
Synthesis of α-Chloroarylacetic Acid via Electrochemical Carboxylation of α,α-Dichloroarylmethane Derivatives


 ,
,  and
and
Abstract
:
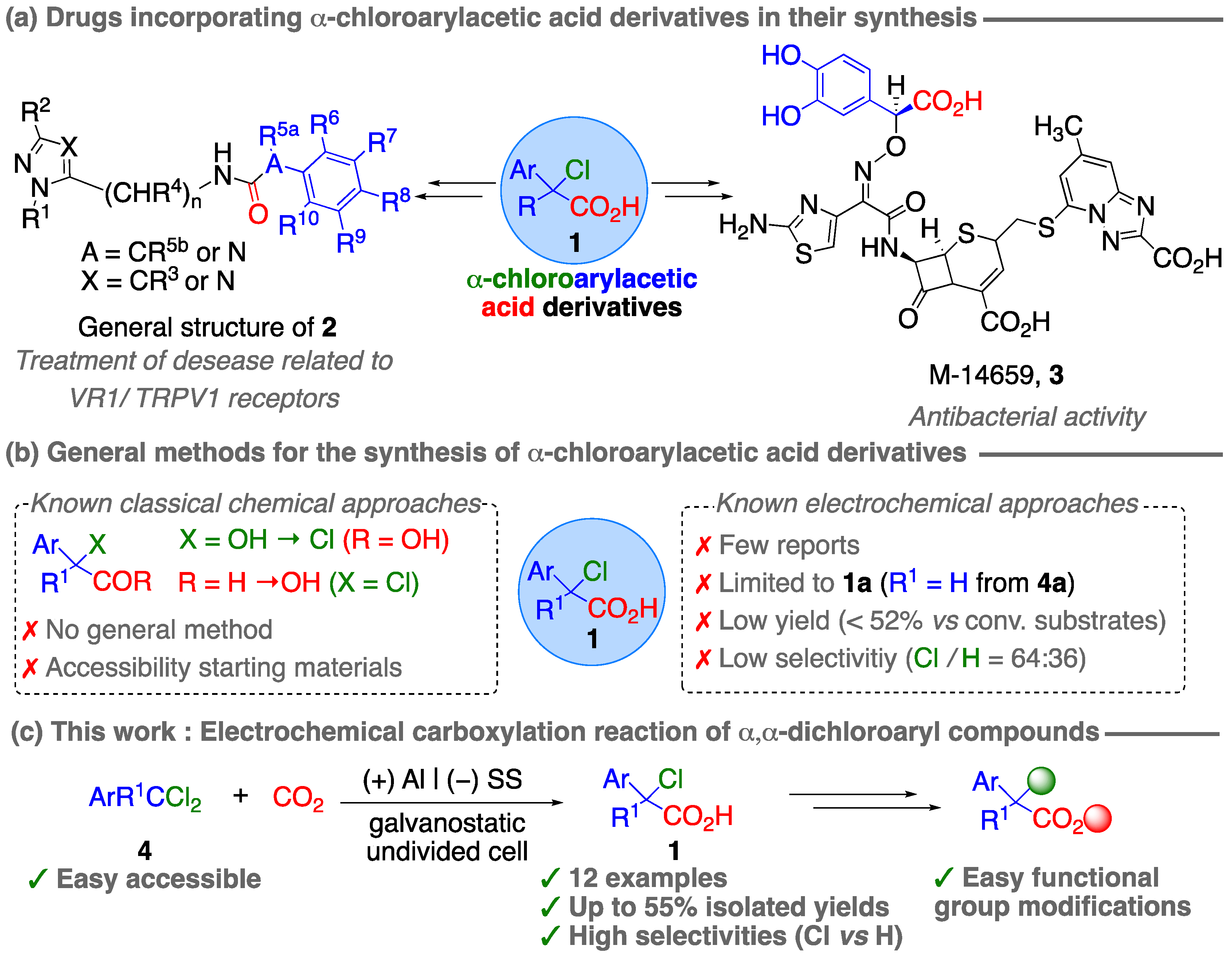
1. Introduction
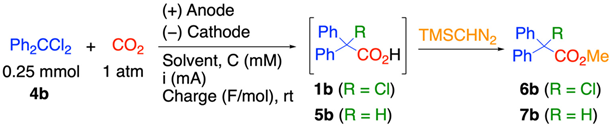
2. Results
2.1. Influence of Reaction Parameters
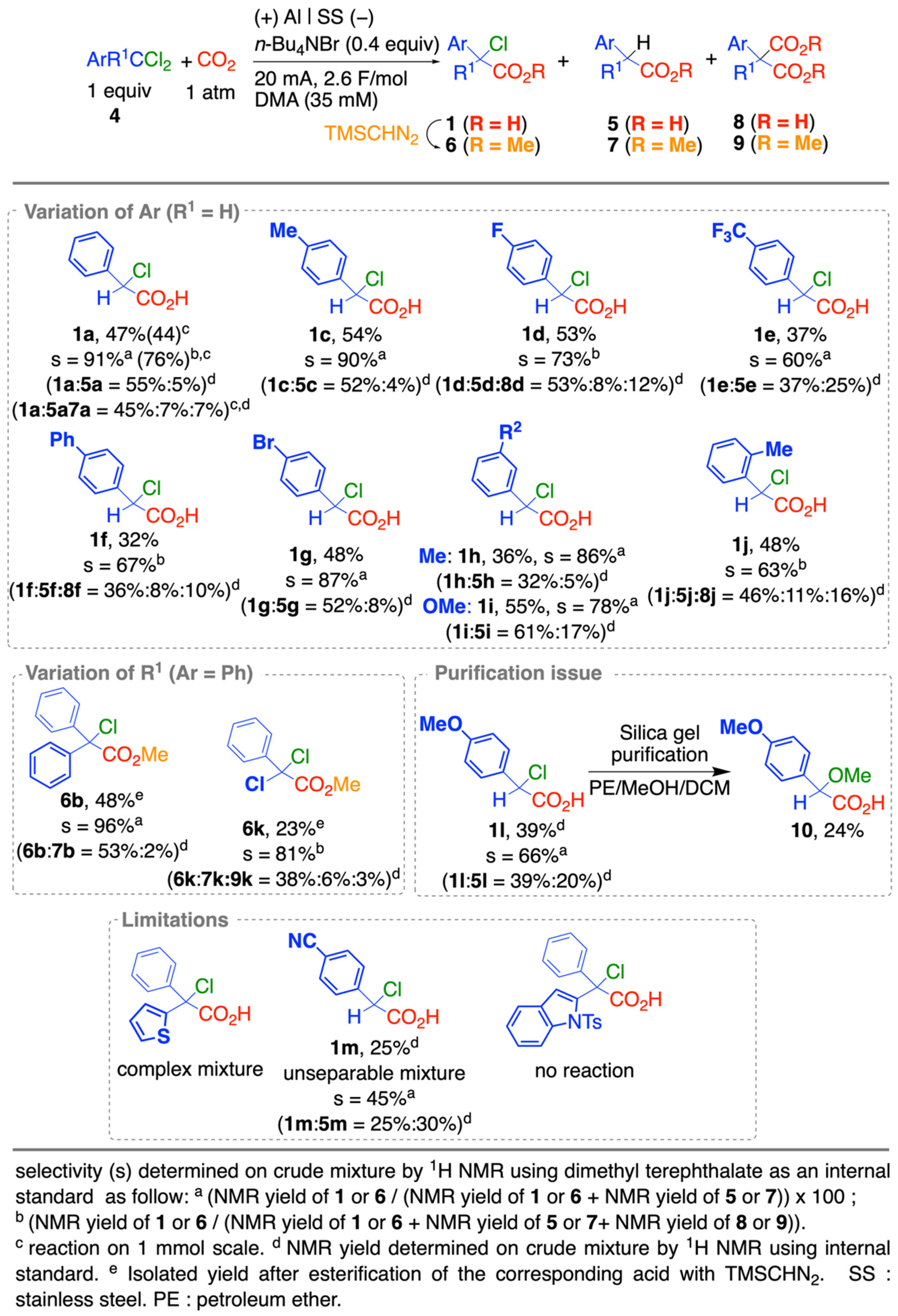
2.2. Scope and Limitations
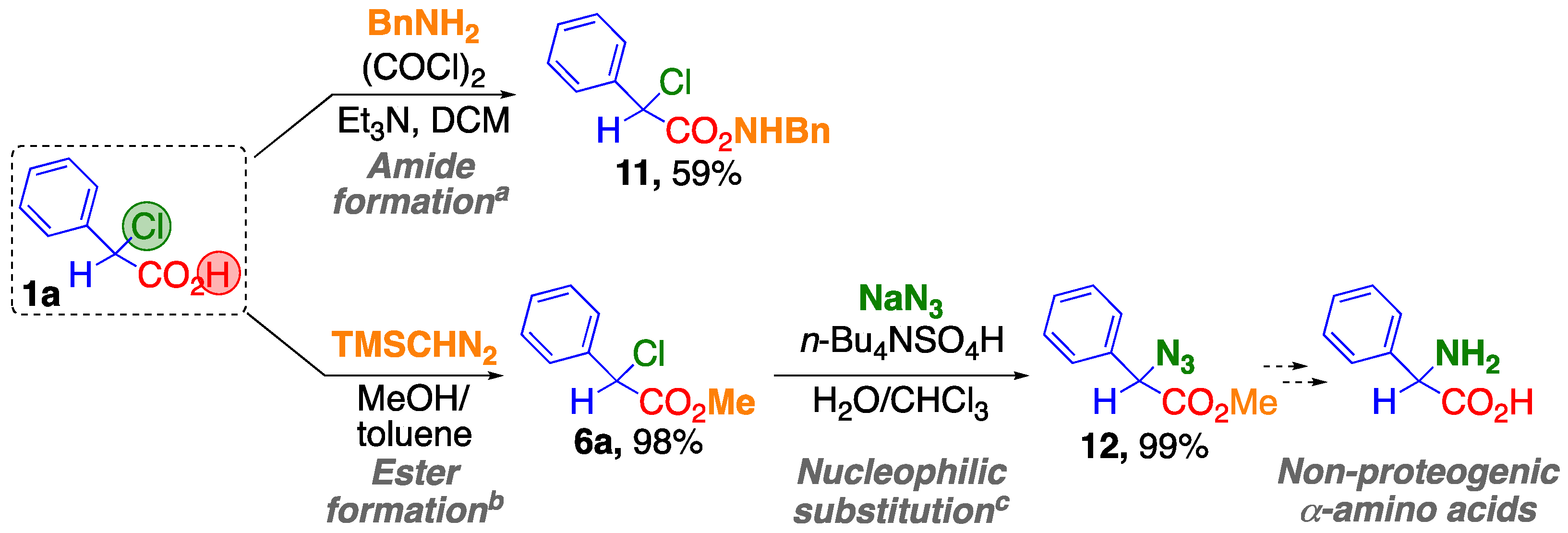
2.3. Versatile Transformations of the α-Chlorophenylacetic Acid 1a
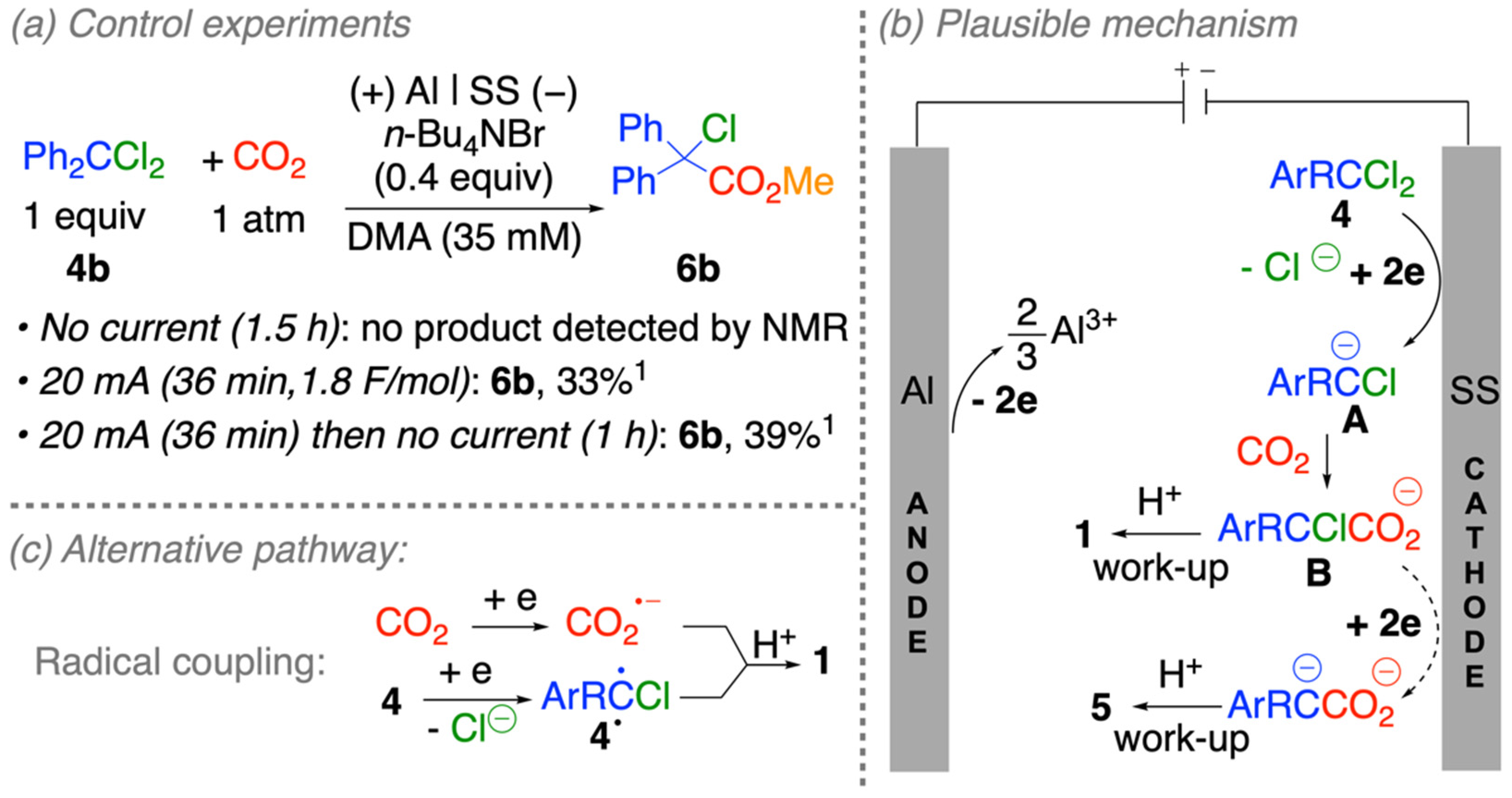
2.4. Mechanistic Considerations
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of α,α-Dichloro Benzyl Derivatives 4 [25]
3.3. General Procedure A for the Synthesis of α-Chloroarylacetic Acid Derivatives 1
1 mmol Procedure
3.4. Synthesis of Methyl Methyl 2-Chloro-2-phenylacetate 6a
3.5. General Procedure B for the Synthesis of Methyl 2-Chloro-2-phenylacetate Derivatives 6b,k
3.6. Procedure for the Synthesis of 2-Methoxy-2-(4-methoxyphenyl)acetic Acid 10
3.7. Procedure for the Synthesis of N-Benzyl-2-chloro-2-phenylacetamide 11
3.8. Procedure for the Synthesis of Methyl 2-Azido-2-phenylacetate 12
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Frank, R.; Bahrenberg, G.; Christoph, T.; Schiene, K.; DeVry, J.; Damann, N.; Frormann, S.; Lesch, B.; Lee, J.; Kim, Y.-S.; et al. Preparation of Substituted Phenylamine Derivatives for Use as Vanilloid Receptor Ligands. PCT International Application WO2010/127856. 11 November 2010. [Google Scholar]
- Murakami, K.; Ohashi, M.; Matsunaga, A.; Yamamoto, I.; Nohira, H. Asymmetric transformation of a racemic α-(phthalimidooxy)arylacetic ester by a combination of preferential crystallization and simultaneous racemization. Chirality 1993, 5, 41–48. [Google Scholar] [CrossRef]
