
Ubiquitin-Dependent and Independent Proteasomal Degradation in Host-Pathogen Interactions



Abstract
:
1. Introduction
2. Ub-Mediated Proteasomal Degradation
3. Specific Requirements for Efficient Proteasomal Degradation
4. Ub-Dependent versus Ub-Independent Protein Degradation
5. Human Pathogens Hijacking the UPS

{kind=link}
{kind=link}
| Species | Pathogenic Factor | Host Protein | Targeted Pathway/Effect | Ref. |
|---|---|---|---|---|
| IAV | Itch | virus endocytosis | [83] | |
| EBV | EBNA-1 | abolishing MHC class I-restricted cytotoxic T lymphocyte responses | [85] | |
| cholera | toxin | Derlin-1, HRD1 | hijacking the retrotranslocation from ER to the cytosol | [91] |
| HCMV | US11 | TMEM129 | degradation of MHC-I signaling molecules | [93] |
| mouse γ-herpesvirus 68 | mK3 | TMEM129 | degradation of MHC-I signaling molecules | [95] |
| HCMV | US2 | TRC8 | degradation of MHC-I signaling molecules | [96] |
| HIV-1 | Vpu | βTrCP | triggering CD4 degradation | [98] |
| HIV-1 | Vpu | BST-2 | virion release | [99,100] |
| HIV-1 | Vpu | NF-κB, AP1 | suppression of NF-κB activation | [101] |
| HIV-1 | Vif | APOBEC3G/F | antagonization of the APOBEC3 family | [103] |
| HIV-1 | Vif | STAT1/3 | inhibition of the production of type I interferons | [104] |
| HIV-1/2 | Vpr | CRL4A (DCAF1) | enhancing lentiviral reverse transcription | [105] |
| HSV-2 | UL56 | Nedd4 | viral egress | [108] |
| varicella–zoster virus | ORF0 | ITCH | viral egress | [109] |
| HCMV | UL42 | ITCH | viral egress | [109] |
| human herpesvirus 6A | U24 | ITCH | viral egress | [109] |
| HSV-1 | ICP0 | PMLNB | abolishing the silencing of the viral genome | [110] |
| HSV-1 | ICP0 | USP7 | abolishing ICP0 proteasomal degradation | [135] |
| HSV-1 | UL36 | TRAF3 IκBα | inhibition of IFN-β production suppression of NF-κB activation | [113,114] |
| MuHV-4 | mLANA | MYC | antagonizing SCF(Fbw7)-mediated proteasomal degradation of Myc | [112] |
| SARS-CoV2 | PLpro | ISG15 | antagonizing IRF3 and NF-κB signaling | [115] |
| SARS-CoV | PLpro | polyUB chains | antagonizing IRF3 and NF-κB signaling | [115] |
| SARS-CoV | Orf9b | MAVS, TRAF3, TRAF6, DLK1 | counteracting antiviral response abolishing mitochondrial fission | [117] |
| S. enterica | SteD | MHC-II | abrogation of antigen presentation | [84] |
| S. enterica | SSeL | IκBα | suppression of NF-κB activation | [119] |
| S. enterica | AvrA | IκBα, β-catenin | suppression of NF-κB activation | [118] |
| S. enterica | AvrA | Beclin-1 | suppression of autophagy | [121] |
| S. enterica | SopA | TRIM56, TRIM65 | inhibition of the production of type I interferons | [122] |
| S. flexneri | IpaH1.4 | LUBAC | suppression of NF-κB activation | [123,124] |
| S. flexneri | IpaH7.8 | glomulin | induction of macrophage cell death | [125] |
| S. flexneri | IpaH0722 | TRAF2 | suppression of NF-κB activation | [126] |
| S. flexneri | IpaH9.8 | GBP | protection of bacterial motility | [127] |
| L. pneumophila | AnkB | LCV | providing supply of amino acids | [130] |
| L. pneumophila | SdeA | Ub | impairing mitophagy, TNF signaling, proteasomal degradation of host proteins | [132] |
| L. pneumophila | LubX | Cdc2-like kinase 1 | Unknown | [133] |
| L. pneumophila | LubX | Legionella SidH | temporal control of infection | [134] |
6. Plant Pathogens and the UPS
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hershko, A.; Ciechanover, A.; Varshavsky, A. The ubiquitin system. Nat. Med. 2000, 6, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef]
- Fernández-Cruz, I.; Reynaud, E. Proteasome Subunits Involved in Neurodegenerative Diseases. Arch. Med. Res. 2021, 52, 1–14. [Google Scholar] [CrossRef]
- Coux, O.; Zieba, B.A.; Meiners, S. The proteasome system in health and disease. In Advances in Experimental Medicine and Biology; Barrio, R., Sutherland, J., Rodriguez, M., Eds.; Springer Nature Switzerland AG Springer: Berlin/Heidelberg, Germany, 2020; Volume 1233, pp. 55–100. [Google Scholar]
- Osei-Amponsa, V.; Walters, K.J. Proteasome substrate receptors and their therapeutic potential. Trends Biochem. Sci. 2022, 47, 950–964. [Google Scholar] [CrossRef]
- Kolla, S.D.D.; Ye, M.; Mark, K.G.; Rapé, M. Assembly and function of branched ubiquitin chains. Trends Biochem. Sci. 2022, 47, 759–771. [Google Scholar] [CrossRef]
- Tracz, M.M.; Bialek, W.; Białek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell. Mol. Biol. Lett. 2021, 26, 1. [Google Scholar] [CrossRef]
- Greene, E.R.; Dong, K.C.; Martin, A. Understanding the 26S proteasome molecular machine from a structural and conformational dynamics perspective. Curr. Opin. Struct. Biol. 2020, 61, 33–41. [Google Scholar] [CrossRef]
