyloid precursor protein (APP). Further biological markers of this disease are the intracellular neurofibrillary tangles caused by the excessive phosphorylation of the tau protein. The only FDA-approved AD medications are the acetylcholinesterase inhibitors (AChEIs) donepezil, galantamine, and rivastigmine, and the NMDA antagonist memantine (Figure 4) [33]. Despite the many clinical trials, no new drug has received FDA approval in the last 20 years, and, among the reasons of the clinical trial failures, the inadequate comprehension of the pathophysiology of the AD is a widely accepted explanation [34,35].
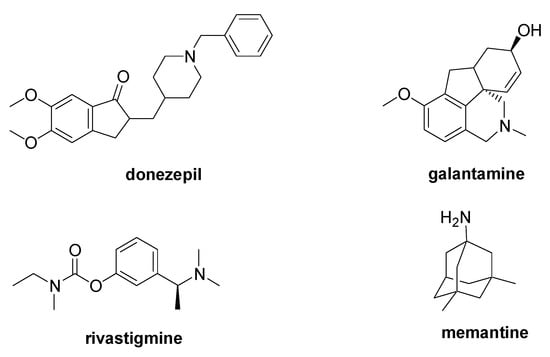
Figure 4.
Chemical structure of the FDA-approved drugs for AD therapy.
In general, cerebral regions of AD patients, specifically the hippocampus and the cerebral cortex, display higher Tyr-NO2 levels, and there is a positive correlation between nNOS expression and neurofibrillary tangles in neurons, as well as amyloid plaque accumulations and nitrergic neurons [36,37,38]. Moreover, the activity of gamma-secretase is affected by nitrotyrosination [39]. Together with β-site APP cleaving enzyme type 1 (BACE1), gamma-secretase is responsible for the production of Aβ1–40 or Aβ1–42 peptides. In AD patients, it has been demonstrated that the nitrotyrosination of this enzyme leads to the imbalanced production of Aβ1–42, which is more prone to aggregate and toxic species [40]. Furthermore, Aβ1–42 nitrotyrosination was also demonstrated to increase aggregates’ stability and toxicity [41]. In AD, the nitrotyrosination of important neuronal metabolic enzymes is also observed, such as lactate dehydrogenase and triosephosphate isomerase (TPI), with a reduction in their activity and important metabolic changes [42]. It was reported that nitro-TPI contributes to intracellular neurofibrillary tangle formation [42].
Besides nitrotyrosination, the extensive S-nitrosylation of proteins is also crucial for synaptic function and neuronal survival and it is associated with AD development [43,44]. Indeed, SNO protein modifications may induce further protein misfolding and neuronal and synaptic damage, leading to mitochondrial stress. In safe cells, these modifications are reversible thanks to the presence of antioxidants such as glutathione and de- and trans-nitrosylating enzymes [45,46]. However, this balance is compromised in the developing AD due to the excessive NO released from the overstimulated nNOS, which leads to massive SNO-proteins. For example, the S-nitrosylation at Cys 83/157 of Cyclic-dependent kinase 5 (CDK5), which is responsible for the cleavage of p35 to p25, upregulates the kinase activity, leading to dendritic spine loss and neuronal apoptosis [47,48]. It was observed that S-nitrosylation of the insulin-degrading enzyme (IDE) inhibits its activity to degrade Aβ [49], while S-nitrosylation of vesicular acetylcholine transporter (VAChT) and vesicular glutamate transporter 1 (VGLUT1) worsens the ACh turnover [50].
In addition, aberrant transnitrosylation reactions, i.e., transfer of the NO group from one protein to another, are highly implicated in AD synaptic loss due to activation of the alternative biochemical network to the physiologic functions of the involved enzymes [51].
2.2. NO-Mediated PTM and Parkinson Disease
PD is a degenerative condition of the brain associated with motor symptoms (rigidity, tremor, bradykinesia, and postural instability) and non-motor disorders (apathy, depression, cognitive dysfunction, and sleep disorders). The main cause of PD is the loss of dopaminergic neurons in the substantia nigra [52] due to the aggregation of the α-synuclein, a protein that regulates the trafficking and release of neurotransmitter vesicles [53]. Currently, there is no therapy to modify the course of PD, with its treatment only being palliative to alleviate the motor and non-motor symptoms that occur during the disease’s development. The administration of the levodopa (Figure 5) is considered the principal therapeutic approach to restore dopamine levels [54], and it can be combined with carbidopa or benserazide (Figure 5), two decarboxylase inhibitors useful to increase levodopa bioavailability [55]. Moreover, the simultaneous treatment with monoamine oxidase B inhibitors such as rasagiline, safinamide, and selegiline is recommended to increase dopamine levels (Figure 5) [55]. Entacapone and tolcapone, two catechol-O-methyltransferase inhibitors, are also used to promote the gastrointestinal absorption of levodopa [55].
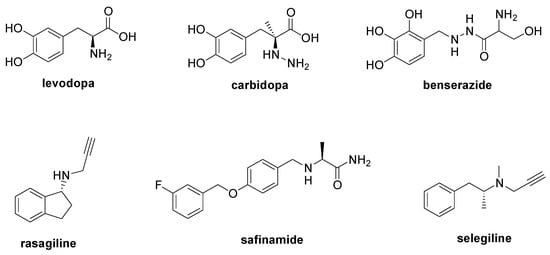
Figure 5.
Chemical structures of approved drugs used in the management of PD.
Although the loss of dopaminergic neurons is a well-established mechanism involved in PD, the reasons leading to the initiation of this process are still unknown. Different studies have reported the accumulation of 3-nitrotyrosinated proteins and a neuronal upregulation of nNOS in cells isolated from PD patients [56,57,58].
