
Synthesis and Thermal Studies of Two Phosphonium Tetrahydroxidohexaoxidopentaborate(1-) Salts: Single-Crystal XRD Characterization of [iPrPPh3][B5O6(OH)4]·3.5H2O and [MePPh3][B5O6(OH)4]·B(OH)3·0.5H2O †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
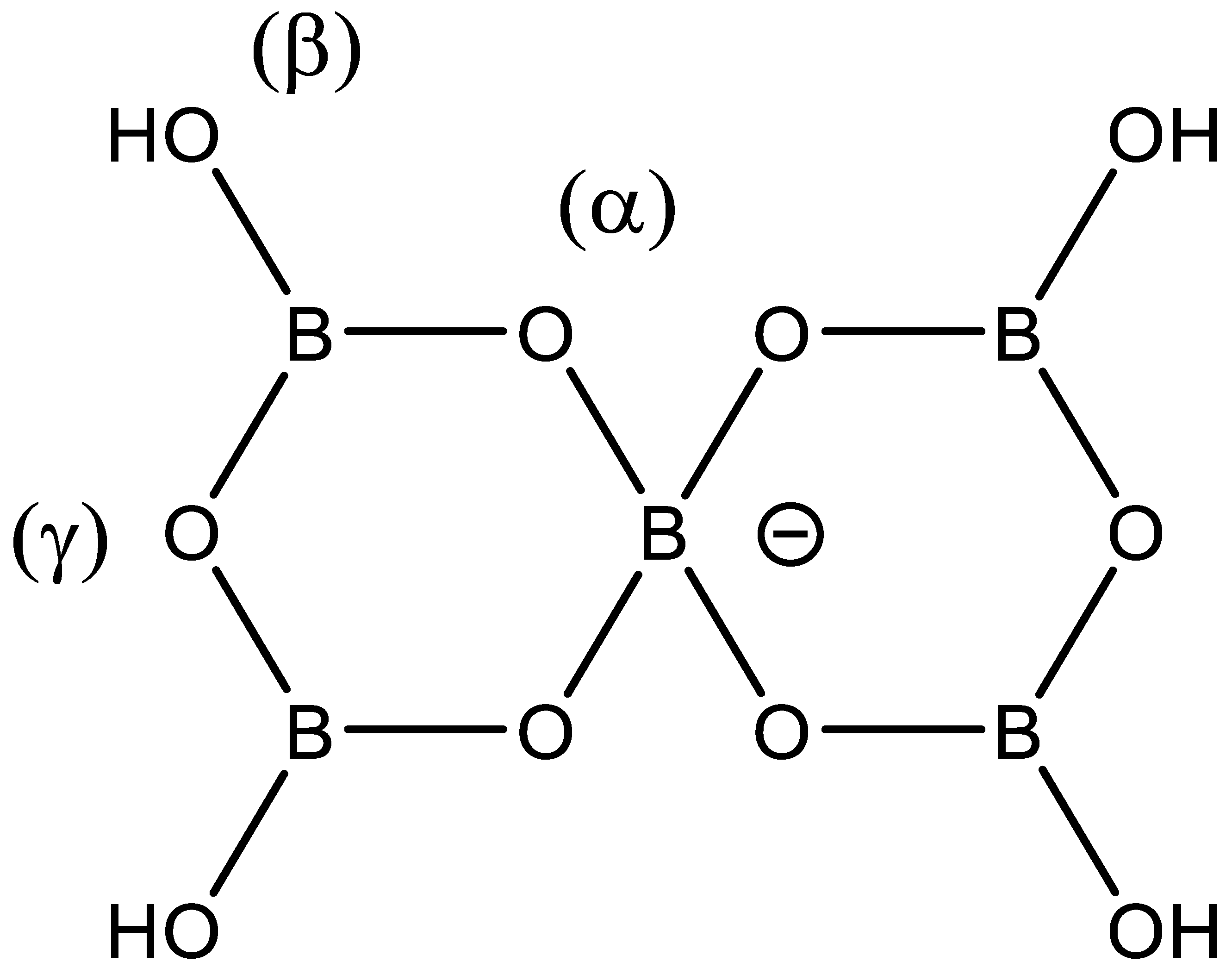
:1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Thermal Studies
2.3. Spectroscopic Studies
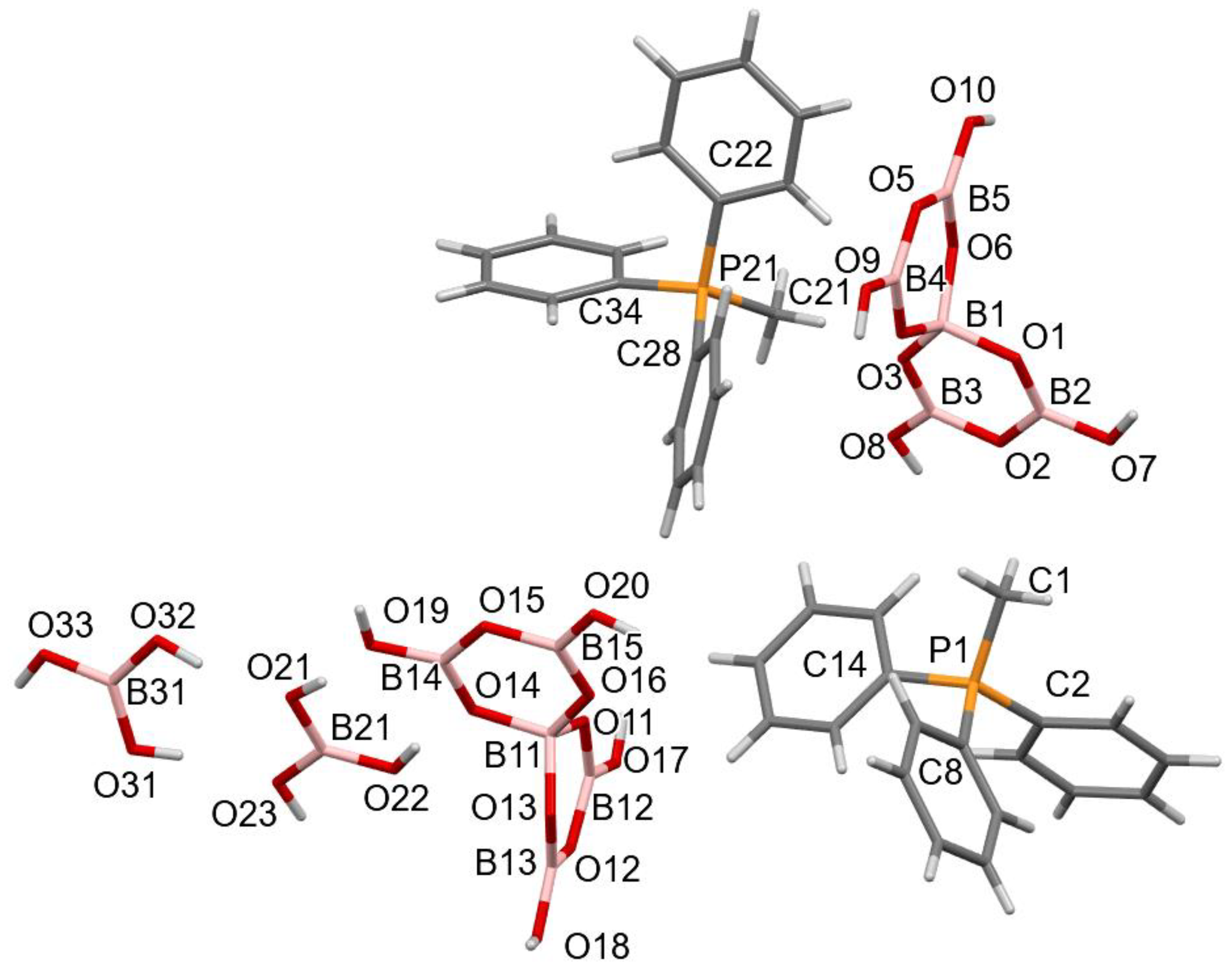
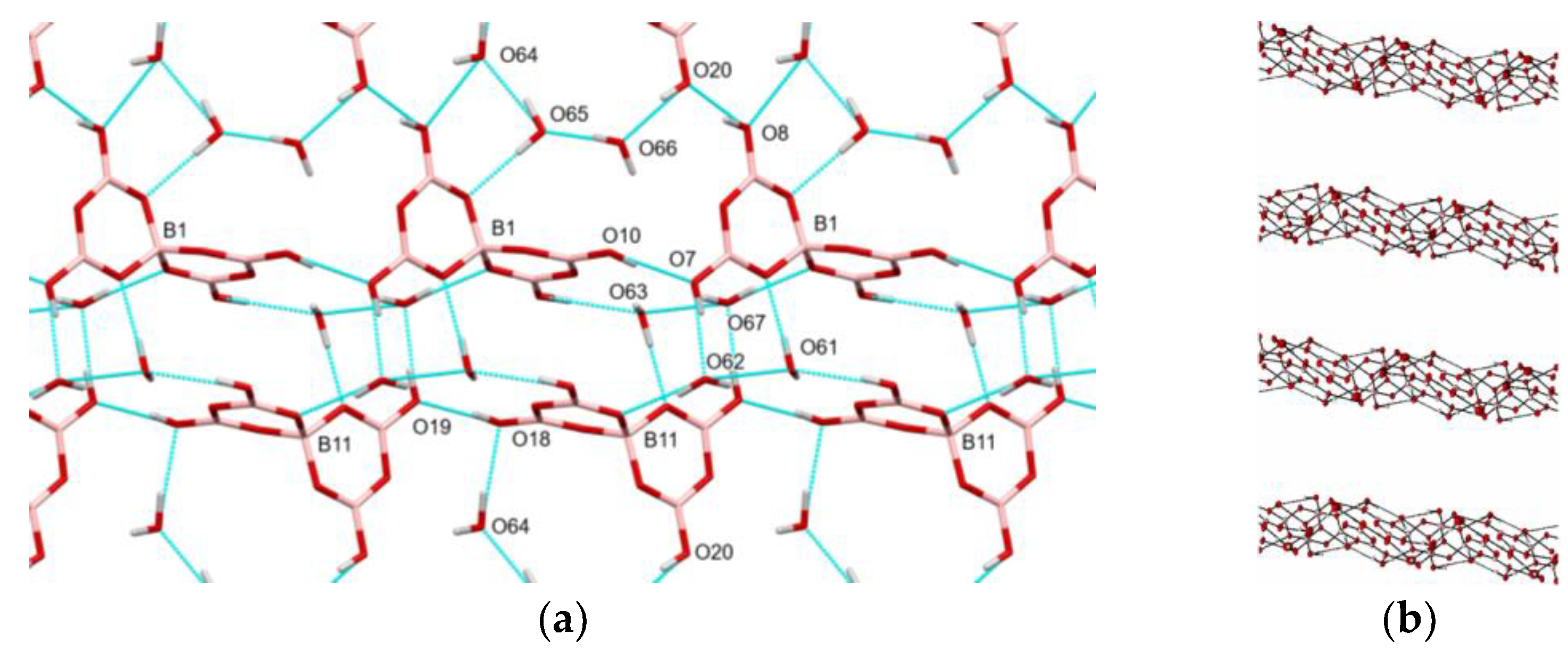
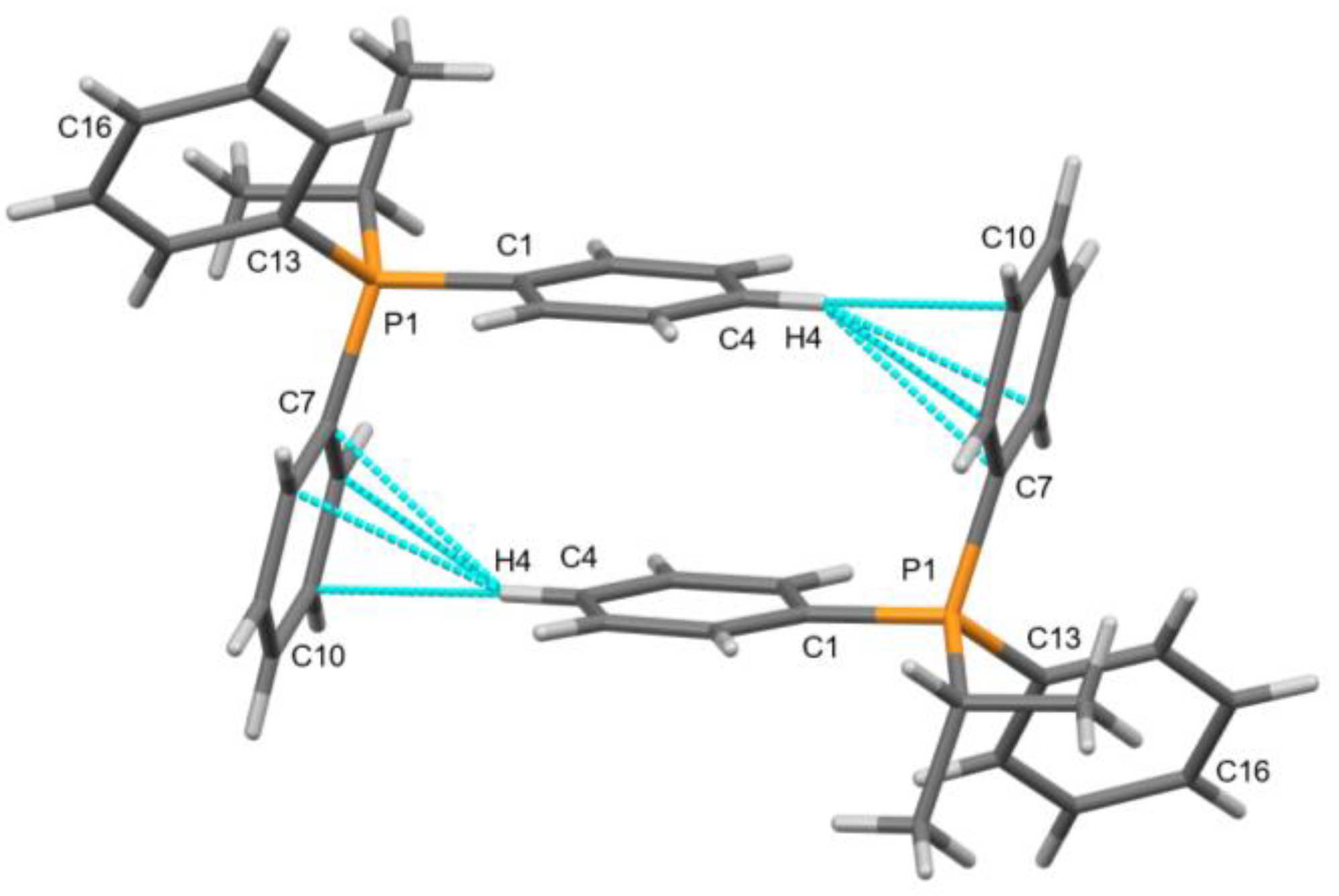
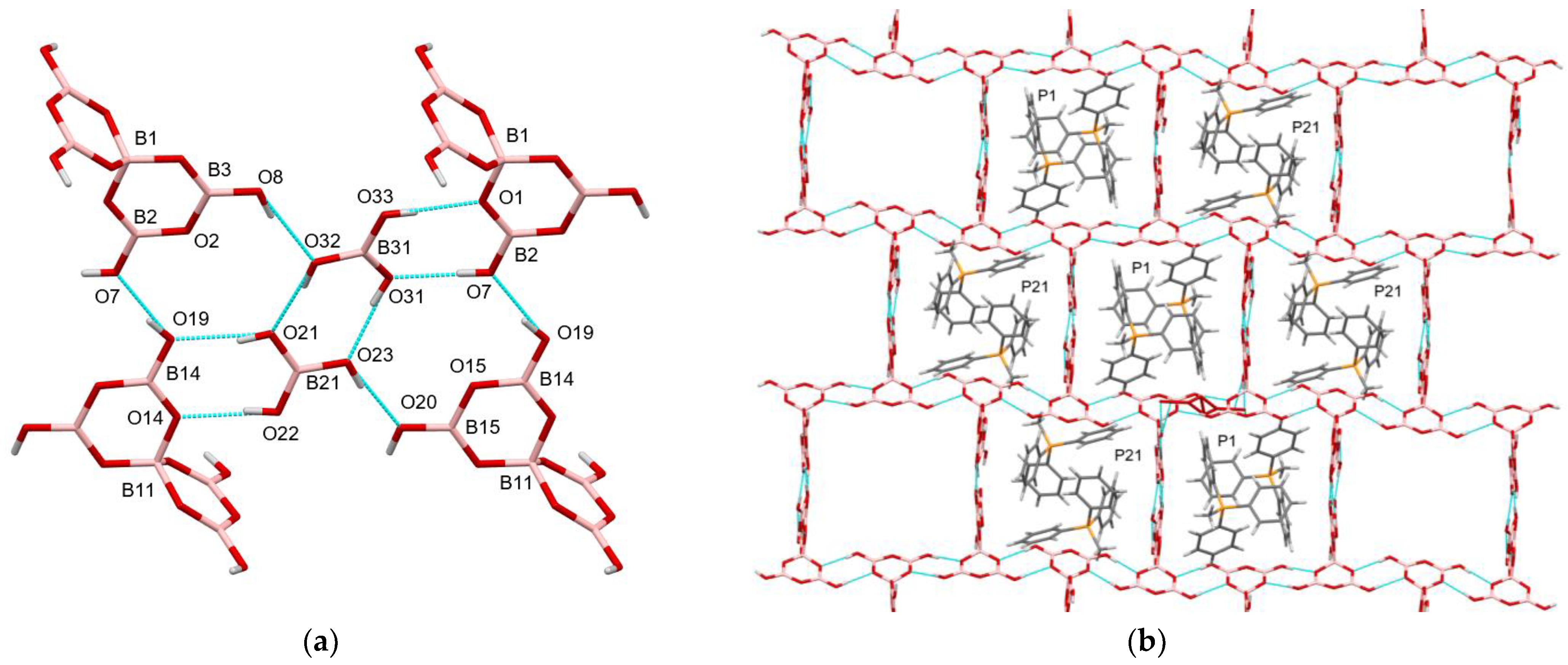
2.4. X-ray Crystallography
3. Materials and Experimental Methods
3.1. General
3.2. X-ray Crystallography
3.3. Preparation of [iPrPPh3][B5O6(OH)4].3.5H2O (1)
3.4. Synthesis of [MePPh3][B5O6(OH)4]·B(OH)3·0.5H2O (2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Farmer, J.B. Metal borates. Adv. Inorg. Chem Radiochem. 1982, 25, 187–237. [Google Scholar]
- Heller, G. A survey of structural types of borates and polyborates. Top. Curr. Chem. 1986, 131, 39–98. [Google Scholar]
- Belokonova, E.L. Borate crystal chemistry in terms of extended OD theory and symmetry analysis. Crystallogr. Rev. 2005, 11, 151–198. [Google Scholar] [CrossRef]
- Topnikova, A.P.; Belokoneva, E.L. The structure and classification of complex borates. Russ. Chem. Rev. 2019, 88, 204–228. [Google Scholar] [CrossRef]
- Burns, P.C.; Grice, J.D.; Hawthorne, F.C. Borate minerals I. Polyhedral clusters and fundamental building blocks. Can. Mineral. 1995, 33, 1131–1151. [Google Scholar]
