
Short-Range Charge Transfer in DNA Base Triplets: Real-Time Tracking of Coherent Fluctuation Electron Transfer


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Photoinduced Charge Transfer
2.2. Reduced Density Gradient
2.3. Charge Density Difference Analysis
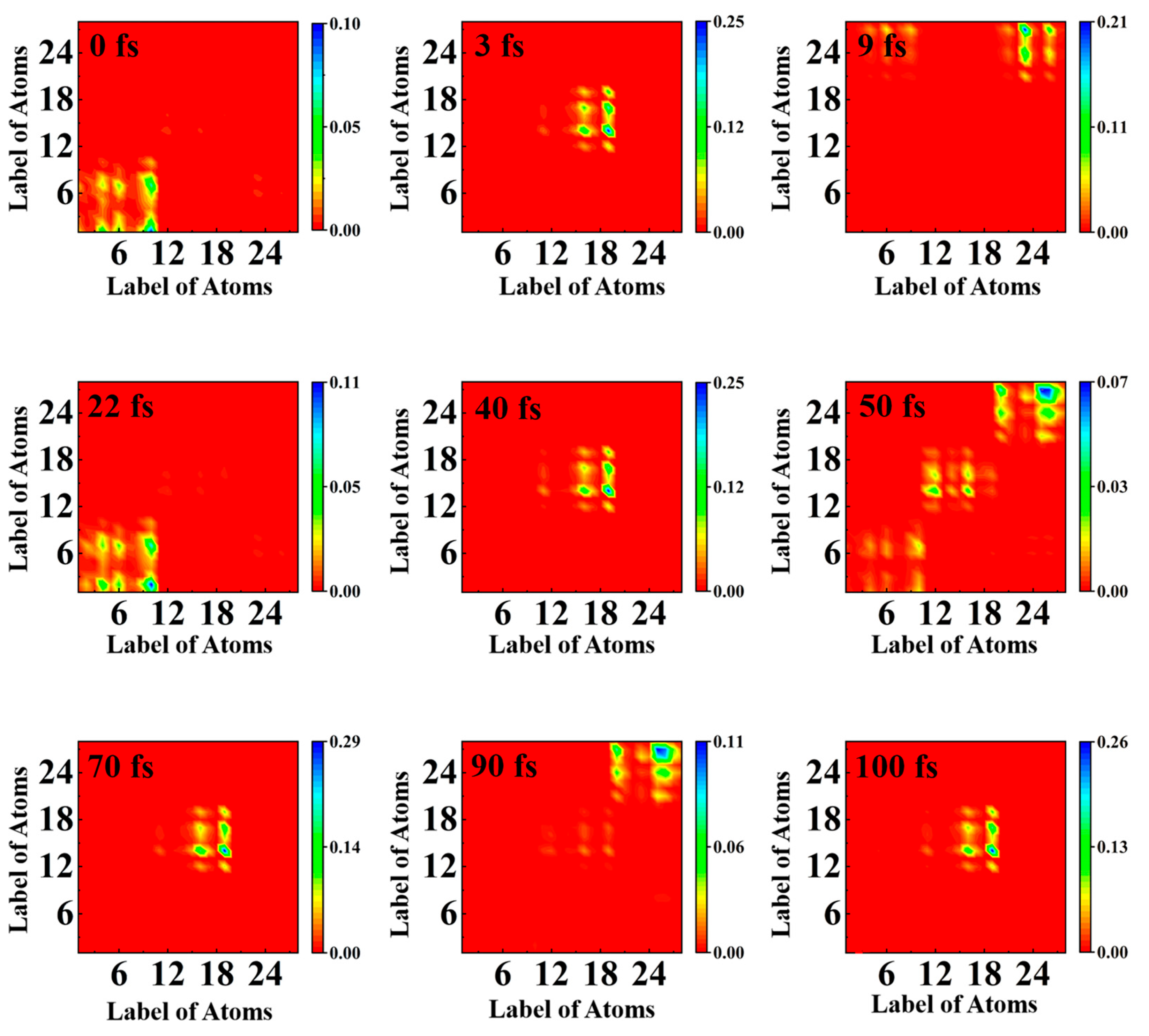
2.4. Transition Density Matrix Analysis
2.5. Hole–Electron Analysis and Marcus–Levich–Jortner Theory
3. Conclusions
4. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kawai, K.; Hayashi, M.; Majima, T. Hole Transfer in LNA and 5-Me-2′-deoxyzebularine-Modified DNA. J. Am. Chem. Soc. 2012, 134, 9406–9409. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Zhao, H.; Reboud, J.L.; Cooper, J.M. Cycling of Rational Hybridization Chain Reaction to Enable Enzyme-Free DNA-Based Clinical Diagnosis. ACS Nano 2018, 12, 7213–7219. [Google Scholar] [CrossRef] [PubMed]
- Karakaidos, P.; Karagiannis, D.; Rampias, T. Resolving DNA Damage: Epigenetic Regulation of DNA Repair. Molecules 2020, 25, 2496. [Google Scholar] [CrossRef] [PubMed]
- Lewis, F.D.; Wu, T.F.; Zhang, Y.F.; Letsinger, R.L.; Greenfield, S.R.; Wasielewski, M.R. Distance-dependent electron transfer in DNA hairpins. Science 1997, 277, 5326. [Google Scholar] [CrossRef] [PubMed]
- Peluso, A.; Caruso, T.; Landi, A.; Capobianco, A. The Dynamics of Hole Transfer in DNA. Molecules 2019, 24, 4044. [Google Scholar] [CrossRef] [PubMed]
- Menacher, F.; Rubner, M.; Berndl, S.; Wagenknecht, H.A. Thiazole Orange and Cy3: Improvement of Fluorescent DNA Probes with Use of Short Range Electron Transfer. J. Org. Chem. 2008, 73, 4263–4266. [Google Scholar] [CrossRef] [PubMed]
- Marquetand, P.; Nogueira, J.; Mai, S.; Plasser, F.; González, L. Challenges in Simulating Light-Induced Processes in DNA. Molecules 2017, 22, 49. [Google Scholar] [CrossRef]
- Szabla, R.; Kruse, H.; Stadlbauer, P.; Sponer, J.; Sobolewski, A.L. Sequential electron transfer governs the UVinduced self-repair of DNA photolesions. Chem. Sci. 2018, 9, 3131–3140. [Google Scholar] [CrossRef]
- Giese, B.; Wessely, S.; Spormann, M.; Lindemann, U.; Meggers, E.; Beyerle, M.E.M. On the Mechanism of Long-Range Electron Transfer through DNA. Angew. Chem. Int. Ed. 1999, 38, 7. [Google Scholar] [CrossRef]
- Giese, B.; Amaudrut, J.; Köhler, A.K.; Spormann, M.; Wessely, S. Direct observation of hole transfer through DNA by hopping between adenine bases and by tunnelling. Nature 2001, 412, 6844. [Google Scholar] [CrossRef]