It was demonstrated that α-synuclein nitrotyrosination induces its aggregation and inhibits its interaction with dopamine vesicles [59]. Moreover, the nitration of tyrosine hydrolase (TH) seems to be implicated in PD onset. This enzyme is involved in catecholamine synthesis from tyrosines, and its activity is impaired through nitration, lessening dopamine availability [60]. On the contrary, it was reported that TH nitrosylation at Cys 279 enhances its enzymatic activity both in vitro and in vivo, confirming the important role of NO in the subtle regulation of proteins involved in PD progression [61]. It was reported that the overproduced NO is responsible for the S-nitrosylation of different, other proteins, such as the disulfide isomerase and microtubule-associated protein 1b, as well as of CDK5, impairing axo-dendritic function and neurite length [62]. Moreover, the S-nitrosylation of parkin, a protein involved in the degradation of specific substrates, reduces its activity, and consequently neurotoxic proteins can accumulate, leading to ER stress [62]. These proteins’ PTMs alter network connectivity, which is associated with cognitive decline and neuronal death.
2.3. NO-Mediated PTM and Neurological Disorders
NO plays an important role in neurodevelopment, and its altered signaling appears implicated in the progression of a variety of neurodevelopmental and neuropsychiatric diseases. NO can either facilitate or suppress synaptic plasticity, depending on the brain area, concentration, and cellular environment [11,63]; therefore, both overactivation and downregulation of nNOS can be implied in the development of such diseases.
Recently, interesting studies have put in light connections between the specific genetic mutation of the Shank3 gene occurring in autism, a condition associated with deficits in communication and social skills, and excessive NO synthesis, which is responsible for aberrant protein nitrosation and S-nitrosylation [64,65,66,67]. Elevated levels of different SNO proteins functionally involved in the synaptic vesicle cycle, neurotransmission, and glutamatergic pathway, such as protein phosphatase catalytic subunit α-Ppp3ca, syntaxin-1a, vesicle-associated membrane protein 3, and others, were found in Shank3 KO mouse models [66,67]. Collectively, these observations provide insights into the specific pathological role of dysregulated NO production in autism spectrum disorders.
Epilepsy is the most common neurological disease and it was reported that nNOS-derived NO is neurotoxic in the epileptic brain, due to the formation of peroxynitrite after its reaction with the superoxide radical, triggering PTZ kindling epilepsy-induced neural damage [68]. Indirect evidence suggests that the inhibition of nNOS in pilocarpine-induced temporal lobe epilepsy mice can protect against hippocampal neuronal injuries by increasing neuropeptide Y expression, which has been implicated in energy homeostasis and neuroprotection [69]. Moreover, overexpression of NO and lipid peroxidation was reported in the brain of pentylenetetrazol (PTZ)-induced epilepsy rats, and antioxidant treatment normalized their levels [70]. Specific NO-mediated PTMs are implicated in epilepsy; for example, NO is responsible for type 1 ryanodine receptor (RyR1) S-nitrosylation, inducing Ca2+ release from the endoplasmic reticulum through the Ca2 + release channel, and worsening the disease progression [71].
In an animal model of epilepsy, the kainate receptor, specifically glutamate ionotropic receptor kainate type subunit 2 (GluK2), undergoes S-nitrosylation, and SNO-GluK2 further potentiates calcium influx [72].
3. Inhibitors of nNOS as Modulators of CNS Disease Development
A potential therapeutic approach to rebalance the dysregulated NO signaling occurring in neuronal diseases is the inhibition of nNOS activity [73]. This can be achieved using two different pharmacologic tools: nNOS inhibitors and inhibitors of the PSD95-nNOS interaction.
3.1. nNOS Inhibitors
In the last two decades, several compounds able to inhibit nNOS activity have been reported [74]. Since they compete with the natural substrate L-Arg for the catalytic binding site, the very first generation of NOS inhibitors were aminoacidic analogs of L-Arg, but they were unselective, blocking all the three NOS isoforms with potential severe side-effects. Therefore, non-aminoacidic substrate analogs were developed containing some structural requirements (Figure 6). In particular, they show a molecular moiety able to mimic the guanidino group of L-Arg, which is anchored to the highly conserved Glu residue in the enzyme catalytic site by means of bidentate H-bondings [75]. This moiety is linked to a rigid central core, which is usually aromatic and/or heterocyclic, and can give interactions with the heme protoporphyrin [76]. Then, there is a final tail, which usually contains a polar and/or ionizable group able to extend out of the catalytic site, where there are the major aminoacidic differences between the three NOS isoforms [77]. Among the other derivatives, sterically hindered amidines [78], thienylcarbamidines [79], indazoles [80,81,82], aminopyridines [83], and aminoquinolines [84] gave promising results as nNOS inhibitors, although further efforts are required to modulate these compounds’ pharmacokinetic properties. Indeed, a limiting factor in the clinical evaluation of nNOS inhibitors is their high polarity and basicity, which prevent their blood–brain barrier permeability.
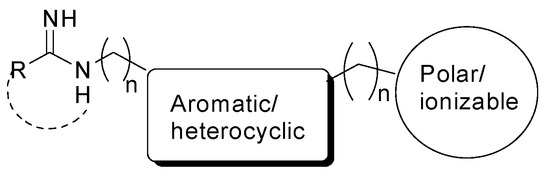
Figure 6.
General pharmacophoric structure of an nNOS inhibitor.
Thanks to its tissue specificity and good pharmacokinetics, 7-Nitroindazole (7-NI, Figure 7) has been studied in different models of neuronal diseases. It has been reported that short-term 7-NI treatment can prevent the acute degeneration of dopaminergic neurons caused by different neurotoxins [85,86]. Moreover, this compound is able to slow down the progressive neurodegeneration in a Parkinson’s disease in vivo model [87]. Very recently, 7-NI was also used to demonstrate the pathological role of NO in autism development, and the in vivo administration of this compound to a Shank3 mouse (M1) model of autism disease led to the recovery of a normal behavioral phenotype [67].