- Deshmukh, F.K.; Ben-Nissan, G.; Olshina, M.A.; Füzesi-Levi, M.G.; Polkinghorn, C.; Arkind, G.; Leushkin, Y.; Fainer, I.; Fleishman, S.J.; Tawfik, D.; et al. Allosteric regulation of the 20S proteasome by the Catalytic Core Regulators (CCRs) family. Nat. Commun. 2023, 14, 3126. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, E.; Htet, Z.M.; Bard, J.A.M.; Dong, K.C.; Martin, A. Ubiquitin modulates 26S proteasome conformational dynamics and promotes substrate degradation. Sci. Adv. 2022, 8, eadd9520. [Google Scholar] [CrossRef] [PubMed]
- Pack, C.-G.; Yukii, H.; Toh-e, A.; Kudo, T.; Tsuchiya, H.; Kaiho, A.; Sakata, E.; Murata, S.; Yokosawa, H.; Sako, Y.; et al. Quantitative live-cell imaging reveals spatio-temporal dynamics and cytoplasmic assembly of the 26S proteasome. Nat. Commun. 2014, 5, 3396. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Mao, Y. Structure, Dynamics and Function of the 26S Proteasome. In Macromolecular Protein Complexes III: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2021; Volume 96, pp. 1–151. [Google Scholar]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, X.; Elsasser, S.; Stocks, B.B.; Tian, G.; Lee, H.; Shi, Y.; Zhang, N.; De Poot, S.A.H.; Tuebing, F.; et al. Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 2016, 351, aad9421. [Google Scholar] [CrossRef]
- Chojnacki, M.; Mansour, W.; Hameed, D.S.; Singh, R.K.; El Oualid, F.; Rosenzweig, R.; Nakasone, M.A.; Yu, Z.; Glaser, F.; Kay, L.E.; et al. Polyubiquitin-Photoactivatable Crosslinking Reagents for Mapping Ubiquitin Interactome Identify Rpn1 as a Proteasome Ubiquitin-Associating Subunit. Cell Chem. Biol. 2017, 24, 443–457.e6. [Google Scholar] [CrossRef]
- De la Peña, A.H.; Goodall, E.A.; Gates, S.N.; Lander, G.C.; Martin, A. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis–driven translocation. Science 2018, 362, eaav0725. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Mansour, W.; Nakasone, M.A.; Von Delbrück, M.; Yu, Z.; Krutauz, D.; Reis, N.; Kleifeld, O.; Sommer, T.; Fushman, D.; Glickman, M.H. Disassembly of Lys11 and mixed linkage polyubiquitin conjugates provides insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. J. Biol. Chem. 2015, 290, 4688–4704. [Google Scholar] [CrossRef]
- Bech-Otschir, D.; Helfrich, A.; Enenkel, C.; Consiglieri, G.; Seeger, M.; Holzhütter, H.G.; Dahlmann, B.; Kloetzel, P.M. Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat. Struct. Mol. Biol. 2009, 16, 219–225. [Google Scholar] [CrossRef]
- Kim, H.T.; Goldberg, A.L. The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J. Biol. Chem. 2017, 292, 9830–9839. [Google Scholar] [CrossRef] [PubMed]
- Peth, A.; Kukushkin, N.; Bossé, M.; Goldberg, A.L. Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J. Biol. Chem. 2013, 288, 7781–7790. [Google Scholar] [CrossRef] [PubMed]
- Reichard, E.L.; Chirico, G.G.; Dewey, W.J.; Nassif, N.D.; Bard, K.E.; Millas, N.E.; Kraut, X.D.A. Substrate ubiquitination controls the unfolding ability of the proteasome. J. Biol. Chem. 2016, 291, 18547–18561. [Google Scholar] [CrossRef]
- Cundiff, M.D.; Hurley, C.M.; Wong, J.D.; Boscia, J.A.; Bashyal, A.; Rosenberg, J.; Reichard, E.L.; Nassif, N.D.; Brodbelt, J.S.; Kraut, D.A. Ubiquitin receptors are required for substrate-mediated activation of the proteasome’s unfolding ability. Sci. Rep. 2019, 9, 14506. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, D.; Walinda, E.; Fukada, H.; Sugase, K.; Shirakawa, M. Ubiquitylation Directly Induces Fold Destabilization of Proteins. Sci. Rep. 2016, 6, 39453. [Google Scholar] [CrossRef]
- Ji, Z.; Li, H.; Peterle, D.; Paulo, J.A.; Ficarro, S.B.; Wales, T.E.; Marto, J.A.; Gygi, S.P.; Engen, J.R.; Rapoport, T.A. Translocation of polyubiquitinated protein substrates by the hexameric Cdc48 ATPase. Mol. Cell 2022, 82, 570–584.e8. [Google Scholar] [CrossRef]
- Ciechanover, A. Reprint of “A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes”. Biochem. Biophys. Res. Commun. 2012, 425, 565–570. [Google Scholar] [CrossRef]
- Sun, H.; Mali, S.M.; Singh, S.K.; Meledin, R.; Brik, A.; Kwon, Y.T.; Kravtsova-Ivantsiv, Y.; Bercovich, B.; Ciechanover, A. Diverse fate of ubiquitin chain moieties: The proximal is degraded with the target, and the distal protects the proximal from removal and recycles. Proc. Natl. Acad. Sci. USA 2019, 116, 7805–7812. [Google Scholar] [CrossRef]
- Zhang, S.; Zou, S.; Yin, D.; Zhao, L.; Finley, D.; Wu, Z.; Mao, Y. USP14-regulated allostery of the human proteasome by time-resolved cryo-EM. Nature 2022, 605, 567–574. [Google Scholar] [CrossRef]
- Fujisawa, R.; Rivera, C.P.; Labib, K.P.M. Multiple UBX proteins reduce the ubiquitin threshold of the mammalian p97-UFD1-NPL4 unfoldase. Elife 2022, 11, e76763. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.-J.J.; Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Haakonsen, D.L.; Rape, M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019, 29, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, 365, eaax1033. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Tsuchiya, H.; Yamagata, A.; Okatsu, K.; Tanaka, K.; Saeki, Y.; Fukai, S. Structural insights into ubiquitin recognition and Ufd1 interaction of Npl4. Nat. Commun. 2019, 10, 5708. [Google Scholar] [CrossRef]