- Grice, J.D.; Burns, P.C.; Hawthorne, F.C. Borate minerals II. A hierarchy of structures based upon the borate fundamental building block. Can. Mineral. 1999, 37, 731–762. [Google Scholar]
- Christ, C.L.; Clark, J.R. A crystal-chemical classification of borate structures with emphasis on hydrated borates. Phys. Chem. Miner. 1977, 2, 59–87. [Google Scholar] [CrossRef]
- Touboul, M.; Penin, N.; Nowogrocki, G. Borates: A survey of main trends concerning crystal chemistry, polymorphism and dehydration process of alkaline and pseudo-alkaline borates. Solid State Sci. 2003, 5, 1327–1342. [Google Scholar] [CrossRef]
- Mutailipu, M.; Poeppelmeier, K.R.; Pan, S. Borates: A rich source for optical materials. Chem. Rev. 2021, 121, 1130–1202. [Google Scholar] [CrossRef]
- Beckett, M.A. Recent Advances in crystalline hydrated borates with non-metal or transition-metal complex cations. Coord. Chem. Rev. 2016, 323, 2–14. [Google Scholar] [CrossRef]
- Schubert, D.M.; Smith, R.A.; Visi, M.Z. Studies of crystalline non-metal borates. Glass Technol. 2003, 44, 63–70. [Google Scholar]
- Schubert, D.M.; Knobler, C.B. Recent studies of polyborate anions. Phys. Chem. Glasses Eur. J. Glass Sci. Technol. B 2009, 50, 71–78. [Google Scholar]
- Schubert, D.M. Borates in industrial use. Struct. Bond. 2003, 105, 1–40. [Google Scholar]
- Schubert, D.M. Boron oxide, boric acid, and borates. In Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 1–68. [Google Scholar]