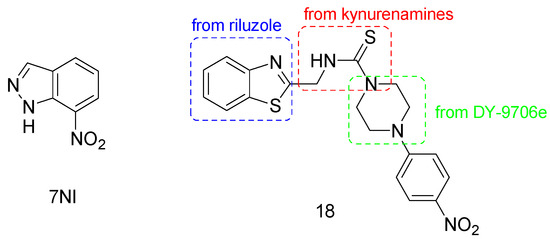
Figure 7.
Chemical structure of nNOS inhibitors.
Recent promising results in the management of PD neurodegeneration were obtained by the new nNOS inhibitor 18 (Figure 7) [88]. It is a benzothiazole derivative containing a piperazine-1-carbothioamide moiety, designed by means of a hybridization approach of compounds directed against molecular targets involved in the progression of PD, i.e., pramipexole and riluzole, kynurenamine-based nNOS inhibitors, and the calmodulin inhibitor DY-9706e [89,90]. It was found that 18 allows the recovery of motor and non-motor functions in an induced unilaterally lesioned rat model of PD. Moreover, this compound modulated the oxidative stress markers, leading to increased levels of dopamine and lessening glutamate and nitrite ion levels.
3.2. PSD95-nNOS Interaction Inhibitors
The subcellular localization and activity of nNOS are influenced by the interaction of its PDZ domain with different proteins bearing this moiety as well (Figure 3) [10]. In particular, the PSD95-nNOS interaction is crucial for synaptic plasticity [24] and it is considered a potential biological target to modulate the overactivation of nNOS occurring in neuronal diseases [91]. In fact, the PSD95 can also bind the NMDARs by means of its PDZ1 domain, and if they are overstimulated, as in many neurodegenerative diseases, they allow the influx of high amounts of Ca2+ into neuronal cells, triggering the nNOS interaction with PSD95’s PDZ2 and its consequent overactivation. In particular, the nNOS-PDZ domain interacts with the PSD95-PDZ2 domain by means of its β-hairpin motif (Figure 2 and Figure 3) [11,92]. Therefore, peptides and small molecules have been designed to prevent the PSD95-nNOS interaction, either by binding the PDZ2 domain of PSD95 or by targeting the nNOS-PDZ domain.
AVLX-144 (Figure 8) is a peptide-based molecule able to bind the PSD95’s PDZ1 and PDZ2 domains simultaneously, blocking the protein binding to both NMDAR and nNOS, with a Ki of 4.6 nM. AVLX-144 is able to permeate the BBB and has proven to be neuroprotective in mice with focal ischemic brain damage [93]. Moreover, it has also been used in a model of cortical spreading depression, a condition associated with stroke and migraine aura, inducing an improvement in neuronal depolarization [94]. Further, AVLX-144 significantly reduced neurodegeneration, cytoskeletal degradation, peroxynitrite formation, and astrogliosis in an in vivo model of epilepsy [95]. Currently, this compound is under clinical development by Avilex Pharma (Copenhagen, Denmark).
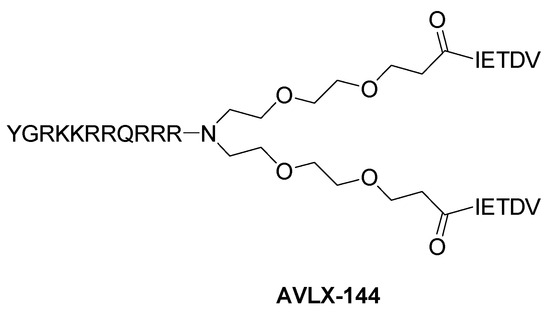
Figure 8.
Structure of the peptide-based PSD95-nNOS interaction inhibitor AVLX-144.
To limit the potential drawbacks due to the potential interactions of these inhibitors with the PDZ domain of other proteins, new cyclic peptides based on the nNOS β-hairpin motif were designed, aiming to optimize the binding with the PSD-95 through more specific interactions [96]. Through the identification of the critical residues in the cyclic nNOS β-hairpin peptide responsible for maintaining high affinity to the PSD-95-PDZ2, two nonproteinogenic AA cyclic peptides were developed with an approximately 100-fold increased binding affinity compared with the wild-type peptide.
Small molecules directed toward the nNOS β-hairpin motif have also been reported, in order to specifically avoid its interaction with PSD95. Starting from the observation that the nNOS β-finger adopts a suitable conformation to bind the PDZ2 domain of PSD95 only if an internal salt bridge between Asp62 on the nNOS PDZ domain and Arg121 on the nNOS β-finger domain is formed, a series of compounds showing hydrophobic ring A and hydrophilic ring B bearing a carboxyl group were prepared (Figure 9) [97]. Ring A forms hydrophobic interactions with Leu107 or Phe111 on the β-finger, hindering the conformational change of nNOS PDZ, while the carboxyl group on ring B interacts with Arg121, disrupting the intra-nNOS salt bridge. In particular, compound ZL006 (Figure 9) was the most potent nNOS-PSD95 inhibitor (IC50 = 0.082 µM), not affecting NMDA receptor function, nNOS expression, or nNOS catalytic activity [97]. ZL006 inhibited NMDA-receptor-mediated NO synthesis and was neuroprotective in neurons and animal ischemic stroke models [97]. Moreover, ZL006 attenuates hemorrhage-induced thalamic pain in mice, it reduces Aβ1–42-induced neuronal damage and oxidative stress, and it lessens neuronal injury and apoptotic cell death in MPP+-cultured cortical neurons [98,99,100].