- Paraskevas, S.M.; Paraskevas, M.S. Chlorination and oxidation of some aldehydes by H2O2 and diphenic acid · CuCl2 complex. Catal. Commun. 2004, 5, 687–690. [Google Scholar] [CrossRef]
- Carnell, A.J.; Kirk, R.; Smith, M.; McKenna, S.; Lian, L.Y.; Gibson, R. Inhibition of human alpha-methylacyl CoA racemase (AMACR): A target for prostate cancer. ChemMedChem 2013, 8, 1643–1647. [Google Scholar] [CrossRef]
- Otero, M.D.; Batanero, B.; Barba, F. CO2 anion–radical in organic carboxylations. Tetrahedron Lett. 2006, 47, 2171–2173. [Google Scholar] [CrossRef]
- Murtaza, A.; Qamar, M.A.; Saleem, K.; Hardwick, T.; Zia Ul, H.; Shirinfar, B.; Ahmed, N. Renewable Electricity Enables Green Routes to Fine Chemicals and Pharmaceuticals. Chem. Rec. 2022, 22, e202100296. [Google Scholar] [CrossRef] [PubMed]
- Senboku, H.; Katayama, A. Electrochemical carboxylation with carbon dioxide. Curr. Opin. Green Sustain. Chem. 2017, 3, 50–54. [Google Scholar] [CrossRef]
- De Sarkar, S.; Maiti, D.; Halder, A.; Mahanty, K. Recent Developments in the Electroreductive Functionalization of Carbon–Halogen Bonds. Synthesis 2022, 55, 400–416. [Google Scholar] [CrossRef]
- Yu, Z.; Shi, M. Recent advances in the electrochemically mediated chemical transformation of carbon dioxide. Chem. Commun. 2022, 58, 13539–13555. [Google Scholar] [CrossRef]
- Wang, S.; Feng, T.; Wang, Y.; Qiu, Y. Recent Advances in Electrocarboxylation with CO2. Chem. Asian J. 2022, 17, e202200543. [Google Scholar] [CrossRef]
- Liu, X.-F.; Zhang, K.; Tao, L.; Lu, X.-B.; Zhang, W.-Z. Recent advances in electrochemical carboxylation reactions using carbon dioxide. Green Chem. Eng. 2022, 3, 125–137. [Google Scholar] [CrossRef]