- Pan, M.; Yu, Y.; Ai, H.; Zheng, Q.; Xie, Y.; Liu, L.; Zhao, M. Mechanistic insight into substrate processing and allosteric inhibition of human p97. Nat. Struct. Mol. Biol. 2021, 28, 614–625. [Google Scholar] [CrossRef]
- Williams, C.; Dong, K.C.; Arkinson, C.; Martin, A. The Ufd1 cofactor determines the linkage specificity of polyubiquitin chain engagement by the AAA+ ATPase Cdc48. Mol. Cell 2023, 83, 759–769.e7. [Google Scholar] [CrossRef]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef]
- Varshavsky, A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011, 20, 1298–1345. [Google Scholar] [CrossRef]
- Verma, R.; McDonald, H.; Yates, J.R.; Deshaies, R.J. Selective degradation of ubiquitinated Sic1 by purified 26S proteasome yields active S phase cyclin-Cdk. Mol. Cell 2001, 8, 439–448. [Google Scholar] [CrossRef]
- Han, J.H.; Batey, S.; Nickson, A.A.; Teichmann, S.A.; Clarke, J. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Biol. 2007, 8, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Hagai, T.; Levy, Y. Ubiquitin not only serves as a tag but also assists degradation by inducing protein unfolding. Proc. Natl. Acad. Sci. USA 2010, 107, 2001–2006. [Google Scholar] [CrossRef] [PubMed]
- Dang, F.; Nie, L.; Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021, 28, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Hagai, T.; Azia, A.; Tóth-Petróczy, Á.; Levy, Y. Intrinsic disorder in ubiquitination substrates. J. Mol. Biol. 2011, 412, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Matouschek, A. Substrate selection by the proteasome through initiation regions. Protein Sci. 2019, 28, 1222–1232. [Google Scholar] [CrossRef]
- Sahu, I.; Mali, S.M.; Sulkshane, P.; Xu, C.; Rozenberg, A.; Morag, R.; Sahoo, M.P.; Singh, S.K.; Ding, Z.; Wang, Y.; et al. The 20S as a stand-alone proteasome in cells can degrade the ubiquitin tag. Nat. Commun. 2021, 12, 6173. [Google Scholar] [CrossRef]
- Yu, H.; Matouschek, A. Recognition of Client Proteins by the Proteasome. Annu. Rev. Biophys. 2017, 46, 149–173. [Google Scholar] [CrossRef]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Mattiroli, F.; Sixma, T.K. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat. Struct. Mol. Biol. 2014, 21, 308–316. [Google Scholar] [CrossRef]
- Li, Y.; Xie, P.; Lu, L.; Wang, J.; Diao, L.; Liu, Z.; Guo, F.; He, Y.; Liu, Y.; Huang, Q.; et al. An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat. Commun. 2017, 8, 347. [Google Scholar] [CrossRef]
- Fischer, E.S.; Scrima, A.; Böhm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Orlicky, S.; Lin, Z.; Willems, A.; Neculai, D.; Ceccarelli, D.; Mercurio, F.; Shilton, B.H.; Sicheri, F.; Tyers, M. Suprafacial Orientation of the SCFCdc4 Dimer Accommodates Multiple Geometries for Substrate Ubiquitination. Cell 2007, 129, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Min, M.; Mayor, U.; Lindon, C. Ubiquitination site preferences in anaphase promoting complex/cyclosome (APC/C) substrates. Open Biol. 2013, 3, 130097. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 358–366. [Google Scholar] [CrossRef]
- Carroll, E.C.; Greene, E.R.; Martin, A.; Marqusee, S. Site-specific ubiquitination affects protein energetics and proteasomal degradation. Nat. Chem. Biol. 2020, 16, 866–875. [Google Scholar] [CrossRef]
- Carroll, E.C.; Latorraca, N.R.; Lindner, J.M.; Maguire, B.C.; Pelton, J.G.; Marqusee, S. Mechanistic basis for ubiquitin modulation of a protein energy landscape. Proc. Natl. Acad. Sci. USA 2021, 118, 126a. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Myers, N.; Moscovitz, O.; Sharon, M.; Prilusky, J.; Shaul, Y. Thermo-resistant intrinsically disordered proteins are efficient 20S proteasome substrates. Mol. Biosyst. 2012, 8, 368–373. [Google Scholar] [CrossRef]
- Ukmar-Godec, T.; Fang, P.; Ibáñez de Opakua, A.; Henneberg, F.; Godec, A.; Pan, K.T.; Cima-Omori, M.S.; Chari, A.; Mandelkow, E.; Urlaub, H.; et al. Proteasomal degradation of the intrinsically disordered protein tau at single-residue resolution. Sci. Adv. 2020, 6, eaba3916. [Google Scholar] [CrossRef]
- Myers, N.; Olender, T.; Savidor, A.; Levin, Y.; Reuven, N.; Shaul, Y. The Disordered Landscape of the 20S Proteasome Substrates Reveals Tight Association with Phase Separated Granules. Proteomics 2018, 18, 1800076. [Google Scholar] [CrossRef]
- Jariel-Encontre, I.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta—Rev. Cancer 2008, 1786, 153–177. [Google Scholar] [CrossRef]
