
In Vitro and In Silico Biological Studies of 4-Phenyl-2-quinolone (4-PQ) Derivatives as Anticancer Agents

 ,
,  ,
, 
 ,
,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
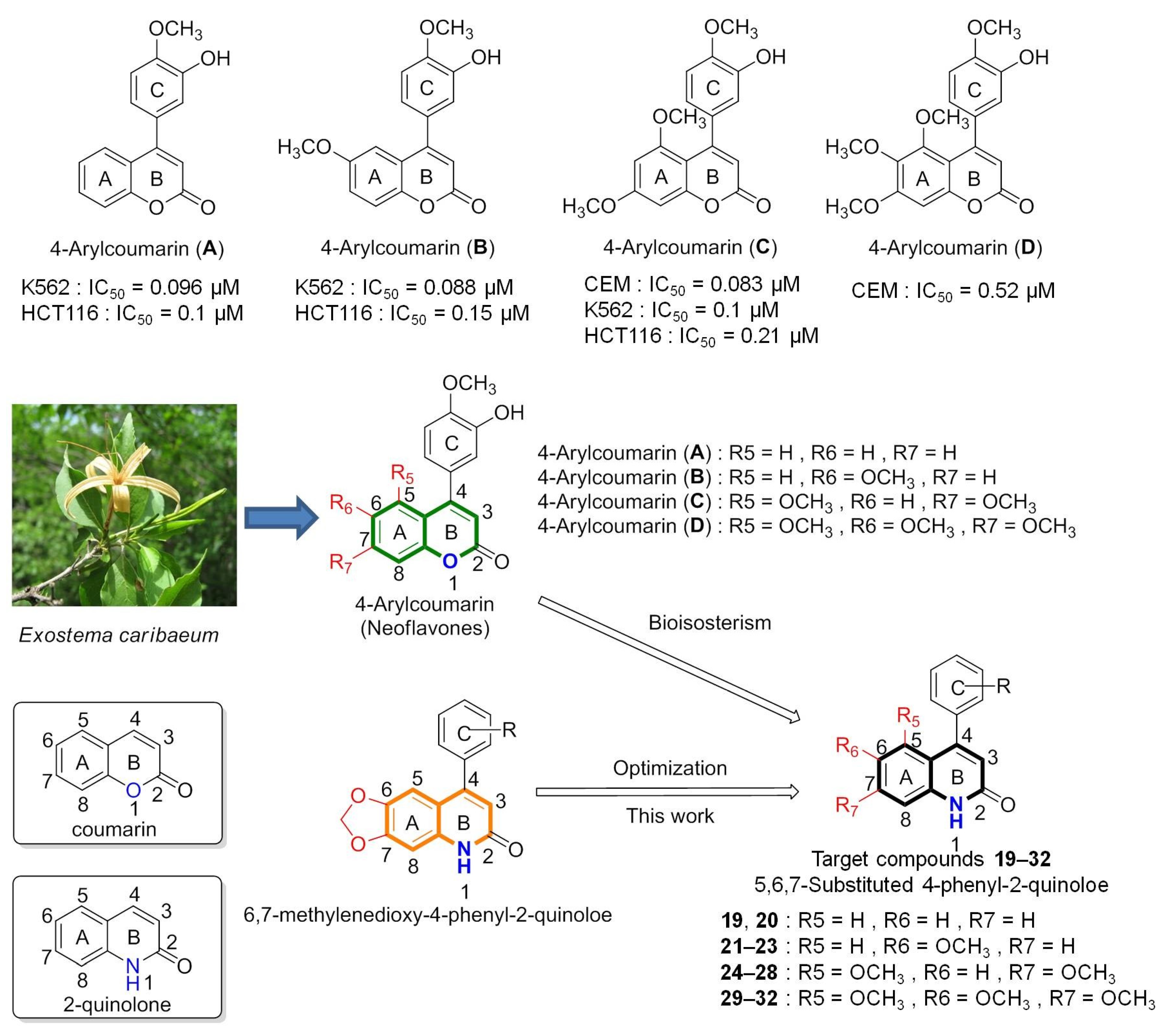