- Xie, S.L.; Gao, X.T.; Wu, H.H.; Zhou, F.; Zhou, J. Direct Electrochemical Defluorinative Carboxylation of gem-Difluoroalkenes with Carbon Dioxide. Org. Lett. 2020, 22, 8424–8429. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Gambino, S.; Filardo, G.; Greco, G.; Gulotta, A. Electrochemical carboxylation of benzal chloride. Tetrahedron Lett. 1984, 25, 4307–4308. [Google Scholar] [CrossRef]
- Silvestri, G.; Gambino, S.; Filardo, G.; Tiitta, M.; Sjöström, M.; Wold, S.; Berglind, R.; Karlsson, B. Use of Sacrificial Anodes in Synthetic Electrochemistry. Processes Involving Carbon Dioxide. Acta Chem. Scand. 1991, 45, 987–992. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemical Methods since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Meyer, T.H.; Choi, I.; Tian, C.; Ackermann, L. Powering the Future: How Can Electrochemistry Make a Difference in Organic Synthesis? Chem 2020, 6, 2484–2496. [Google Scholar] [CrossRef]
- Schotten, C.; Nicholls, T.P.; Bourne, R.A.; Kapur, N.; Nguyen, B.N.; Willans, C.E. Making electrochemistry easily accessible to the synthetic chemist. Green Chem. 2020, 22, 3358–3375. [Google Scholar] [CrossRef]
- Pollok, D.; Waldvogel, S.R. Electro-organic synthesis—A 21(st) century technique. Chem. Sci. 2020, 11, 12386–12400. [Google Scholar] [CrossRef]
- Zhu, C.; Ang, N.W.J.; Meyer, T.H.; Qiu, Y.; Ackermann, L. Organic Electrochemistry: Molecular Syntheses with Potential. ACS Cent. Sci. 2021, 7, 415–431. [Google Scholar] [CrossRef]
- Leech, M.C.; Lam, K. A practical guide to electrosynthesis. Nat. Rev. Chem. 2022, 6, 275–286. [Google Scholar] [CrossRef]