- Schubert, D.M. Hydrated zinc borates and their industrial use. Molecules 2019, 24, 2419. [Google Scholar] [CrossRef]
- Becker, P. Borate materials in nonlinear optics. Adv. Mater. 1998, 10, 979–992. [Google Scholar] [CrossRef]
- Xin, S.-S.; Zhou, M.-H.; Beckett, M.A.; Pan, C.-Y. Recent advances in crystalline oxidopolyborate complexes of d-block or p-block metals: Structural aspects, synthesis, and physical properties. Molecules 2021, 26, 3815. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Horton, P.N.; Jones, C.L. Polyborate anions partnered with large non-metal cations: Triborate(1-), pentaborate(1-) and heptaborate(2-) salts. Eur. J. Inorg. Chem. 2017, 4510–4518. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Timmis, J.L.; Varma, K.S. Synthesis, thermal properties and structural characterization of the tetraphenylphosphonium pentaborate salt, [PPh4][B5O6(OH)4].1.5H2O. Inorg. Chim. Acta. 2012, 383, 199–203. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Knox, D.A.; Timmis, J.L. Structural (XRD) and thermal (DSC, TGA) and BET analysis of materials derived from non-metal cation pentaborate salts. Dalton Trans. 2010, 39, 3944–3951. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Timmis, J.L.; Varma, K.S. Templated heptaborate and pentaborate salts of cyclo-alkylammonium cations: Structural and thermal properties. Dalton Trans. 2012, 41, 4396–4403. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Timmis, J.L. Triborate and pentaborate salts of non-metal cations derived from N-substituted piperazines: Synthesis and structural (XRD) and thermal properties. RSC Adv. 2013, 3, 15181–15191. [Google Scholar] [CrossRef]