- Chen, H.; Meena; McLaughlin, L.W. A Janus-Wedge DNA Triplex with A-W1-T and G-W2-C Base Triplets. J. Am. Chem. Soc. 2008, 130, 13190–13191. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Fujitsuka, M.; Majima, T. Sequence-Dependent Photocurrent Generation through Long-Distance Excess-Electron Transfer in DNA. Angew. Chem. Int. Ed. 2016, 55, 8715–8717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, P.; He, X.H.; Han, K.L. Self-Consistent Polarization of the Boundary in the Redistributed Charge and Dipole Scheme for Combined Quantum-Mechanical and Molecular-Mechanical Calculations. J. Chem. Theory Comput. 2016, 3, 1378–1398. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Terakawa, T.; Qi, Z.; Steinfeld, J.B.; Redding, S.; Kwon, Y.H.; Gaines, W.A.; Zhao, W.X.; Sung, P.; Greene, E.C. Base triplet stepping by the Rad51/RecA family of recombinases. Science 2015, 349, 6251. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Lu, H.T.; Zhang, X.; Yu, C.; Tohyama, T.; Lu, R.F. High-Harmonic Generation Approaching the Quantum Critical Point of Strongly Correlated Systems. Phys. Rev. Lett. 2022, 128, 047401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.N.; Yin, S.H.; Ma, Y.H.; Yang, S.Q. Excess-electron capture and energy transfer to bulk water for aqueous DNA nucleotide. Phys. Chem. Chem. Phys. 2023, 25, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, M.; Majima, T. Charge transfer dynamics in DNA revealed by time-resolved spectroscopy. Chem. Sci. 2017, 8, 1752–1762. [Google Scholar] [CrossRef]
- Landi, A.; Borrelli, R.; Capobianco, A.; Peluso, A. Transient and Enduring Electronic Resonances Drive Coherent Long Distance Charge Transport in Molecular Wires. J. Phys. Chem. Lett. 2019, 10, 1845–1851. [Google Scholar] [CrossRef]
- Lee, H.T.; Carr, C.E.; Khutsishvili, I.; Marky, L.A. Effect of Loop Length and Sequence on the Stability of DNA Pyrimidine Triplexes with TAT Base Triplets. J. Phys. Chem. B 2017, 121, 9175–9184. [Google Scholar] [CrossRef]
- Carr, C.E.; Ganugula, R.; Shikiya, R.; Soto, A.M.; Marky, L.A. Effect of dC→d(m5C) substitutions on the folding of intramolecular triplexes with mixed TAT and C+GC base triplets. Biochimie 2018, 146, 156–165. [Google Scholar] [CrossRef]
- Keppler, M.D.; James, P.L.; Neidle, S. DNA sequence specificity of triplex-binding ligands. J. Biochem. 2003, 270, 4982–4992. [Google Scholar] [CrossRef]
- Reiling-Steffensmeier, C.; Marky, L.A. Chapter Sixteen-The Complementarity of the Loop to the Stem in DNA Pseudoknots Gives Rise to Local TAT Base-Triplets. Methods Enzymol. 2016, 567, 413. [Google Scholar] [PubMed]
- Natali, M.; Campagna, S.; Scandola, F. Photoinduced electron transfer across molecular bridges: Electron- and hole-transfer superexchange pathways. Chem. Soc. Rev. 2014, 43, 4005–4018. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Horsley, J.R.; Abell, A.D. A controllable mechanistic transition of charge transfer in helical peptides: From hopping to superexchange. RSC Adv. 2017, 7, 42370–42378. [Google Scholar] [CrossRef]
- Grozema, F.C.; Tonzani, S.; Berlin, Y.A.; Schatz, G.C.; Siebbeles, L.D.A.; Ratner, M.A. Effect of Structural Dynamics on Charge Transfer in DNA Hairpins. J. Am. Chem. Soc. 2008, 130, 5157–5166. [Google Scholar] [CrossRef] [PubMed]