- Chaussard, J.; Folest, J.-C.; Nedelec, J.-Y.; Perichon, J.; Sibille, S.; Troupel, M. Use of Sacrificial Anodes in Electrochemical Functionalization of Organic Halides. Synthesis 1990, 369–381. [Google Scholar] [CrossRef]
- Oudeyer, S.; Léonel, E.; Paugam, J.P.; Nédélec, J.-Y. Copper-catalyzed electrosynthesis of 1-acyl-2,2-diphenylcyclopropanes and their behaviour in acidic medium. Tetrahedron 2003, 59, 1073–1081. [Google Scholar] [CrossRef]
- Oudeyer, S.; Léonel, E.; Paugam, J.P.; Nédélec, J.Y. Epoxide formation by indirect electroreductive coupling between aldehydes or ketones and activated gem-dichloro compounds. Synthesis 2004, 389–400. [Google Scholar] [CrossRef]
- Oudeyer, S.; Léonel, E.; Paugam, J.P.; Sulpice-Gaillet, C.; Nédélec, J.-Y. Formation of polysubstituted chlorocyclopropanes from electrophilic olefins and activated trichloromethyl compounds. Tetrahedron 2006, 62, 1583–1589. [Google Scholar] [CrossRef]
- Fergus, S.; Eustace, S.J.; Hegarty, A.F. Nitrile ylide dimerization: Investigation of the carbene reactivity of nitrile ylides. J. Org. Chem. 2004, 69, 4663–4669. [Google Scholar] [CrossRef]
- Rolla, F. Sodium borohydride reactions under phase-transfer conditions: Reduction of azides to amines. J. Org. Chem. 2002, 47, 4327–4329. [Google Scholar] [CrossRef]
- Green, J.E.; Bender, D.M.; Jackson, S.; O’Donnell, M.J.; McCarthy, J.R. Mitsunobu approach to the synthesis of optically active α,α-disubstituted amino acids. Org. Lett. 2009, 11, 807–810. [Google Scholar] [CrossRef]
- Sengmany, S.; Léonel, E.; Paugam, J.P.; Nédélec, J.Y. Cyclopropane formation by copper-catalyzed indirect electroreductive coupling of activated olefins and activated α,α,α-trichloro or gem-dichloro compounds. Synthesis 2002, 533–537. [Google Scholar] [CrossRef]