- Beckett, M.A.; Brellocks, B.; Chizhevsky, I.T.; Damhus, T.; Hellwich, K.-H.; Kennedy, J.D.; Laitinen, R.; Powell, W.H.; Rabinovich, D.; Vinas, C.; et al. Nomenclature for boranes and related species (IUPAC Recommendations 2019). Pure Appl. Chem. 2020, 92, 355–381. [Google Scholar] [CrossRef]
- Visi, M.Z.; Knobler, C.B.; Owen, J.J.; Khan, M.I.; Schubert, D.M. Structures of self-assembled nonmetal borates derived from α,ω-diaminoalkanes. Cryst. Growth Des. 2006, 6, 538–545. [Google Scholar] [CrossRef]
- Anderson, J.L.; Eyring, E.M.; Whittaker, M.P. Temperature jump rate studies of polyborate formation in aqueous boric acid. J. Phys. Chem. 1964, 68, 1128–1132. [Google Scholar] [CrossRef]
- Salentine, G. High-field 11B NMR of alkali borate. Aqueous polyborate equilibria. Inorg. Chem. 1983, 22, 3920–3924. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Q.; Lan, Y.; Wang, D.; Zhang, L.; Tang, X.; Yang, S.; Luo, Z.; Tian, G. Speciation of borate in aqueous solution studied experimentally by potentiometry and Raman spectroscopy and computationally by DFT calculations. New J. Chem. 2023, 47, 8499–8506. [Google Scholar] [CrossRef]
- Corbett, P.T.; Leclaire, J.; Vial, L.; West, K.R.; Wietor, J.-L.; Sanders, J.K.M.; Otto, S. Dynamic combinatorial chemistry. Chem. Rev. 2006, 106, 3652–3711. [Google Scholar] [CrossRef]