- Erales, J.; Coffino, P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta—Mol. Cell Res. 2014, 1843, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Abi Habib, J.; De Plaen, E.; Stroobant, V.; Zivkovic, D.; Bousquet, M.P.; Guillaume, B.; Wahni, K.; Messens, J.; Busse, A.; Vigneron, N.; et al. Efficiency of the four proteasome subtypes to degrade ubiquitinated or oxidized proteins. Sci. Rep. 2020, 10, 15765. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Myers, N.; Steinberger, S.; Adler, J.; Riutin, M.; Broennimann, K.; Reuven, N.; Shaul, Y. The C-Terminus of the PSMA3 Proteasome Subunit Preferentially Traps Intrinsically Disordered Proteins for Degradation. Cells 2022, 11, 3231. [Google Scholar] [CrossRef] [PubMed]
- Makaros, Y.; Raiff, A.; Timms, R.T.; Wagh, A.R.; Gueta, M.I.; Bekturova, A.; Guez-Haddad, J.; Brodsky, S.; Opatowsky, Y.; Glickman, M.H.; et al. Ubiquitin-independent proteasomal degradation driven by C-degron pathways. Mol. Cell 2023, 83, 1921–1935.e7. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Chen, Y.; Cai, H. An intelligent genetic algorithm designed for global optimization of multi-minima functions. Appl. Math. Comput. 2006, 178, 355–371. [Google Scholar] [CrossRef]
- Ha, S.W.; Ju, D.; Xie, Y. The N-terminal domain of Rpn4 serves as a portable ubiquitin-independent degron and is recognized by specific 19S RP subunits. Biochem. Biophys. Res. Commun. 2012, 419, 226–231. [Google Scholar] [CrossRef]
- Gödderz, D.; Schäfer, E.; Palanimurugan, R.; Dohmen, R.J. The N-terminal unstructured domain of yeast odc functions as a transplantable and replaceable ubiquitin-independent degron. J. Mol. Biol. 2011, 407, 354–367. [Google Scholar] [CrossRef]
- Manfredonia, A.J.; Kraut, D.A. The 26S Proteasome Switches between ATP-Dependent and -Independent Mechanisms in Response to Substrate Ubiquitination. Biomolecules 2022, 12, 750. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Myers, N.; Eliav, R.; Adamovich, Y.; Hagai, T.; Adler, J.; Navon, A.; Shaul, Y. NADH Binds and stabilizes the 26S proteasomes independent of ATP. J. Biol. Chem. 2014, 289, 11272–11281. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Myers, N.; Adler, J.; Shaul, Y. Degradation of intrinsically disordered proteins by the nadh 26s proteasome. Biomolecules 2020, 10, 1642. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Waku, T. New addiction to the NRF2-related factor NRF3 in cancer cells: Ubiquitin-independent proteolysis through the 20S proteasome. Cancer Sci. 2020, 111, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Shlomai, A.; Tsvetkov, P.; Umansky, K.B.; Reuven, N.; Estall, J.L.; Spiegelman, B.M.; Shaul, Y. The Protein Level of PGC-1α, a Key Metabolic Regulator, Is Controlled by NADH-NQO1. Mol. Cell. Biol. 2013, 33, 2603–2613. [Google Scholar] [CrossRef]
- Liao, Y.; Sumara, I.; Pangou, E. Non-proteolytic ubiquitylation in cellular signaling and human disease. Commun. Biol. 2022, 5, 114. [Google Scholar] [CrossRef]
- Becker, S.H.; Li, H.; Heran Darwin, K. Biology and Biochemistry of Bacterial Proteasomes. In Macromolecular Protein Complexes II: Structure and Function; Springer Nature Switzerland AG: Berlin/Heidelberg, Germany, 2019; pp. 339–358. [Google Scholar] [CrossRef]
- Franklin, T.G.; Pruneda, J.N. Bacteria make surgical strikes on host ubiquitin signaling. PLoS Pathog. 2021, 17, e1009341. [Google Scholar] [CrossRef]
- Bassères, E.; Coppotelli, G.; Pfirrmann, T.; Andersen, J.B.; Masucci, M.; Frisan, T. The ubiquitin C-terminal hydrolase UCH-L1 promotes bacterial invasion by altering the dynamics of the actin cytoskeleton. Cell. Microbiol. 2010, 12, 1622–1633. [Google Scholar] [CrossRef]
- Sheng, X.; You, Q.; Zhu, H.; Chang, Z.; Li, Q.; Wang, H.; Wang, C.; Wang, H.; Hui, L.; Du, C.; et al. Bacterial effector NleL promotes enterohemorrhagic E. coli-induced attaching and effacing lesions by ubiquitylating and inactivating JNK. PLoS Pathog. 2017, 13, e1006534. [Google Scholar] [CrossRef]
- Haglund, K.; Dikic, I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J. Cell Sci. 2012, 125, 265–275. [Google Scholar] [CrossRef]
- Su, W.-C.; Chen, Y.-C.; Tseng, C.-H.; Hsu, P.W.-C.; Tung, K.-F.; Jeng, K.-S.; Lai, M.M.C. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. USA 2013, 110, 17516–17521. [Google Scholar] [CrossRef]
- Alix, E.; Godlee, C.; Cerny, O.; Blundell, S.; Tocci, R.; Matthews, S.; Liu, M.; Pruneda, J.N.; Swatek, K.N.; Komander, D.; et al. The Tumour Suppressor TMEM127 Is a Nedd4-Family E3 Ligase Adaptor Required by Salmonella SteD to Ubiquitinate and Degrade MHC Class II Molecules. Cell Host Microbe 2020, 28, 54–68.e7. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the epstein-barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, M.A.; Zich, J.; Takeuchi, J.; Zhang, M.; Govaerts, C.; Coffino, P. Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J. 2006, 25, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Heessen, S.; Leonchiks, A.; Issaeva, N.; Sharipo, A.; Selivanova, G.; Masucci, M.G.; Dantuma, N.P. Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc. Natl. Acad. Sci. USA 2002, 99, 1532–1537. [Google Scholar] [CrossRef] [PubMed]