- Lamy, E.; Nadjo, L.; Saveant, J.M. Standard potential and kinetic parameters of the electrochemical reduction of carbon dioxide in dimethylformamide. J. Electroanal. Chem. 1977, 78, 403–407. [Google Scholar] [CrossRef]
- An, J.; Tang, X.; Moore, J.; Lewis, W.; Denton, R.M. Phosphorus(V)-catalyzed deoxydichlorination reactions of aldehydes. Tetrahedron 2013, 69, 8769–8776. [Google Scholar] [CrossRef]
- Kelly, B.D.; Lambert, T.H. Aromatic cation activation of alcohols: Conversion to alkyl chlorides using dichlorodiphenylcyclopropene. J. Am. Chem. Soc. 2009, 131, 13930–13931. [Google Scholar] [CrossRef]
- Timperley, C.M.; Bird, M.; Gore, S.J.; Lindsay, C.D.; Rice, H.; Tattersall, J.E.H.; Whitmore, C.L.; Green, A.C. 3-Quinuclidinyl-alpha-methoxydiphenylacetate: A multi-targeted ligand with antimuscarinic and antinicotinic effects designed for the treatment of anticholinesterase poisoning. Toxicol. Lett. 2020, 325, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Dong, H.; Ma, Y.; Shao, K.; Li, Y.; Wu, X.; Wang, S.; Shao, Y.; Zhao, W. Structure-activity relationships study of neolamellarin A and its analogues as hypoxia inducible factor-1 (HIF-1) inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 2327–2331. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Aliyu, M.A.; Gao, Z.; Li, T.; Dong, W.; Li, J.; Shi, E.; Tang, W. General Synthesis of Chiral alpha,alpha-Diaryl Carboxamides by Enantioselective Palladium-Catalyzed Cross-Coupling. Org. Lett. 2020, 22, 4974–4978. [Google Scholar] [CrossRef] [PubMed]
- Stockhammer, L.; Weinzierl, D.; Bogl, T.; Waser, M. Enantioselective alpha-Chlorination Reactions of in Situ Generated C1 Ammonium Enolates under Base-Free Conditions. Org. Lett. 2021, 23, 6143–6147. [Google Scholar] [CrossRef]