2.1. Chemistry
2.2. Anticancer Activities and SAR Studies of New 4-PQ Derivatives
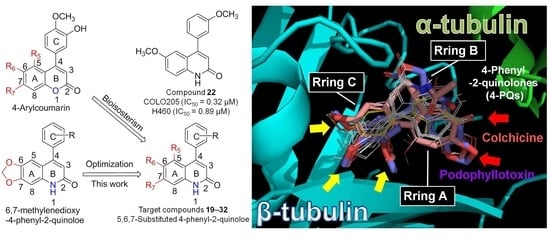
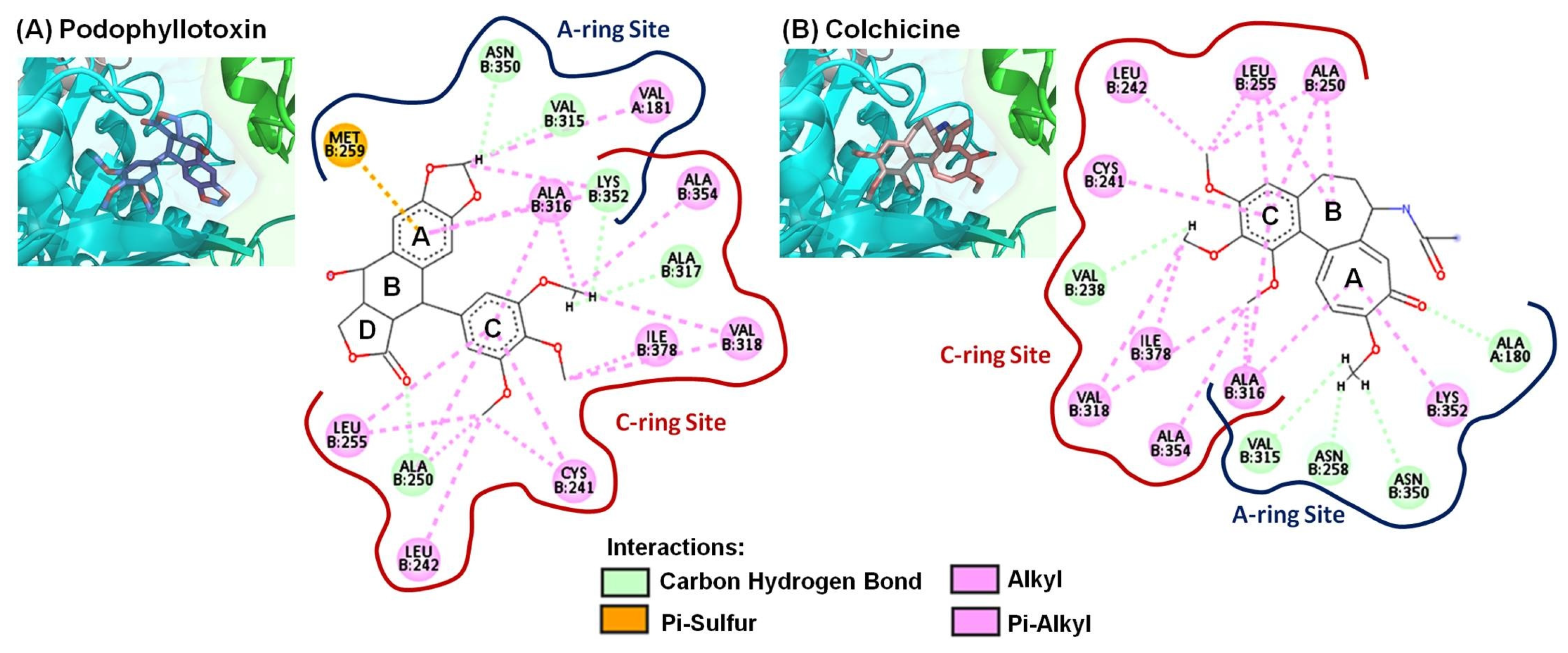
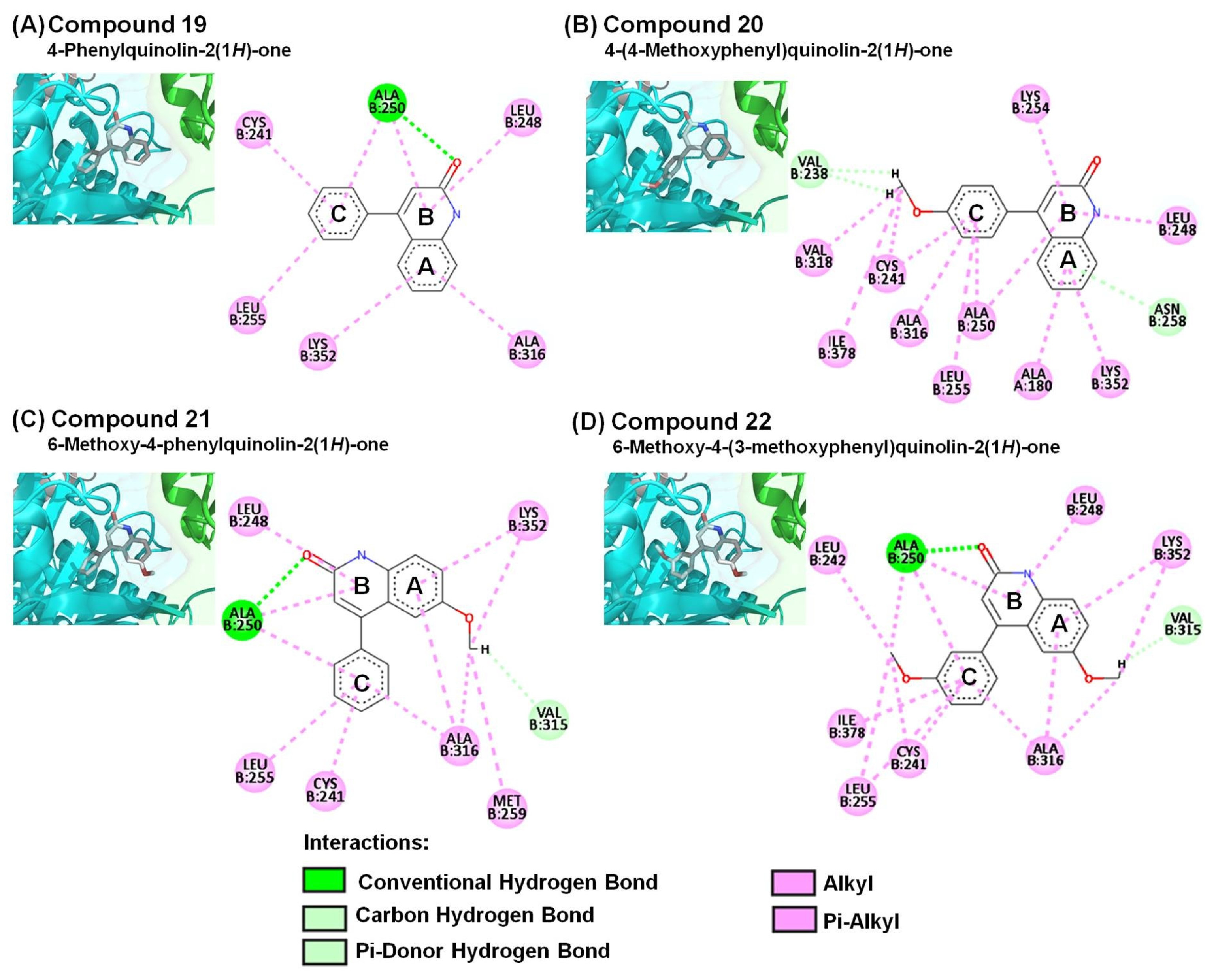