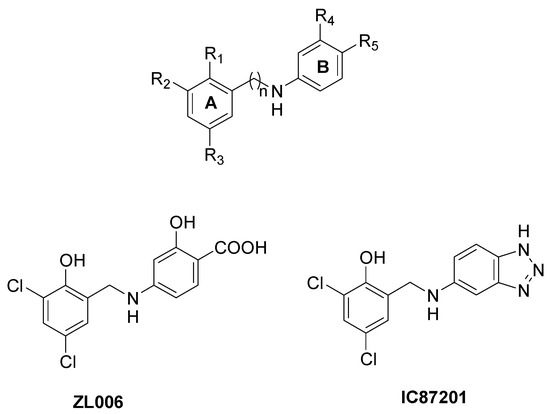
Figure 9.
Chemical structure of small molecules able to selectively bind the nNOS β-finger and of ZL006 and IC87201.
By means of a high-throughput screening approach, another nNOS-PSD95 inhibitor was discovered (IC87201, Figure 9), although its interaction mode has not been fully elucidated [101,102]. This compound has shown great effects in animal models of ischemic stroke, depression, pain, and regenerative repair after stroke [101,103,104].
4. Conclusions
The dysregulated activation of nNOS in neurons is critical in the development of different conditions affecting the SNC. As a downstream event of NMDAR overactivation, an excessive production of NO by nNOS is responsible for a number of proteins’ PTMs, which can lead to aberrant biochemical pathways, impairing SNC functions. In this review, we revise evidence of how nitrotyrosination and cysteine nitrosylation, which are the two major NO-dependent PTMs, are related to the development of different SNC pathological conditions. Moreover, the promising effects that the pharmacological regulation of nNOS activity could have in the management of such diseases are also discussed. However, despite this evidence and due to the multifaceted role of NO both in SNC physiology and pathology, it remains crucial and challenging to understand the chronology of NO-induced PTM in the disease progression. Indeed, the nNOS’ physiologic activity should be preserved due to its role in neuroplasticity, long-term potentiation, and memory, while its overactivation should be tuned by means of molecules endowed with protein and tissue selectivity. In this regard, nNOS-mediated PPIs are supposed to be safer with respect to nNOS inhibitors, since the latter have been associated with side-effects due to their poor isoform selectivity and induction of iNOS [105,106]. In fact, blocking nNOS-PSD95 interactions should avoid any unwanted inhibition of eNOS.
However, more knowledge of the pathophysiology of proteins’ PTMs is needed to hopefully find new effective and safe treatments able to restore an appropriate NO signaling in SNC diseases.
Author Contributions
Conceptualization, C.M. and R.A.; formal analysis, C.M.; resources, C.M.; data curation, C.M.; writing—original draft preparation, C.M.; writing—review and editing, C.M. and R.A.; visualization, C.M. and R.A.; supervision, C.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Murad, F. Discovery of some of the biological effects of nitric oxide and its role in cell signaling. Biosci. Rep. 2004, 24, 452–474. [Google Scholar] [CrossRef]
- Knott, A.B.; Bossy-Wetzel, E. Nitric oxide in health and disease of the nervous system. Antioxid. Redox Signal. 2009, 11, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Picón-Pagès, P.; Garcia-Buendia, J.; Muñoz, F.J. Functions and dysfunctions of nitric oxide in brain. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.A.; da Silva, R.S.; Miranda, K.M.; Switzer, C.H.; Wink, D.A.; Fukuto, J.M. Biological nitric oxide signalling: Chemistry and terminology. Br. J. Pharmacol. 2013, 169, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef]
- Hobbs, A.J. Soluble guanylate cyclase: The forgotten sibling. Trends Pharmacol. Sci. 1997, 18, 484–491. [Google Scholar] [CrossRef]
- Kass, D.A.; Takimoto, E.; Nagayama, T.; Champion, H.C. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc. Res. 2007, 75, 303–314. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Bradley, S.A.; Steinert, J.R. Nitric Oxide-Mediated Posttranslational Modifications: Impacts at the Synapse. Oxid. Med. Cell. Longev. 2016, 2016, 5681036. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, D.Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230. [Google Scholar] [CrossRef]
- Zhu, L.J.; Li, F.; Zhu, D.Y. nNOS and Neurological, Neuropsychiatric Disorders: A 20-Year Story. Neurosci. Bull. 2023, 39, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; Austin, S.A. Neurovascular Protective Function of Endothelial Nitric Oxide—Recent Advances. Circ. J. 2016, 80, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Bechade, C.; Colasse, S.; Diana, M.A.; Rouault, M.; Bessis, A. NOS2 expression is restricted to neurons in the healthy brain but is triggered in microglia upon inflammation. Glia 2014, 62, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Bechade, C.; Pascual, O.; Triller, A.; Bessis, A. Nitric oxide regulates astrocyte maturation in the hippocampus: Involvement of NOS2. Mol. Cell. Neurosci. 2011, 46, 762–769. [Google Scholar] [CrossRef]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef]
- Eissa, N.; Sadeq, A.; Sasse, A.; Sadek, B. Role of Neuroinflammation in Autism Spectrum Disorder and the Emergence of Brain Histaminergic System. Lessons Also for BPSD? Front. Pharmacol. 2020, 11, 886. [Google Scholar] [CrossRef]
- Thiel, V.E.; Audus, K.L. Nitric oxide and blood-brain barrier integrity. Antioxid. Redox Signal. 2001, 3, 273–278. [Google Scholar] [CrossRef]