- Renaud, N.; Berlin, Y.A.; Lewis, F.D.; Ratner, M.A. Between Superexchange and Hopping: An Intermediate Charge-Transfer Mechanism in Poly(A)-Poly(T) DNA Hairpins. J. Am. Chem. Soc. 2013, 135, 3953–3963. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, M.; Majima, T. Hole and excess electron transfer dynamics in DNA. Phys. Chem. Chem. Phys. 2012, 14, 11234–11244. [Google Scholar] [CrossRef]
- Menzel, J.P.; Groot, H.J.M.; Buda, F. Photoinduced Electron Transfer in Donor-Acceptor Complexes: Isotope Effect and Dynamic Symmetry Breaking. J. Phys. Chem. Lett. 2019, 10, 6504–6511. [Google Scholar] [CrossRef]
- Banyasz, A.; Ketola, T.; Martínez-Fernández, L.; Improta, R.; Markovitsi, D. Adenine radicals generated in alternating AT duplexes by direct absorption of low-energy UV radiation. Faraday Discuss. 2018, 207, 181–197. [Google Scholar] [CrossRef]
- Lewis, F.D.; Zhu, H.H.; Daublain, P.; Fiebig, T.; Raytchev, M.; Wang, Q.; Shafirovich, V. Crossover from Superexchange to Hopping as the Mechanism for Photoinduced Charge Transfer in DNA Hairpin Conjugates. J. Am. Chem. Soc. 2006, 128, 791–800. [Google Scholar] [CrossRef]
- Yu, F.B.; Li, P.; Li, G.Y.; Zhao, G.J.; Chu, T.S.; Han, K.L. A Near-IR Reversible Fluorescent Probe Modulated by Selenium for Monitoring Peroxynitrite and Imaging in Living Cells. J. Am. Chem. Soc. 2011, 133, 11030–11033. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.Y.; Yang, S.Q. Notable effect of water on excess electron attachment to aqueous DNA deoxyribonucleosides. Phys. Chem. Chem. Phys. 2019, 21, 8925–8932. [Google Scholar] [CrossRef] [PubMed]
- Uzawa, Y.; Wang, Z. Coherent multiple charge transfer in a superconducting NbN tunnel junction. Phys. Rev. Lett. 2005, 95, 017002. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Yu, C.; Jiang, S.C.; Zhang, T.; Gao, J.C.; Shi, S.; Pi, H.Q.; Weng, H.M.; Lu, R.F. Role of Shift Vector in High-Harmonic Generation from Noncentrosymmetric Topological Insulators under Strong Laser Fields. Phys. Rev. X 2022, 12, 021030. [Google Scholar] [CrossRef]
- Falke, S.M.; Rozzi, C.A.; Brida, D.; Maiuri, M.; Amato, M.; Ephraim, S.; DeSio, A.; Rubio, A.; Cerullo, G.; Molinari, E.; et al. Coherent ultrafast charge transfer in an organic photovoltaic blend. Science 2014, 344, 1001. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.X.; Zhou, Q.; Cao, B.F.; Li, B.; Wang, Z.R.; Zhang, X.L.; Yin, H.; Shi, Y. Theoretical reconsideration of the mechanism of the excited state proton transfer of indigo carmine in water. J. Mol. Liq. 2022, 347, 118365. [Google Scholar] [CrossRef]
- Newton, M.D. Bridge-Mediated Electron Transfer and Multiple Reaction Coordinates. Isr. J. Chem. 2004, 44, 83. [Google Scholar] [CrossRef]