- Golas, P.L.; Tsarevsky, N.V.; Matyjaszewski, K. Structure–Reactivity Correlation in “Click” Chemistry: Substituent Effect on Azide Reactivity. Macromol. Rapid Commun. 2008, 29, 1167–1171. [Google Scholar] [CrossRef]
- Munaretto, L.S.; Dos Santos, C.Y.; Gallo, R.D.C.; Okada, C.Y., Jr.; Deflon, V.M.; Jurberg, I.D. Visible-Light-Mediated Strategies to Assemble Alkyl 2-Carboxylate-2,3,3-Trisubstituted beta-Lactams and 5-Alkoxy-2,2,4-Trisubstituted Furan-3(2H)-ones Using Aryldiazoacetates and Aryldiazoketones. Org. Lett. 2021, 23, 9292–9296. [Google Scholar] [CrossRef]
- Léonel, E.; Paugam, J.-P.; Heintz, M.; Nédélec, J.-Y. A Simple and Efficient Procedure for the Preparation of Benzal Chlorides and Benzal Bromides. Synth. Commun. 1999, 29, 4015–4024. [Google Scholar] [CrossRef]
- Polat, E.; Cakici, M. Deoxygenative Chlorination of Aldehydes and Alcohols with Dichloromethyl Methyl Ether and TiCl4. Eur. J. Org. Chem. 2022, 2022, e202201106. [Google Scholar] [CrossRef]
- Zhao, Z.; Kulkarni, K.G.; Murphy, G.K. Synthesis of Aryldihalomethanes by Denitrogenative Dihalogenation of Benzaldehyde Hydrazones. Adv. Synth. Catal. 2017, 359, 2222–2228. [Google Scholar] [CrossRef]
- Tao, J.; Tran, R.; Murphy, G.K. Dihaloiodoarenes: α,α-dihalogenation of phenylacetate derivatives. J. Am. Chem. Soc. 2013, 135, 16312–16315. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Anode/Cathode | Solvent | Conc. (mM) | Charge (F/mol) | i (mA) | NMR Yield 6b/7b (%) 1 | Selectivity, s (%) 2 |
| 1 | Al/NiFoam | ACN | 83 | 2.6 | 20 | 23/12 | 66 |
| 2 | Co/NiFoam | ACN | 83 | 2.6 | 20 | 13/11 | 54 |