- Kim, H.G.; Moon, M.; Choi, J.G.; Park, G.; Kim, A.J.; Hur, J.; Lee, K.T.; Oh, M.S. Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology 2014, 40, 23–32. [Google Scholar] [CrossRef]
- Caruso, G.; Grasso, M.; Fidilio, A.; Torrisi, S.A.; Musso, N.; Geraci, F.; Tropea, M.R.; Privitera, A.; Tascedda, F.; Puzzo, D.; et al. Antioxidant Activity of Fluoxetine and Vortioxetine in a Non-Transgenic Animal Model of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 809541. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Justo, A.F.O.; Suemoto, C.K. The modulation of neuroinflammation by inducible nitric oxide synthase. J. Cell. Commun. Signal. 2022, 2, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Merino-Gracia, J.; Costas-Insua, C.; Canales, M.Á.; Rodríguez-Crespo, I. Insights into the c-terminal peptide binding specificity of the pdz domain of neuronal nitric-oxide synthase: Characterization of the interaction with the tight junction protein claudin-3. J. Biol. Chem. 2016, 291, 11581–11595. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Rothe, F.; Canzler, U.; Wolf, G. Subcellular localization of the neuronal isoform of nitric oxide synthase in the rat brain: A critical evaluation. Neuroscience 1998, 83, 259–269. [Google Scholar] [CrossRef]
- Park, J.H.; Straub, V.A.; O’Shea, M. Anterograde signaling by nitric oxide: Characterization and in vitro reconstitution of an identified nitrergic synapse. J. Neurosci. 1998, 18, 5463–5476. [Google Scholar] [CrossRef]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013, 7, 190. [Google Scholar] [CrossRef]
- Paul, V.; Ekambaram, P. Involvement of nitric oxide in learning & memory processes. Indian J. Med. Res. 2011, 133, 471–478. [Google Scholar]
- Gallo, E.F.; Iadecola, C. Neuronal nitric oxide contributes to neuroplasticity-associated protein expression through cGMP, protein kinase G, and extracellular signal-regulated kinase. J. Neurosci. 2011, 31, 6947–6955. [Google Scholar] [CrossRef]
- Moosavi, M.; Abbasi, L.; Zarifkar, A.; Rastegar, K. The role of nitric oxide in spatial memory stages, hippocampal ERK and CaMKII phosphorylation. Pharmacol. Biochem. Behav. 2014, 122, 164–172. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Sandín, S.; Alonso, M.T.; García-Sancho, J. Calcium homoeostasis modulator 1 (CALHM1) reduces the calcium content of the endoplasmic reticulum (ER) and triggers ER stress. Biochem. J. 2011, 437, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Atri, A. Current and future treatments in Alzheimer’s disease. Semin. Neurol. 2019, 39, 227–240. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, M.; Gopinath, P.; Govindaraju, T. Role of Post-translational Modifications in Alzheimer’s Disease. Chembiochem 2020, 21, 1052–1079. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical Analysis of Protein Nitrotyrosine and Dityrosine in the Alzheimer Brain Indicates Region-Specific Accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef]
- Quinn, J.; Davis, F.; Woodward, W.R.; Eckenstein, F. Beta-amyloid plaques induce neuritic dystrophy of nitric oxide-producing neurons in a transgenic mouse model of Alzheimer’s disease. Exp. Neurol. 2001, 168, 203–212. [Google Scholar] [CrossRef]
- Bandookwala, M.; Sengupta, P. 3-Nitrotyrosine: A versatile oxidative stress biomarker for major neurodegenerative diseases. Int. J. Neurosci. 2020, 130, 1047–1062. [Google Scholar] [CrossRef]
- Guix, F.X.; Wahle, T.; Vennekens, K.; Snellinx, A.; Chávez-Gutiérrez, L.; Ill-Raga, G.; Ramos-Fernandez, E.; Guardia-Laguarta, C.; Lleó, A.; Arimon, M.; et al. Modification of γ-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer’s disease. EMBO Mol. Med. 2012, 4, 660–673. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Guivernau, B.; Bonet, J.; Valls-Comamala, V.; Bosch-Morató, M.; Godoy, J.A.; Inestrosa, N.C.; Perálvarez-Marín, A.; Fernández-Busquets, X.; Andreu, D.; Oliva, B.; et al. Amyloid-β peptide nitrotyrosination stabilizes oligomers and enhances NMDAR-mediated toxicity. J. Neurosci. 2016, 36, 11693–11703. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Ill-Raga, G.; Bravo, R.; Nakaya, T.; de Fabritiis, G.; Coma, M.; Pietro Miscione, G.; Villà-Freixa, J.; Suzuki, T.; Fernàndez-Busquets, X.; et al. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain 2009, 132, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Thongboonkerd, V.; Klein, J.B.; Lynn, B.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem. 2003, 85, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.-K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.-i.; Lipton, S.A. Aberrant protein Snitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef]
- Anand, P.; Stamler, J.S. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. 2012, 90, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, G.; Rizza, S.; Montagna, C.; Filomeni, G. Established principles and emerging concepts on the interplay between mitochondrial physiology and S-(De) nitrosylation: Implications in cancer and neurodegeneration. Int. J. Cell Biol. 2012, 2012, 361872. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335. [Google Scholar] [CrossRef]