- Daskalogianni, C.; Apcher, S.; Candeias, M.M.; Naski, N.; Calvo, F.; Fåhraeus, R. Gly-Ala Repeats Induce Position- and Substrate-specific Regulation of 26 S Proteasome-dependent Partial Processing. J. Biol. Chem. 2008, 283, 30090–30100. [Google Scholar] [CrossRef]
- Christianson, J.C.; Jarosch, E.; Sommer, T. Mechanisms of substrate processing during ER-associated protein degradation. Nat. Rev. Mol. Cell Biol. 2023, 1–20. [Google Scholar] [CrossRef]
- Rodighiero, C.; Tsai, B.; Rapoport, T.A.; Lencer, W.I. Role of ubiquitination in retro—Translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep. 2002, 3, 1222–1227. [Google Scholar] [CrossRef]
- Bernardi, K.M.; Williams, J.M.; Kikkert, M.; van Voorden, S.; Wiertz, E.J.; Ye, Y.; Tsai, B. The E3 Ubiquitin Ligases Hrd1 and gp78 Bind to and Promote Cholera Toxin Retro-Translocation. Mol. Biol. Cell. 2010, 21, 140–151. [Google Scholar] [CrossRef]
- van den Boomen, D.J.H.; Timms, R.T.; Grice, G.L.; Stagg, H.R.; Skødt, K.; Dougan, G.; Nathan, J.A.; Lehner, P.J. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. USA 2014, 111, 11425–11430. [Google Scholar] [CrossRef]
- van de Weijer, M.L.; Bassik, M.C.; Luteijn, R.D.; Voorburg, C.M.; Lohuis, M.A.M.; Kremmer, E.; Hoeben, R.C.; LeProust, E.M.; Chen, S.; Hoelen, H.; et al. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat. Commun. 2014, 5, 3832. [Google Scholar] [CrossRef]
- Herr, R.A.; Harris, J.; Fang, S.; Wang, X.; Hansen, T.H. Role of the RING-CH domain of viral ligase mK3 in ubiquitination of non-lysine and lysine MHC I residues. Traffic 2009, 10, 1301–1317. [Google Scholar] [CrossRef] [PubMed]
- Stagg, H.R.; Thomas, M.; van den Boomen, D.; Wiertz, E.J.H.J.; Drabkin, H.A.; Gemmill, R.M.; Lehner, P.J. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 2009, 186, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-L.; van den Boomen, D.J.H.; Tomasec, P.; Weekes, M.P.; Antrobus, R.; Stanton, R.J.; Ruckova, E.; Sugrue, D.; Wilkie, G.S.; Davison, A.J.; et al. Plasma Membrane Profiling Defines an Expanded Class of Cell Surface Proteins Selectively Targeted for Degradation by HCMV US2 in Cooperation with UL141. PLoS Pathog. 2015, 11, e1004811. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A Novel Human WD Protein, h-βTrCP, that Interacts with HIV-1 Vpu Connects CD4 to the ER Degradation Pathway through an F-Box Motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The Interferon-Induced Protein BST-2 Restricts HIV-1 Release and Is Downregulated from the Cell Surface by the Viral Vpu Protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef]
- Sauter, D.; Hotter, D.; Van Driessche, B.; Stürzel, C.M.; Kluge, S.F.; Wildum, S.; Yu, H.; Baumann, B.; Wirth, T.; Plantier, J.-C.; et al. Differential Regulation of NF-κB-Mediated Proviral and Antiviral Host Gene Expression by Primate Lentiviral Nef and Vpu Proteins. Cell Rep. 2015, 10, 586–599. [Google Scholar] [CrossRef]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef]
- Jäger, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 2012, 481, 371–375. [Google Scholar] [CrossRef]
- Gargan, S.; Ahmed, S.; Mahony, R.; Bannan, C.; Napoletano, S.; O’Farrelly, C.; Borrow, P.; Bergin, C.; Stevenson, N.J. HIV-1 Promotes the Degradation of Components of the Type 1 IFN JAK/STAT Pathway and Blocks Anti-viral ISG Induction. EBioMedicine 2018, 30, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Romani, B.; Cohen, É.A. Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr. Opin. Virol. 2012, 2, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yao, X. Posttranslational Modifications of HIV-1 Integrase by Various Cellular Proteins during Viral Replication. Viruses 2013, 5, 1787–1801. [Google Scholar] [CrossRef]
- Zheng, Y.; Ao, Z.; Wang, B.; Jayappa, K.D.; Yao, X. Host Protein Ku70 Binds and Protects HIV-1 Integrase from Proteasomal Degradation and Is Required for HIV Replication. J. Biol. Chem. 2011, 286, 17722–17735. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, Y.; Koshizuka, T.; Goshima, F.; Kimura, H.; Nishiyama, Y. Herpes Simplex Virus Type 2 UL56 Interacts with the Ubiquitin Ligase Nedd4 and Increases Its Ubiquitination. J. Virol. 2008, 82, 5220–5233. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Kobayashi, T.; Ishioka, K.; Suzutani, T. Herpesviruses possess conserved proteins for interaction with Nedd4 family ubiquitin E3 ligases. Sci. Rep. 2018, 8, 4447. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Parsy, M.-L.; Orr, A. Analysis of the Functions of Herpes Simplex Virus Type 1 Regulatory Protein ICP0 That Are Critical for Lytic Infection and Derepression of Quiescent Viral Genomes. J. Virol. 2009, 83, 4963–4977. [Google Scholar] [CrossRef]
- Rodríguez, M.C.; Dybas, J.M.; Hughes, J.; Weitzman, M.D.; Boutell, C. The HSV-1 ubiquitin ligase ICP0: Modifying the cellular proteome to promote infection. Virus Res. 2020, 285, 198015. [Google Scholar] [CrossRef]