- Sola, J.; Lafuente, M.; Atcher, J.; Alfonso, I. Constitutional self-selection from dynamic combinatorial libraries in aqueous solution through supramolecular interactions. Chem. Commun. 2014, 50, 4564–4566. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular synthons: Validation and ranking of intermolecular interaction energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Freyhardt, C.C.; Wiebcke, M.; Felsche, J.; Engelhardt, G. Clathrates and three dimensional host structures of hydrogen bonded pentaborate [B5O6(OH)4]− ions: Pentaborates with cations NMe4+, NEt4+, NPhMe3+ and pipH+ (pipH+ = piperidinium). Z. Naturforsch B. 1993, 48, 978–985. [Google Scholar]
- Beckett, M.A.; Coles, S.J.; Horton, P.N.; Rixon, T.A. Structural (XRD) characterization and an analysis of H-bonding motifs in some tetrahydroxidohexaoxidopentaborate(1-) salts of N-substituted guanidinium cations. Molecules 2023, 28, 3273. [Google Scholar] [CrossRef] [PubMed]
- Beckett, M.A.; Horton, P.N.; Coles, S.J.; Kose, D.A.; Kreuziger, A.-M. Structural and thermal studies of non-metal cation pentaborate salts with cations derived from 1,5-diazobicyclo[4.3.0]non-5-ene, 1,8-diazobicyclo[5.4.0]undec-7-ene and 1,8-bis(dimethylamino)naphthalene. Polyhedron 2012, 38, 157–161. [Google Scholar] [CrossRef]
- Yang, Y.; Fu, D.S.; Li, G.F.; Zhang, Y. Synthesis, crystal structure, and variable-temperature-luminescent property of the organically templated pentaborate [C10N2H9][B5O6(OH)4]·H3BO3·H2O. Z. Anorg. Chem. 2013, 639, 722–727. [Google Scholar] [CrossRef]