- Qu, J.; Nakamura, T.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-nitrosylation of Cdk5. Prion 2012, 6, 364–370. [Google Scholar] [CrossRef]
- Cordes, C.M.; Bennett, R.G.; Siford, G.L.; Hamel, F.G. Redox regulation of insulin degradation by insulin-degrading enzyme. PLoS ONE 2011, 6, e18138. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Z.; Tan, H.; Zhu, S.; Sun, Y.; Li, M.X.; Wang, J.F. Nitrosylation of vesicular transporters in brain of amyloid precursor protein/presenilin 1 double transgenic mice. J. Alzheimer’s Dis. 2017, 55, 1683–1692. [Google Scholar] [CrossRef]
- Lipton, S.A. Hidden networks of aberrant protein transnitrosylation contribute to synapse loss in Alzheimer’s disease. Free Radic. Biol. Med. 2022, 193, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P. Understanding the molecular basis of Parkinson’s disease, identification of biomarkers and routes to therapy. Expert. Rev. Proteom. 2010, 2010, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A. Levodopa Therapy for Parkinson’s Disease: Pharmacokinetics and Pharmacodynamics. Mov. Disord. 2015, 30, 64–72. [Google Scholar] [CrossRef]
- Dhanawat, M.; Mehta, D.K.; Gupta, S.; Das, R. Understanding the Pathogenesis Involved in Parkinson’s Disease and Potential Therapeutic Treatment Strategies. Cent. Nerv. Syst. Agents Med. Chem. 2020, 20, 88–102. [Google Scholar] [CrossRef]
- Gatto, E.M.; Riob’o, N.A.; Carreras, M.C.; Chernavsky, A.; Rubio, A.; Satz, M.L.; Poderoso, J.J. Overexpression of neutrophil neuronal nitric oxide synthase in Parkinson’s disease. Nitric Oxide 2000, 4, 534–539. [Google Scholar] [CrossRef]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Joniec, I.; Ciesielska, A.; Kurkowska-Jastrzebska, I.; Przybylkowski, A.; Czlonkowska, A.; Czlonkowski, A. Age- and sex-differences in the nitric oxide synthase expression and dopamine concentration in the murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 2009, 1261, 7–19. [Google Scholar] [CrossRef]
- Burai, R.; Ait Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the role of site-specific nitration of α-synuclein in the pathogenesis of Parkinson’s disease via protein semisynthesis and mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef]
- Blanchard-Fillion, B.; Souza, J.M.; Friel, T.; Jiang, G.C.T.; Vrana, K.; Sharov, V.; Barrón, L.; Schöneich, C.; Quijano, C.; Alvarez, B.; et al. Nitration and Inactivation of Tyrosine Hydroxylase by Peroxynitrite. J. Biol. Chem. 2001, 276, 46017–46023. [Google Scholar] [CrossRef]
- Wang, Y.; Sung, C.C.; Chung, K.K. Novel enhancement mechanism of tyrosine hydroxylase enzymatic activity by nitric oxide through S-nitrosylation. Sci. Rep. 2017, 7, 44154. [Google Scholar] [CrossRef] [PubMed]
- Stykel, M.G.; Ryan, S.D. Nitrosative stress in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Zoupa, E.; Pitsikas, N. The Nitric Oxide (NO) Donor Sodium Nitroprusside (SNP) and Its Potential for the Schizophrenia Therapy: Lights and Shadows. Molecules 2021, 26, 3196. [Google Scholar] [CrossRef] [PubMed]
- Kartawy, M.; Khaliulin, I.; Amal, H. Systems biology reveals S-nitrosylation dependent regulation of mitochondrial functions in mice with Shank3 mutation associated with autism spectrum disorder. Brain Sci. 2021, 11, 677. [Google Scholar] [CrossRef]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Amal, H.; Barak, B.; Bhat, V.; Gong, G.; Joughin, B.A.; Wang, X.; Wishnok, J.S.; Feng, G.; Tannenbaum, S.R. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatr. 2018, 25, 1835–1848. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Ojha, S.K.; Kartawy, M.; Hamoudi, W.; Choudhary, A.; Stern, S.; Aran, A.; Amal, H. The NO Answer for Autism Spectrum Disorder. Adv. Sci. 2023, 10, 2205783. [Google Scholar] [CrossRef]
- Zhu, X.; Dong, J.; Han, B.; Huang, R.; Zhang, A.; Xia, Z.; Chang, H.; Chao, J.; Yao, H. Neuronal nitric oxide synthase contributes to PTZ kindling epilepsy-induced hippocampal endoplasmic reticulum stress and oxidative damage. Front. Cell. Neurosci. 2017, 11, 377. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, Y.; Yang, J.; Zhang, C.; He, Y.; Qi, H.; Zeng, Y.; Zhang, A.; Liu, X.; Zhu, X. Inhibition of neuronal nitric oxide synthase protects against hippocampal neuronal injuries by increasing neuropeptide Y expression in temporal lobe epilepsy mice. Free Radic. Biol. Med. 2022, 188, 45–61. [Google Scholar] [CrossRef]
- Bashkatova, V.; Narkevich, V.; Vitskova, G.; Vanin, A. The influence of anticonvulsant and antioxidant drugs on nitric oxide level and lipid peroxidation in the rat brain during penthylenetetrazole-induced epileptiform model seizures. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 487–492. [Google Scholar] [CrossRef]