- Rodrigues, L.; Popov, N.; Kaye, K.M.; Simas, J.P. Stabilization of Myc through Heterotypic Poly-Ubiquitination by mLANA Is Critical for γ-Herpesvirus Lymphoproliferation. PLoS Pathog. 2013, 9, e1003554. [Google Scholar] [CrossRef]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Inhibits Beta Interferon Production by Deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-κB Activation in DNA Sensing Signal Pathway. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, T.; Hofmann, K. Bacterial dubs: Deubiquitination beyond the seven classes. Biochem. Soc. Trans. 2019, 47, 1857–1866. [Google Scholar] [CrossRef]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef]
- Ye, Z.; Petrof, E.O.; Boone, D.; Claud, E.C.; Sun, J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am. J. Pathol. 2007, 171, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G.; Faustin, B.; Welsh, K.; Loeffler, M.; Krajewska, M.; Hasegawa, P.; Mukherjee, S.; Orth, K.; Krajewski, S.; Godzik, A.; et al. Salmonella Secreted Factor L Deubiquitinase of Salmonella typhimurium Inhibits NF-κB, Suppresses IκBα Ubiquitination and Modulates Innate Immune Responses. J. Immunol. 2008, 180, 5045–5056. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Thomas, M.; Sachse, M.; Santos, A.J.M.; Figueira, R.; Holden, D.W. The Salmonella deubiquitinase Ssel inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 2012, 8, e1002743. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, Y.G.; Lin, Z.; Lu, R.; Xia, Y.; Meng, C.; Pan, Z.; Xu, X.; Jiao, X.; Sun, J. Salmonella Enteritidis Effector AvrA Suppresses Autophagy by Reducing Beclin-1 Protein. Front. Immunol. 2020, 11, 686. [Google Scholar] [CrossRef]
- Fiskin, E.; Bhogaraju, S.; Herhaus, L.; Kalayil, S.; Hahn, M.; Dikic, I. Structural basis for the recognition and degradation of host TRIM proteins by Salmonella effector SopA. Nat. Commun. 2017, 8, 14004. [Google Scholar] [CrossRef]
- De Jong, M.F.; Liu, Z.; Chen, D.; Alto, N.M. Shigella flexneri suppresses NF-κB activation by inhibiting linear ubiquitin chain ligation. Nat. Microbiol. 2016, 1, 16084. [Google Scholar] [CrossRef]
- Noad, J.; Von Der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol. 2017, 2, 17063. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Mimuro, H.; Kim, M.; Ogawa, M.; Ashida, H.; Toyotome, T.; Franchi, L.; Suzuki, M.; Sanada, T.; Suzuki, T.; et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to Demolish macrophages. Proc. Natl. Acad. Sci. USA 2014, 111, E4254–E4263. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Nakano, H.; Sasakawa, C. Shigella IpaH0722 E3 Ubiquitin Ligase Effector Targets TRAF2 to Inhibit PKC-NF-κB Activity in Invaded Epithelial Cells. PLoS Pathog. 2013, 9, e1003409. [Google Scholar] [CrossRef] [PubMed]
- Wandel, M.P.; Pathe, C.; Werner, E.I.; Ellison, C.J.; Boyle, K.B.; von der Malsburg, A.; Rohde, J.; Randow, F. GBPs Inhibit Motility of Shigella flexneri but Are Targeted for Degradation by the Bacterial Ubiquitin Ligase IpaH9.8. Cell Host Microbe 2017, 22, 507–518.e5. [Google Scholar] [CrossRef]
- Li, P.; Jiang, W.; Yu, Q.; Liu, W.; Zhou, P.; Li, J.; Xu, J.; Xu, B.; Wang, F.; Shao, F. Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature 2017, 551, 378–383. [Google Scholar] [CrossRef]
- Xu, L.; Luo, Z.-Q. Cell biology of infection by Legionella pneumophila. Microbes Infect. 2013, 15, 157–167. [Google Scholar] [CrossRef]
- Price, C.T.D.; Al-Quadan, T.; Santic, M.; Rosenshine, I.; Abu Kwaik, Y. Host Proteasomal Degradation Generates Amino Acids Essential for Intracellular Bacterial Growth. Science 2011, 334, 1553–1557. [Google Scholar] [CrossRef]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.-Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of Ubiquitin Promotes Serine Ubiquitination and Impairs Conventional Ubiquitination. Cell 2016, 167, 1636–1649.e13. [Google Scholar] [CrossRef]
- Kubori, T.; Hyakutake, A.; Nagai, H. Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 2008, 67, 1307–1319. [Google Scholar] [CrossRef]
- Kubori, T.; Shinzawa, N.; Kanuka, H.; Nagai, H. Legionella metaeffector exploits host proteasome to temporally regulate cognate effector. PLoS Pathog. 2010, 6, e1001216. [Google Scholar] [CrossRef]
- Canning, M.; Boutell, C.; Parkinson, J.; Everett, R.D. A RING Finger Ubiquitin Ligase Is Protected from Autocatalyzed Ubiquitination and Degradation by Binding to Ubiquitin-specific Protease USP7. J. Biol. Chem. 2004, 279, 38160–38168. [Google Scholar] [CrossRef] [PubMed]
- Leong, J.X.; Raffeiner, M.; Spinti, D.; Langin, G.; Franz-Wachtel, M.; Guzman, A.R.; Kim, J.; Pandey, P.; Minina, A.E.; Macek, B.; et al. A bacterial effector counteracts host autophagy by promoting degradation of an autophagy component. EMBO J. 2022, 41, e110352. [Google Scholar] [CrossRef] [PubMed]