| 3 | Zn/NiFoam | ACN | 83 | 2.6 | 20 | 4/7 | 36 |
| 4 | Mg/NiFoam | ACN | 83 | 2.6 | 20 | 6/17 | 26 |
| 5 | Al/NiFoam | THF | 83 | 2.6 | 20 | nr/- | - |
| 6 | Al/NiFoam | DMF | 83 | 2.6 | 20 | 17/11 | 61 |
| 7 | Al/NiFoam | DMA | 83 | 2.6 | 20 | 42/20 | 68 |
| 8 | Al/Glassy C | DMA | 83 | 2.6 | 20 | 32/26 | 55 |
| 9 | Al/Graphite | DMA | 83 | 2.6 | 20 | 25/24 | 51 |
| 10 | Al/Ni | DMA | 83 | 2.6 | 20 | 36/10 | 78 |
| 11 | Al/SS | DMA | 83 | 2.6 | 20 | 54/5 | 91 |
| 12 | Al/SS | DMA | 83 | 1.8 | 20 | 33/traces | >99 |
| 13 | Al/SS | DMA | 83 | 3.2 | 20 | 54/11 | 83 |
| 14 | Al/SS | DMA | 83 | 2.6 | 10 | 39/6 | 87 |
| 15 | Al/SS | DMA | 83 | 2.6 | 30 | 50/8 | 86 |
| 16 | Al/SS | DMA | 125 | 2.6 | 20 | 29/7 | 81 |
| 17 | Al/SS | DMA | 35 | 2.6 | 20 | 55(48) 3/<5 | >92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maret, C.; David, N.; Pierrot, D.; Léonel, E.; Levacher, V.; Brière, J.-F.; Oudeyer, S. Synthesis of α-Chloroarylacetic Acid via Electrochemical Carboxylation of α,α-Dichloroarylmethane Derivatives. Molecules 2023, 28, 6704. https://doi.org/10.3390/molecules28186704
Maret C, David N, Pierrot D, Léonel E, Levacher V, Brière J-F, Oudeyer S. Synthesis of α-Chloroarylacetic Acid via Electrochemical Carboxylation of α,α-Dichloroarylmethane Derivatives. Molecules. 2023; 28(18):6704. https://doi.org/10.3390/molecules28186704
Chicago/Turabian StyleMaret, Corentin, Nicolas David, David Pierrot, Eric Léonel, Vincent Levacher, Jean-François Brière, and Sylvain Oudeyer. 2023. "Synthesis of α-Chloroarylacetic Acid via Electrochemical Carboxylation of α,α-Dichloroarylmethane Derivatives" Molecules 28, no. 18: 6704. https://doi.org/10.3390/molecules28186704