- Mikami, Y.; Kanemaru, K.; Okubo, Y.; Nakaune, T.; Suzuki, J.; Shibata, K.; Sugiyama, H.; Koyama, R.; Murayama, T.; Ito, A.; et al. Nitric Oxide-induced Activation of the Type 1 Ryanodine Receptor Is Critical for Epileptic Seizure-induced Neuronal Cell Death. eBioMedicine 2016, 11, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Lu, R.; Dong, G.; Chen, X.; Yun, W.; Zhou, X. The role of S-nitrosylation of kainate-type of ionotropic glutamate receptor 2 in epilepsy induced by kainic acid. J. Neurochem. 2017, 144, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, C.; Amoroso, R. Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders. Neural Regen. Res. 2016, 11, 1731–1734. [Google Scholar] [CrossRef]
- Annedi, S.C. Cell-Permeable Inhibitors of Neuronal Nitric Oxide Synthase Open New Prospects for the Treatment of Neurological Disorders. J. Med. Chem. 2015, 58, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, H.; Flinspach, M.; Poulos, T.L.; Silverman, R.B. Computer modeling of selective regions in the active site of nitric oxide synthases: Implication for the design of isoform-selective inhibitors. J. Med. Chem. 2003, 46, 5700–5711. [Google Scholar] [CrossRef]
- Carrión, M.D.; Rubio-Ruiz, B.; Franco-Montalban, F.; Amoia, P.; Zuccarini, M.C.; De Simone, C.; Camacho, M.E.; Amoroso, R.; Maccallini, C. New amidine-benzenesulfonamides as iNOS inhibitors for the therapy of the triple negative breast cancer. Eur. J. Med. Chem. 2023, 248, 115112. [Google Scholar] [CrossRef]
- Maccallini, C.; Montagnani, M.; Paciotti, R.; Ammazzalorso, A.; De Filippis, B.; Di Matteo, M.; Di Silvestre, S.; Fantacuzzi, M.; Giampietro, L.; Potenza, M.A.; et al. Selective Acetamidine-Based Nitric Oxide Synthase Inhibitors: Synthesis, Docking, and Biological Studies. ACS Med. Chem. Lett. 2015, 6, 635–640. [Google Scholar] [CrossRef]
- Maccallini, C.; Marinelli, L.; Indorf, P.; Cacciatore, I.; Fantacuzzi, M.; Clement, B.; Di Stefano, A.; Amoroso, R. A Novel Prodrug of a nNOS Inhibitor with Improved Pharmacokinetic Potential. ChemMedChem 2020, 15, 2157–2163. [Google Scholar] [CrossRef]
- Annedi, S.C.; Ramnauth, J.; Maddaford, S.P.; Renton, P.; Rakhit, S.; Mladenova, G.; Dove, P.; Silverman, S.; Andrews, J.S.; Felice, M.D.; et al. Discovery of cis-N-(1-(4-(methylamino)cyclohexyl)indolin-6-yl)thiophene-2-carboximidamide: A 1,6-disubstituted indoline derivative as a highly selective inhibitor of human neuronal nitric oxide synthase (nNOS) without any cardiovascular liabilities. J. Med. Chem. 2012, 55, 943–955. [Google Scholar] [CrossRef]
- Moore, P.K.; Bland-Ward, P.A. 7-Nitroindazole: An inhibitor of nitric oxide synthase. Methods Enzymol. 1996, 268, 393–398. [Google Scholar] [CrossRef]
- Maccallini, C.; Di Matteo, M.; Vullo, D.; Ammazzalorso, A.; Carradori, S.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Pandolfi, A.; Supuran, C.T.; et al. Indazole, Pyrazole, and Oxazole Derivatives Targeting Nitric Oxide Synthases and Carbonic Anhydrases. ChemMedChem 2016, 11, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, C.; Gallorini, M.; Sisto, F.; Akdemir, A.; Ammazzalorso, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Carradori, S.; Cataldi, A.; et al. New azolyl-derivatives as multitargeting agents against breast cancer and fungal infections: Synthesis, biological evaluation and docking study. J. Enzyme Inhib. Med. Chem. 2021, 36, 1632–1645. [Google Scholar] [CrossRef]
- Vasu, D.; Li, H.; Hardy, C.D.; Poulos, T.L.; Silverman, R.B. 2-Aminopyridines with a shortened amino sidechain as potent, selective, and highly permeable human neuronal nitric oxide synthase inhibitors. Bioorg. Med. Chem. 2022, 69, 116878. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Reidl, C.T.; Li, H.; Chreifi, G.; Poulos, T.L.; Silverman, R.B. First Contact: 7-Phenyl-2-Aminoquinolines, Potent and Selective Neuronal Nitric Oxide Synthase Inhibitors That Target an Isoform Specific Aspartate. J. Med. Chem. 2020, 63, 4528–4554. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Yokoyama, R.; Shibata, T.; Dawson, V.L.; Dawson, T.M. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridin (MPTP)-induced dopaminergic neurotoxicity. Proc. Natl. Acad. Sci. USA 1996, 93, 4565–4571. [Google Scholar] [CrossRef]
- Yuste, J.E.; Echeverry, M.B.; Ros-Bernal, F.; Gomez, A.; Ros, C.M.; Campuzano, C.M.; Fernandez-Villalba, E.; Herrero, M.T. 7-Nitroindazole down-regulates dopamine/DARPP-32 signaling in neostriatal neurons in a rat model of Parkinson’s disease. Neuropharmacology 2012, 63, 1258–1267. [Google Scholar] [CrossRef]
- Ehara, A.; Nakadate, K.; Sugimoto, H.; Yoshimoto, K.; Ueda, S. Role of neuronal nitric oxide synthase in slowly progressive dopaminergic neurodegeneration in the Zitter rat. Nitric Oxide 2018, 78, 41–50. [Google Scholar] [CrossRef]
- Agrawal, S.; Kumari, R.; Sophronea, T.; Kumari, N.; Luthra, P.M. Design and synthesis of benzo[d]thiazol-2-yl-methyl-4-(substituted)-piperazine-1-carbothioamide as novel neuronal nitric oxide inhibitors and evaluation of their neuroprotecting effect in 6-OHDA-induced unilateral lesioned rat model of Parkinson’s disease. Biomed. Pharmacother. 2022, 156, 113838. [Google Scholar] [CrossRef]