- Freyhardt, C.C.; Wiebcke, M.; Felsche, J.; Englehardt, G. N(nPr4)[B5O6(OH)4][B(OH)3]2 and N(nBu4)[B5O6(OH)4][B(OH)3]2: Clathrates with a diamondoid arrangement of hydrogen bonded pentaborate anions. J. Inclusion Phenom. Mol. Recogn. Chem. 1994, 18, 161–175. [Google Scholar] [CrossRef]
- Ferrillo, R.G.; Granzow, A. Thermogravimetric study of phosphonium halides. Thermochem. Acta 1981, 45, 177–187. [Google Scholar] [CrossRef]
- Brauner, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Timmis, J.L. Characterization of Non-Metal Cation Polyborate Salts and Silicate Solutions. Ph.D. Thesis, Bangor University, Bangor, UK, 2011. [Google Scholar]
- Schubert, U.; Husing, N. Synthesis of Inorganic Materials, 2nd ed.; Wiley VCH: Weinheim, Germany, 2007; Volume Ch 6, pp. 305–352. [Google Scholar]
- Li, J.; Xia, S.; Gao, S. FT-IR and Raman spectroscopic study of hydrated borates. Spectrochim. Acta 1995, 51, 519–532. [Google Scholar]
- Grim, S.O.; McFarlane, W.; Davidoff, E.F.; Marks, T.J. Phosphorus-31 chemical shifts of quaternary phosphonium salts. J. Am. Chem. Soc. 1966, 70, 581–584. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Davies, R.A.; Horton, P.N.; Jones, C.L. Pentaborate(1−) salts templated by substituted pyrrolidinium cations: Synthesis, structural characterization, and modelling of solid-state H-bond interactions by DFT calculations. Dalton Trans. 2015, 44, 7032–7040. [Google Scholar] [CrossRef]