- Bullones-Bolaños, A.; Bernal-Bayard, J.; Ramos-Morales, F. The NEL Family of Bacterial E3 Ubiquitin Ligases. Int. J. Mol. Sci. 2022, 23, 7725. [Google Scholar] [CrossRef]
- Chen, H.; Chen, J.; Li, M.; Chang, M.; Xu, K.; Shang, Z.; Zhao, Y.; Palmer, I.; Zhang, Y.; McGill, J.; et al. A Bacterial Type III Effector Targets the Master Regulator of Salicylic Acid Signaling, NPR1, to Subvert Plant Immunity. Cell Host Microbe 2017, 22, 777–788.e7. [Google Scholar] [CrossRef]
- Cheng, W.; Munkvold, K.R.; Gao, H.; Mathieu, J.; Schwizer, S.; Wang, S.; Yan, Y.-B.; Wang, J.; Martin, G.B.; Chai, J. Structural analysis of pseudomonas syringae AvrPtoB bound to host BAK1 reveals two similar kinase-interacting domains in a type III effector. Cell Host Microbe 2011, 10, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Rosebrock, T.R.; Zeng, L.; Brady, J.J.; Abramovitch, R.B.; Xiao, F.; Martin, G.B. A bacterial E3 ubiquitin ligase targets a host protein kinase to disrupt plant immunity. Nature 2007, 448, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Göhre, V.; Spallek, T.; Häweker, H.; Mersmann, S.; Mentzel, T.; Boller, T.; de Torres, M.; Mansfield, J.W.; Robatzek, S. Plant Pattern-Recognition Receptor FLS2 Is Directed for Degradation by the Bacterial Ubiquitin Ligase AvrPtoB. Curr. Biol. 2008, 18, 1824–1832. [Google Scholar] [CrossRef]
- Gimenez-Ibanez, S.; Hann, D.R.; Ntoukakis, V.; Petutschnig, E.; Lipka, V.; Rathjen, J.P. AvrPtoB Targets the LysM Receptor Kinase CERK1 to Promote Bacterial Virulence on Plants. Curr. Biol. 2009, 19, 423–429. [Google Scholar] [CrossRef]
- Qin, J.; Zhou, X.; Sun, L.; Wang, K.; Yang, F.; Liao, H.; Rong, W.; Yin, J.; Chen, H.; Chen, X.; et al. The Xanthomonas effector XopK harbours E3 ubiquitin-ligase activity that is required for virulence. New Phytol. 2018, 220, 219–231. [Google Scholar] [CrossRef]
- Nomura, K.; DebRoy, S.; Lee, Y.H.; Pumplin, N.; Jones, J.; He, S.Y. A bacterial virulence protein suppresses host innate immunity to cause plant disease. Science 2006, 313, 220–223. [Google Scholar] [CrossRef]
- Yang, L.; Teixeira, P.J.P.L.; Biswas, S.; Finkel, O.M.; He, Y.; Salas-Gonzalez, I.; English, M.E.; Epple, P.; Mieczkowski, P.; Dangl, J.L. Pseudomonas syringae Type III Effector HopBB1 Promotes Host Transcriptional Repressor Degradation to Regulate Phytohormone Responses and Virulence. Cell Host Microbe 2017, 21, 156–168. [Google Scholar] [CrossRef] [PubMed]
- MacLean, A.M.; Orlovskis, Z.; Kowitwanich, K.; Zdziarska, A.M.; Angenent, G.C.; Immink, R.G.H.; Hogenhout, S.A. Phytoplasma Effector SAP54 Hijacks Plant Reproduction by Degrading MADS-box Proteins and Promotes Insect Colonization in a RAD23-Dependent Manner. PLoS Biol. 2014, 12, e1001835. [Google Scholar] [CrossRef]
- Huang, W.; MacLean, A.M.; Sugio, A.; Maqbool, A.; Busscher, M.; Cho, S.T.; Kamoun, S.; Kuo, C.H.; Immink, R.G.H.; Hogenhout, S.A. Parasitic modulation of host development by ubiquitin-independent protein degradation. Cell 2021, 184, 5201–5214.e12. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Chen, S.; Shirsekar, G.; Zhou, B.; Khang, C.H.; Songkumarn, P.; Afzal, A.J.; Ning, Y.; Wang, R.; Bellizzi, M.; et al. The magnaporthe oryzae effector avrpiz-t targets the RING E3 ubiquitin ligase APIP6 to suppress pathogen-associated molecular pattern-triggered immunity in ricesW OA. Plant Cell 2012, 24, 4748–4762. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.I.B.; Armstrong, M.R.; Gilroy, E.M.; Boevink, P.C.; Hein, I.; Taylor, R.M.; Zhendong, T.; Engelhardt, S.; Vetukuri, R.R.; Harrower, B.; et al. Phytophthora infestans effector AVR3a is essential for virulence and manipulates plant immunity by stabilizing host E3 ligase CMPG1. Proc. Natl. Acad. Sci. USA 2010, 107, 9909–9914. [Google Scholar] [CrossRef]
- Ishikawa, K.; Yamaguchi, K.; Sakamoto, K.; Yoshimura, S.; Inoue, K.; Tsuge, S.; Kojima, C.; Kawasaki, T. Bacterial effector modulation of host E3 ligase activity suppresses PAMP-triggered immunity in rice. Nat. Commun. 2014, 5, 5430. [Google Scholar] [CrossRef]
- Tan, L.; Rong, W.; Luo, H.; Chen, Y.; He, C. The Xanthomonas campestris effector protein XopDXcc8004 triggers plant disease tolerance by targeting DELLA proteins. New Phytol. 2014, 204, 595–608. [Google Scholar] [CrossRef]
- Raffeiner, M.; Üstün, S.; Guerra, T.; Spinti, D.; Fitzner, M.; Sonnewald, S.; Baldermann, S.; Börnke, F. The Xanthomonas type-III effector XopS stabilizes CaWRKY40a to regulate defense responses and stomatal immunity in pepper (Capsicum annuum). Plant Cell 2022, 34, 1684–1708. [Google Scholar] [CrossRef]
- Tanaka, S.; Brefort, T.; Neidig, N.; Djamei, A.; Kahnt, J.; Vermerris, W.; Koenig, S.; Feussner, K.; Feussner, I.; Kahmann, R. A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. Elife 2014, 3, e01355. [Google Scholar] [CrossRef]