- Zhou, P.W.; Tang, Z.; Li, P.; Liu, J.Y. Unraveling the Mechanism for Tuning the Fluorescence of Fluorescein Derivatives: The Role of the Conical Intersection and nπ* State. J. Phys. Chem. Lett. 2021, 12, 6478–6485. [Google Scholar] [CrossRef]
- Zhu, L.X.; Zhou, Q.; Wan, Y.F.; Li, Q.; Wan, Y.; Yin, H.; Shi, Y. The dynamical temporal behaviors of guanine–cytosine coherent charge transfer. Phys. Chem. Chem. Phys. 2023, 25, 10661–10670. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Duan, Y.P.; Song, L.L.; Zheng, D.Y.; Zhang, M.X.; Zhao, G.J. Charge-transfer mobility and electrical conductivity of PANI as conjugated organic semiconductors. J. Chem. Phys. 2017, 147, 114905. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.S.; Liu, J.Y.; Zhao, G.J.; Liu, L.; Han, K.L.; Cook, T.R.; Stang, P.J. Photophysical Properties of a Post-Self-Assembly Host/Guest Coordination Cage: Visible Light Driven Core-to-Cage Charge Transfer. J. Phys. Chem. Lett. 2015, 6, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.W.; Han, K.L. Unraveling the Detailed Mechanism of Excited-State Proton Transfer. Acc. Chem. Res. 2018, 51, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, C.H.; Hu, H.; Ye, S.T.; Funk, B.E.; Jain, D.; Hsiung, C.H.; Ning, G.; Liu, Y.; Li, S.; Zhang, X. AggFluor: Fluorogenic Toolbox Enables Direct Visualization of the Multi-Step Protein Aggregation Process in Live Cells. J. Am. Chem. Soc. 2020, 142, 17515–17523. [Google Scholar] [CrossRef] [PubMed]
- Amenabar, I.; Poly, S.; Goikoetxea, M.; Nuansing, W.; Lasch, P.; Hillenbrand, R. Hyperspectral infrared nanoimaging of organic samples based on Fourier transform infrared nanospectroscopy. Nat. Commun. 2017, 8, 14402. [Google Scholar] [CrossRef] [PubMed]
- Redmore, N.P.; Rubtsov, I.V.; Therien, M.J. Synthesis, Electronic Structure, and Electron Transfer Dynamics of (Aryl) ethynyl-Bridged Donor-Acceptor Systems. J. Am. Chem. Soc. 2003, 125, 87698778. [Google Scholar] [CrossRef] [PubMed]
- Campos-Gonzalez-Angulo, J.A.; Ribeiro, R.F.; Yuen-Zhou, J. Resonant catalysis of thermally activated chemical reactions with vibrational polaritons. Nat. Commun. 2019, 10, 4685. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Barbara, P.F.; Meyer, T.J.; Ratner, M.A. Contemporary Issues in Electron Transfer Research. J. Phys. Chem. 1996, 100, 13148–13168. [Google Scholar] [CrossRef]
- Jortner, J. Temperature dependent activation energy for electron transfer between biological molecules. J. Chem. Phys. 1976, 64, 4860. [Google Scholar] [CrossRef]
- Kavarnos, G.; Turro, N. Photosensitization by reversible electron transfer: Theories, experimental evidence, and examples. Chem. Rev. 1986, 86, 401–449. [Google Scholar] [CrossRef]
- Lemaur, V.; Steel, M.; Beljonne, D.; Bredas, J.L.; Cornil, J. Photoinduced Charge Generation and Recombination Dynamics in Model Donor/Acceptor Pairs for Organic Solar Cell Applications: A Full Quantum-Chemical Treatment. J. Am. Chem. Soc. 2005, 127, 6077–6086. [Google Scholar] [CrossRef]