- Beckett, M.A.; Brassington, D.S.; Owen, P.; Hursthouse, M.B.; Light, M.E.; Malik, K.M.A.; Varma, K.S. π-Bonding in B-O ring species: Lewis acidity of Me3B3O3, synthesis of Me3B3O3 amine adducts, and the crystal and molecular structure of Me3B3O3.NH2iBu.MeB(OH)2. J. Organomet. Chem. 1999, 585, 7–11. [Google Scholar] [CrossRef]
- Hosten, E.; Gerber, T.; Betz, R. Crystal structure of methyltriphenylphosphonium iodide, C19H18IP. Z. Kristallogr. NCS 2012, 227, 331–332. [Google Scholar]
- Jaliliana, E.; Lidi, S. Bis(isopropyltriphenylphosphonium)di-μ-iodidobis[iodidocopper(I)]. Acta Cryst. 2010, E66, m432–m433. [Google Scholar]
- Hunter, C.A.; Sanders, J.K.M. The nature of π-π-interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Supramolecular motifs: Concerted multiple phenyl embraces between PPh4+ cations are attractive and ubiquitous. Chem. Eur. J. 1996, 2, 481–486. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic chemistry. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Duisenberg, A.J.M. Indexing in single-crystal diffractometry with an obstinate list of reflections. J. Appl. Cryst. 1992, 25, 92–96. [Google Scholar] [CrossRef]
- Hooft, R.; Nonius, B.V. COLLECT, Data Collection Software. 1998. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 1997, 276, 307–326. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. 1995, A51, 33–37. [Google Scholar] [CrossRef]