- Zhang, C.; Wei, Y.; Xu, L.; Wu, K.C.; Yang, L.; Shi, C.N.; Yang, G.Y.; Chen, D.; Yu, F.F.; Xie, Q.; et al. A Bunyavirus-Inducible Ubiquitin Ligase Targets RNA Polymerase IV for Degradation during Viral Pathogenesis in Rice. Mol. Plant 2020, 13, 836–850. [Google Scholar] [CrossRef]
- Jia, Q.; Liu, N.; Xie, K.; Dai, Y.; Han, S.; Zhao, X.; Qian, L.; Wang, Y.; Zhao, J.; Gorovits, R.; et al. CLCuMuB βC1 Subverts Ubiquitination by Interacting with NbSKP1s to Enhance Geminivirus Infection in Nicotiana benthamiana. PLoS Pathog. 2016, 12, e1005668. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Zhou, C.J.; Wang, Q.; Chen, X.R.; Sun, Q.; Zhao, T.Y.; Ye, J.C.; Wang, Y.; Zhang, Z.Y.; Zhang, Y.L.; et al. Rice black streaked dwarf virus P7-2 forms a SCF complex through binding to Oryza sativa SKP1-like proteins, and interacts with GID2 involved in the gibberellin pathway. PLoS ONE 2017, 12, e0177518. [Google Scholar] [CrossRef]
- Thiel, H.; Hleibieh, K.; Gilmer, D.; Varrelmann, M. The P25 pathogenicity factor of Beet necrotic yellow vein virus targets the sugar beet 26s proteasome involved in the induction of a hypersensitive resistance response via interaction with an F-box protein. Mol. Plant-Microbe Interact. 2012, 25, 1058–1072. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Qin, Q.; Wang, Y.; Pu, Y.; Liu, L.; Wen, X.; Ji, S.; Wu, J.; Wei, C.; Ding, B.; et al. Rice Dwarf Virus P2 Protein Hijacks Auxin Signaling by Directly Targeting the Rice OsIAA10 Protein, Enhancing Viral Infection and Disease Development. PLoS Pathog. 2016, 12, e1005847. [Google Scholar] [CrossRef]
- Gelvin, S.B. Agrobacterium -Mediated Plant Transformation: The Biology behind the “Gene-Jockeying” Tool. Microbiol. Mol. Biol. Rev. 2003, 67, 16–37. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Rojas, C.M.; Tang, Y.; Mysore, K.S. Several components of SKP1/Cullin/F-box E3 ubiquitin ligase complex and associated factors play a role in Agrobacterium-mediated plant transformation. New Phytol. 2012, 195, 203–216. [Google Scholar] [CrossRef]
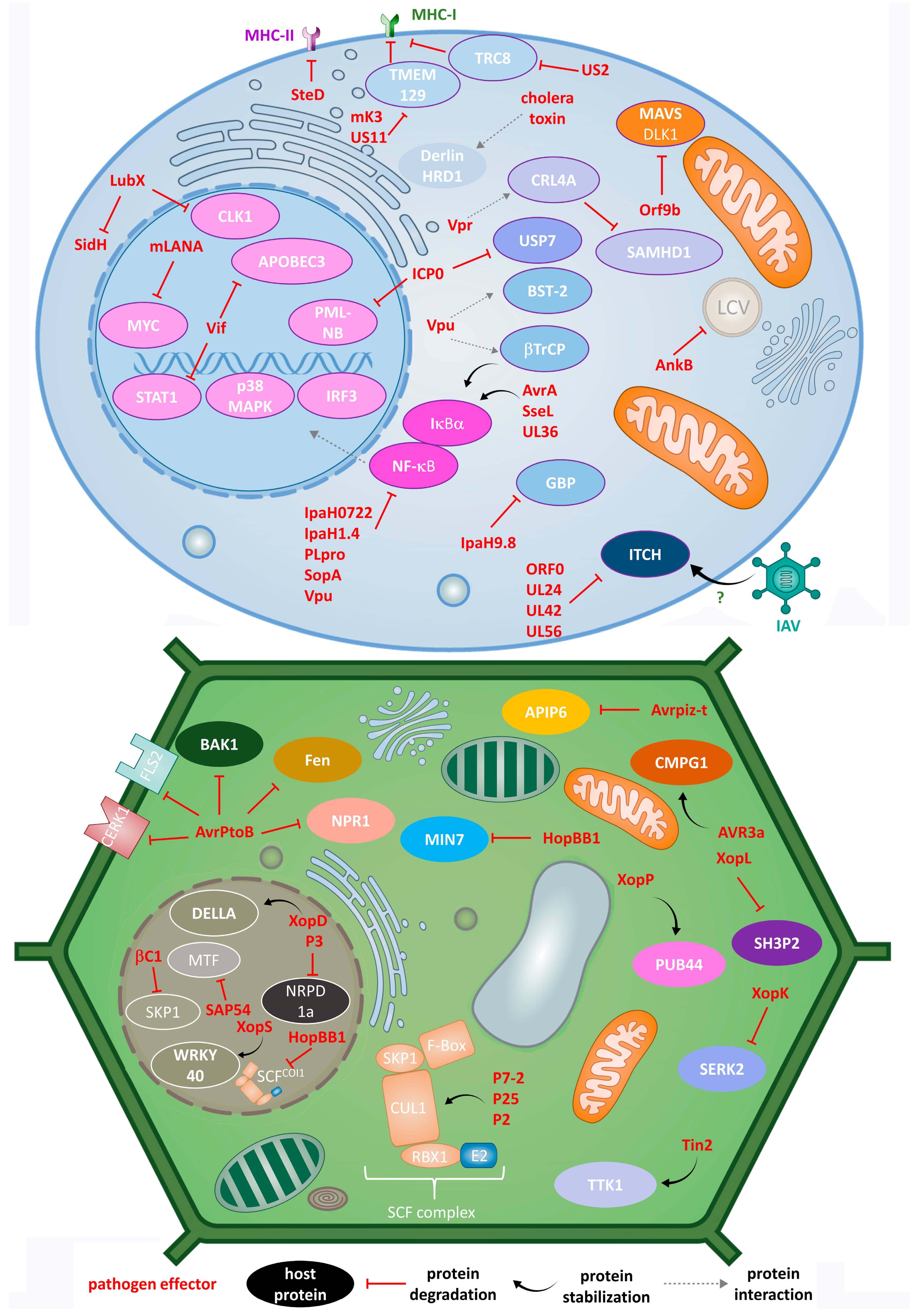
| Species | Pathogenic Effector | Host Protein | Targeted Pathway/Effect | Ref. |
|---|---|---|---|---|
| X. capestris | XopL | SH3P2 | abolishing autophagy | [136] |
| P. syringae | AvrPtoB | NPR1 | deregulation of plant immunity | [138] |
| P. syringae | AvrPtoB | BAK1 | deregulation of plant immunity | [139] |
| P. syringae | AvrPtoB | Fen | deregulation of plant immunity | [140] |
| P. syringae | AvrPtoB | FLS2 | deregulation of plant immunity | [141] |
| P. syringae | AvrPtoB | CERK1 | enhancing bacterial virulence | [142] |
| P. syringae | HopM1 | MIN7 | manipulation of vesicle trafficking | [144] |
| P. syringae | HopBB1 | SCFCOI1 | promotion of host transcriptional repressor degradation to regulate phytohormone responses and virulence | [145] |
| M. oryzae | Avrpiz-t | APIP6 | suppression of PAMP-triggered immunity | [148] |
| P. infestans | SAP54 | MTF | induction of insect colonization | [146] |
| P. infestans | AVR3a | CMPG1 | Prevention of cell death upon infection | [149] |
| X. oryzae | XopP | PUB44 | suppression of PAMP-triggered immunity | [150] |
| X. oryzae | XopK | SERK2 | deregulation of plant immunity | [143] |
| X. oryzae | XopD | DELLA | induction of plant disease tolerance | [151] |
| X. oryzae | XopS | WRKY40a | deregulation of plant immunity | [152] |
| U. maydis | Tin2 | TTK1 | enhancing fungal proliferation | [153] |
| Rice grassy stunt virus | P3 | P3IP1 | degradation of OsNRPD1a, inducing hypomethylation of downstream genes | [154] |
| Cotton leaf curl Multan virus | βC1 | SKP1 | enhancing virus DNA accumulation | [155] |
| Black streaked dwarf virus | P7-2 | SCF-E3 ligase | possible role in the gibberellin signaling pathway | [156] |
| Beet necrotic yellow vein virus | P25 | SCF-E3 ligase | enhancing virus pathogenicity | [157] |
| Rice dwarf virus | P2 | SCF-E3 ligase | enhancing viral infection | [158] |
| A. tumefaciens | SCF-E3 ligase | release of T-DNA | [160] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bialek, W.; Collawn, J.F.; Bartoszewski, R. Ubiquitin-Dependent and Independent Proteasomal Degradation in Host-Pathogen Interactions. Molecules 2023, 28, 6740. https://doi.org/10.3390/molecules28186740
Bialek W, Collawn JF, Bartoszewski R. Ubiquitin-Dependent and Independent Proteasomal Degradation in Host-Pathogen Interactions. Molecules. 2023; 28(18):6740. https://doi.org/10.3390/molecules28186740
Chicago/Turabian StyleBialek, Wojciech, James F. Collawn, and Rafal Bartoszewski. 2023. "Ubiquitin-Dependent and Independent Proteasomal Degradation in Host-Pathogen Interactions" Molecules 28, no. 18: 6740. https://doi.org/10.3390/molecules28186740
APA StyleBialek, W., Collawn, J. F., & Bartoszewski, R. (2023). Ubiquitin-Dependent and Independent Proteasomal Degradation in Host-Pathogen Interactions. Molecules, 28(18), 6740. https://doi.org/10.3390/molecules28186740