- Chayah, M.; Carrion, M.C.; Gallo, M.A.; Jimenez, R.; Duarte, J.; Camacho, M.E. Development of urea and thiourea kynurenamine derivatives: Synthesis, molecular modeling, and biological evaluation as nitric oxide synthase inhibitors. ChemMedChem 2015, 10, 874–882. [Google Scholar] [CrossRef]
- Hashiguchi, A.; Kawano, T.; Yano, S.; Morioka, M.; Hamada, J.; Sato, T.; Shirasaki, Y.; Ushio, Y.; Fukunaga, K. The neuroprotective effect of a novel calmodulin antagonist, 3-[2-[4-(3-chloro-2-methylphenyl)-1-piperazinyl]ethyl]-5,6-dimethoxy-1-(4-imidazolylmethyl)-1H-indazole dihydrochloride 3.5 hydrate, in transient forebrain ischemia. Neuroscience 2003, 121, 379–386. [Google Scholar] [CrossRef]
- Gu, Y.; Zhu, D. nNOS-mediated protein-protein interactions: Promising targets for treating neurological and neuropsychiatric disorders. J. Biomed. Res. 2021, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.; Clausen, B.H.; Møller, M.; Vestergaard, B.; Chi, C.N.; Round, A.; Sørensen, P.L.; Nissen, K.B.; Kastrup, J.S.; Gajhede, M.; et al. A high-affinity, dimeric inhibitor of PSD-95 bivalently interacts with PDZ1-2 and protects against ischemic brain damage. Proc. Natl. Acad. Sci. USA 2012, 109, 3317–3322. [Google Scholar] [CrossRef]
- Kucharz, K.; Søndergaard Rasmussen, I.; Bach, A.; Strømgaard, K.; Lauritzen, M. PSD-95 uncoupling from NMDA receptors by Tat- N-dimer ameliorates neuronal depolarization in cortical spreading depression. J. Cereb. Blood Flow Metab. 2017, 37, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Nobili, P.; Cipelletti, B.; Cagnoli, C.; Zambon, S.; Locatelli, D.; de Curtis, M.; Battaglia, G.S. Targeting PSD95-nNOS interaction by Tat-N-dimer peptide during status epilepticus is neuroprotective in MAM-pilocarpine rat model. Neuropharmacology 2019, 153, 82–97. [Google Scholar] [CrossRef]
- Balboa, J.R.; Essig, D.J.; Ma, S.; Karer, N.; Clemmensen, L.S.; Pedersen, S.W.; Joerger, A.C.; Knapp, S.; Østergaard, S.; Strømgaard, K. Development of a Potent Cyclic Peptide Inhibitor of the nNOS/PSD-95 Interaction. J. Med. Chem. 2023, 66, 976–990. [Google Scholar] [CrossRef]
- Zhou, L.; Li, F.; Xu, H.B.; Luo, C.X.; Wu, H.Y.; Zhu, M.M.; Lu, W.; Ji, X.; Zhou, Q.G.; Zhu, D.Y. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat. Med. 2010, 16, 1439–1443. [Google Scholar] [CrossRef]
- Cai, W.H.; Wu, S.G.; Pan, Z.; Xiao, J.; Li, F.; Cao, J.; Zang, W.; Tao, Y. Disrupting interaction of PSD-95 with nNOS attenuates hemorrhage-induced thalamic pain. Neuropharmacology 2018, 141, 238–248. [Google Scholar] [CrossRef]
- Tao, W.Y.; Yu, L.J.; Jiang, S.; Cao, X.; Chen, J.; Bao, X.Y.; Li, F.; Xu, Y.; Zhu, X.L. Neuroprotective effects of ZL006 in Aβ1-42-treated neuronal cells. Neural Regen. Res. 2020, 15, 2296–2305. [Google Scholar] [CrossRef]
- Hu, W.; Guan, L.S.; Dang, X.B.; Ren, P.Y.; Zhang, Y.L. Small-molecule inhibitors at the PSD-95/nNOS interface attenuate MPP+-induced neuronal injury through Sirt3 mediated inhibition of mitochondrial dysfunction. Neurochem. Int. 2014, 79, 57–64. [Google Scholar] [CrossRef]
- Florio, S.K.; Loh, C.; Huang, S.M.; Iwamaye, A.E.; Kitto, K.F.; Fowler, K.W.; Treiberg, J.A.; Hayflick, J.S.; Walker, J.M.; Fairbanks, C.A.; et al. Disruption of nNOS-PSD95 protein-protein interaction inhibits acute thermal hyperalgesia and chronic mechanical allodynia in rodents. Br. J. Pharmacol. 2009, 158, 494–506. [Google Scholar] [CrossRef]
- Bach, A.; Pedersen, S.W.; Dorr, L.A.; Vallon, G.; Ripoche, I.; Ducki, S.; Lian, L.Y. Biochemical investigations of the mechanism of action of small molecules ZL006 and IC87201 as potential inhibitors of the nNOS-PDZ/PSD-95-PDZ interactions. Sci. Rep. 2015, 5, 12157. [Google Scholar] [CrossRef]
- Doucet, M.V.; Levine, H.; Dev, K.K.; Harkin, A. Small-molecule inhibitors at the PSD-95/nNOS interface have antidepressant like properties in mice. Neuropsychopharmacology 2013, 38, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.X.; Lin, Y.H.; Qian, X.D.; Tang, Y.; Zhou, H.H.; Jin, X.; Ni, H.Y.; Zhang, F.Y.; Qin, C.; Li, F.; et al. Interaction of nNOS with PSD-95 negatively controls regenerative repair after stroke. J. Neurosci. 2014, 34, 13535–13548. [Google Scholar] [CrossRef]
- Luo, C.X.; Zhu, X.J.; Zhou, Q.G.; Wang, B.; Wang, W.; Cai, H.H.; Sun, Y.J.; Hu, M.; Jiang, J.; Hua, Y.; et al. Reduced neuronal nitric oxide synthase is involved in ischemia-induced hippocampal neurogenesis by up-regulating inducible nitric oxide synthase expression. J. Neurochem. 2007, 103, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Dao, V.T.; Elbatreek, M.H.; Fuchß, T.; Grädler, U.; Schmidt, H.H.H.W.; Shah, A.M.; Wallace, A.; Knowles, R. Nitric Oxide Syn-thase Inhibitors into the Clinic at Last. Handb. Exp. Pharmacol. 2021, 264, 169–204. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).