- Bredas, J.L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, G.; Leeuwen, R. Nonequilibrium Many-Body Theory of Quantum Systems: A Modern Introduction; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. General performance of density functionals. J. Phys. Chem. A 2007, 111, 10439–10452. [Google Scholar] [CrossRef]
- Castro, A.; Appel, H.; Oliveira, M.; Rozzi, C.A.; Andrade, X.; Lorenzen, F.; Marques, M.A.L.; Gross, E.K.U.; Rubio, A. Octopus: A tool for the application of time-dependent density functional theory. Phys. Status Solidi B Basic Res. 2006, 243, 2465. [Google Scholar] [CrossRef]
- Andrade, X.; Rodriguez, J.A.; Strubbe, D.A.; Oliveira, M.J.T.; Nogueira, F.; Castro, A.; Muguerza, J.; Arruabarrena, A.; Louie, S.G.; Guzik, A.A. Time-Dependent Density-Functional Theory in Massively Parallel Computer Architectures: The Octopus Project. J. Phys. Condens. Matter. 2012, 24, 233202. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Andrade, X.; Castro, A.; Zueco, D.; Alonso, J.L.; Echenique, P.; Falceto, F.; Rubio, Á. Modified Ehrenfest formalism for efficient large-scale ab initio molecular dynamics. J. Chem. Theory Comput. 2009, 5, 728–742. [Google Scholar] [CrossRef]
- Castro, A.; Marques, M.A.L.; Rubio, A. Propagators for the time-dependent Kohn-Sham equations. J. Chem. Phys. 2004, 121, 3425. [Google Scholar] [CrossRef]
- Eisenmayer, T.J.; Buda, F. Real-time Simulations of Photoinduced Coherent Charge Transfer and Proton-Coupled Electron Transfer. ChemPhysChem 2014, 15, 3258–3263. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Lu, T.; Chen, Q.X. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Electronicstructure, electronic spectrum, and optical nonlinearity. Carbon 2020, 165, 461–467. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Li, Q.; Wan, Y.; Guo, M.; Yan, L.; Yin, H.; Shi, Y. Short-Range Charge Transfer in DNA Base Triplets: Real-Time Tracking of Coherent Fluctuation Electron Transfer. Molecules 2023, 28, 6802. https://doi.org/10.3390/molecules28196802
Zhu L, Li Q, Wan Y, Guo M, Yan L, Yin H, Shi Y. Short-Range Charge Transfer in DNA Base Triplets: Real-Time Tracking of Coherent Fluctuation Electron Transfer. Molecules. 2023; 28(19):6802. https://doi.org/10.3390/molecules28196802
Chicago/Turabian StyleZhu, Lixia, Qi Li, Yongfeng Wan, Meilin Guo, Lu Yan, Hang Yin, and Ying Shi. 2023. "Short-Range Charge Transfer in DNA Base Triplets: Real-Time Tracking of Coherent Fluctuation Electron Transfer" Molecules 28, no. 19: 6802. https://doi.org/10.3390/molecules28196802
APA StyleZhu, L., Li, Q., Wan, Y., Guo, M., Yan, L., Yin, H., & Shi, Y. (2023). Short-Range Charge Transfer in DNA Base Triplets: Real-Time Tracking of Coherent Fluctuation Electron Transfer. Molecules, 28(19), 6802. https://doi.org/10.3390/molecules28196802