- Blessing, R.H. Outlier Treatment in Data Merging. J. Appl. Cryst. 1997, 30, 421–426. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of ShelX. Acta Cryst. 2008, A64, 339–341. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Timmis, J.L. Synthesis and Thermal Studies of Two Phosphonium Tetrahydroxidohexaoxidopentaborate(1-) Salts: Single-Crystal XRD Characterization of [iPrPPh3][B5O6(OH)4]·3.5H2O and [MePPh3][B5O6(OH)4]·B(OH)3·0.5H2O. Molecules 2023, 28, 6867. https://doi.org/10.3390/molecules28196867
Beckett MA, Horton PN, Hursthouse MB, Timmis JL. Synthesis and Thermal Studies of Two Phosphonium Tetrahydroxidohexaoxidopentaborate(1-) Salts: Single-Crystal XRD Characterization of [iPrPPh3][B5O6(OH)4]·3.5H2O and [MePPh3][B5O6(OH)4]·B(OH)3·0.5H2O. Molecules. 2023; 28(19):6867. https://doi.org/10.3390/molecules28196867
Chicago/Turabian StyleBeckett, Michael A., Peter N. Horton, Michael B. Hursthouse, and James L. Timmis. 2023. "Synthesis and Thermal Studies of Two Phosphonium Tetrahydroxidohexaoxidopentaborate(1-) Salts: Single-Crystal XRD Characterization of [iPrPPh3][B5O6(OH)4]·3.5H2O and [MePPh3][B5O6(OH)4]·B(OH)3·0.5H2O" Molecules 28, no. 19: 6867. https://doi.org/10.3390/molecules28196867
APA StyleBeckett, M. A., Horton, P. N., Hursthouse, M. B., & Timmis, J. L. (2023). Synthesis and Thermal Studies of Two Phosphonium Tetrahydroxidohexaoxidopentaborate(1-) Salts: Single-Crystal XRD Characterization of [iPrPPh3][B5O6(OH)4]·3.5H2O and [MePPh3][B5O6(OH)4]·B(OH)3·0.5H2O. Molecules, 28(19), 6867. https://doi.org/10.3390/molecules28196867