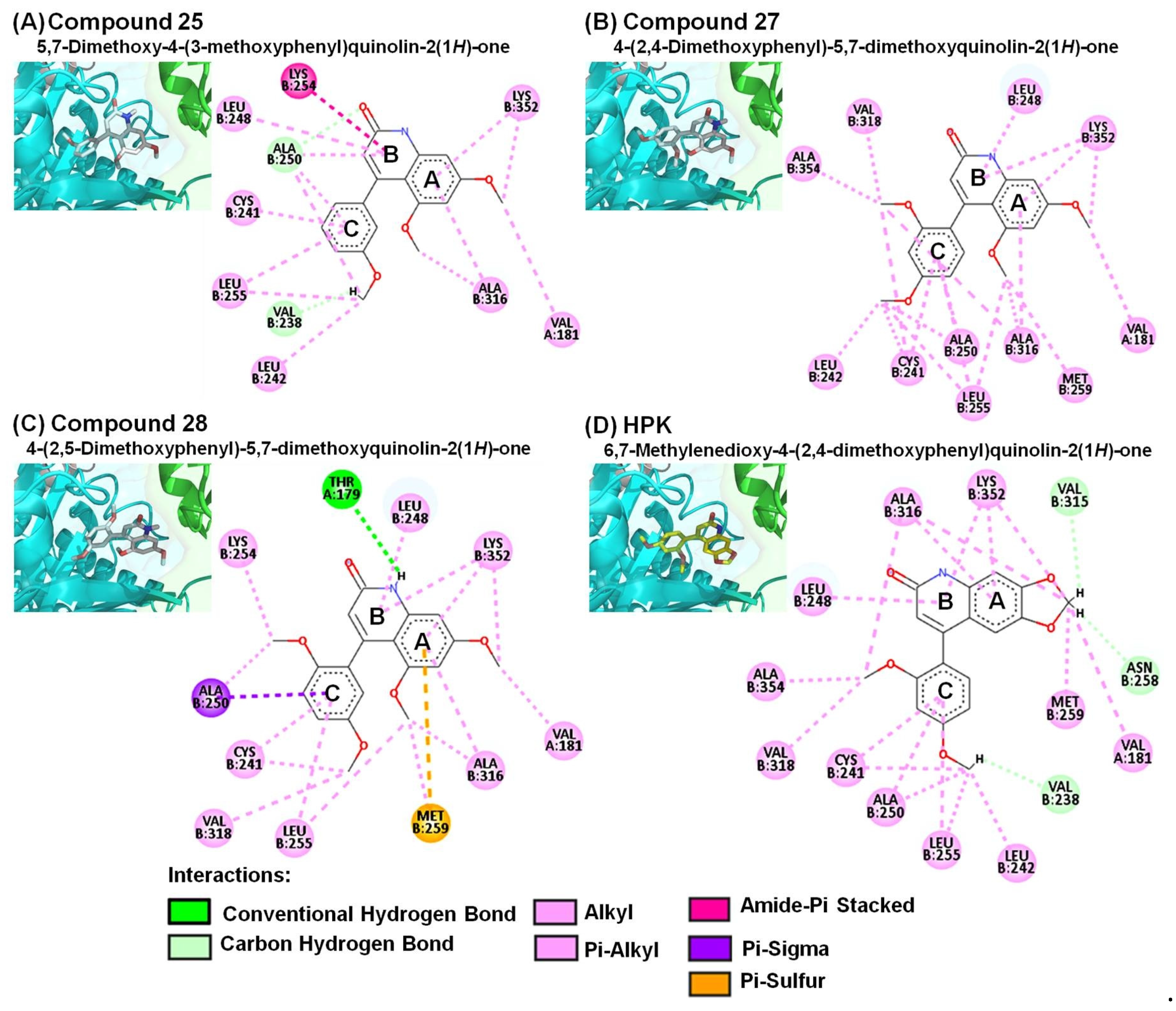
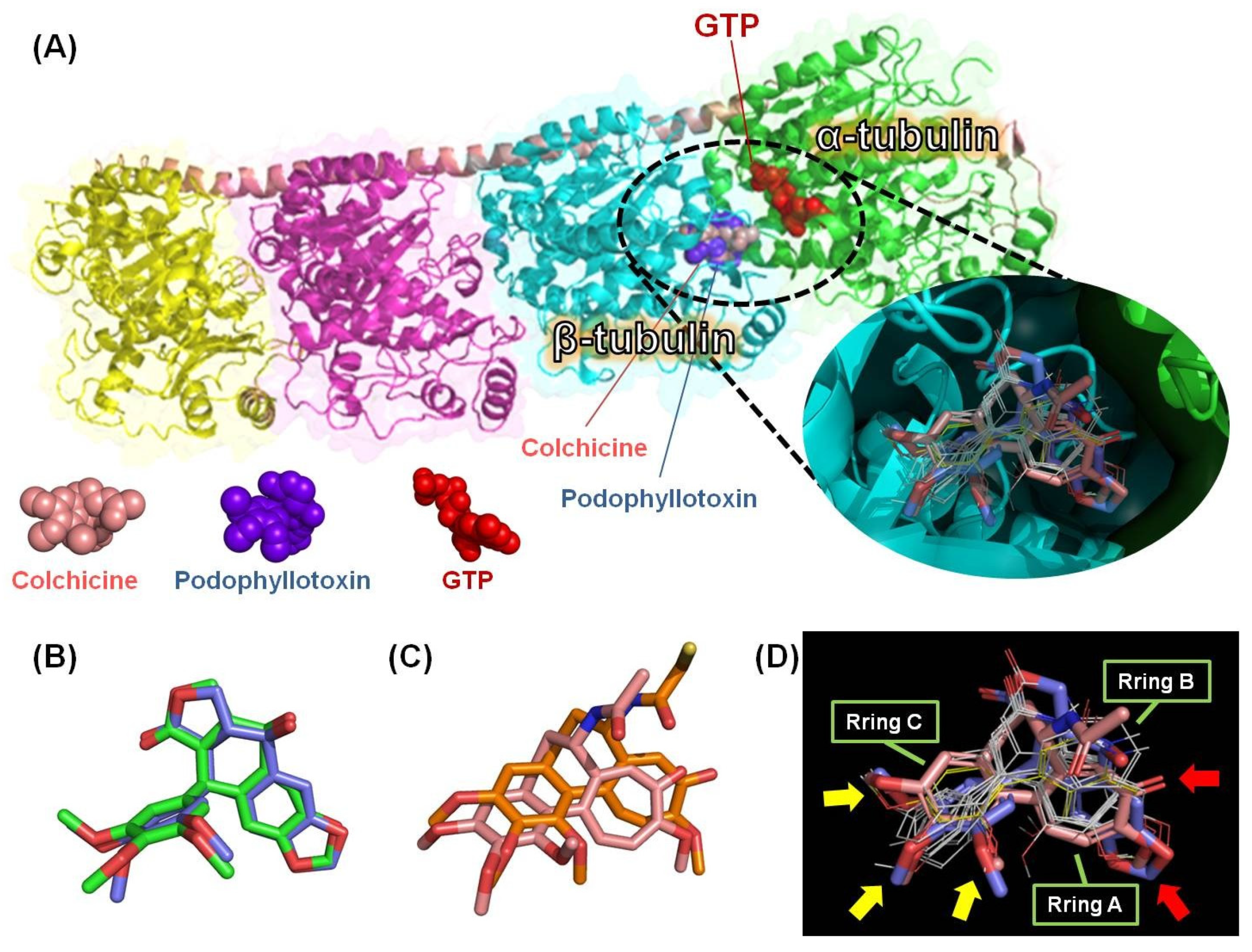
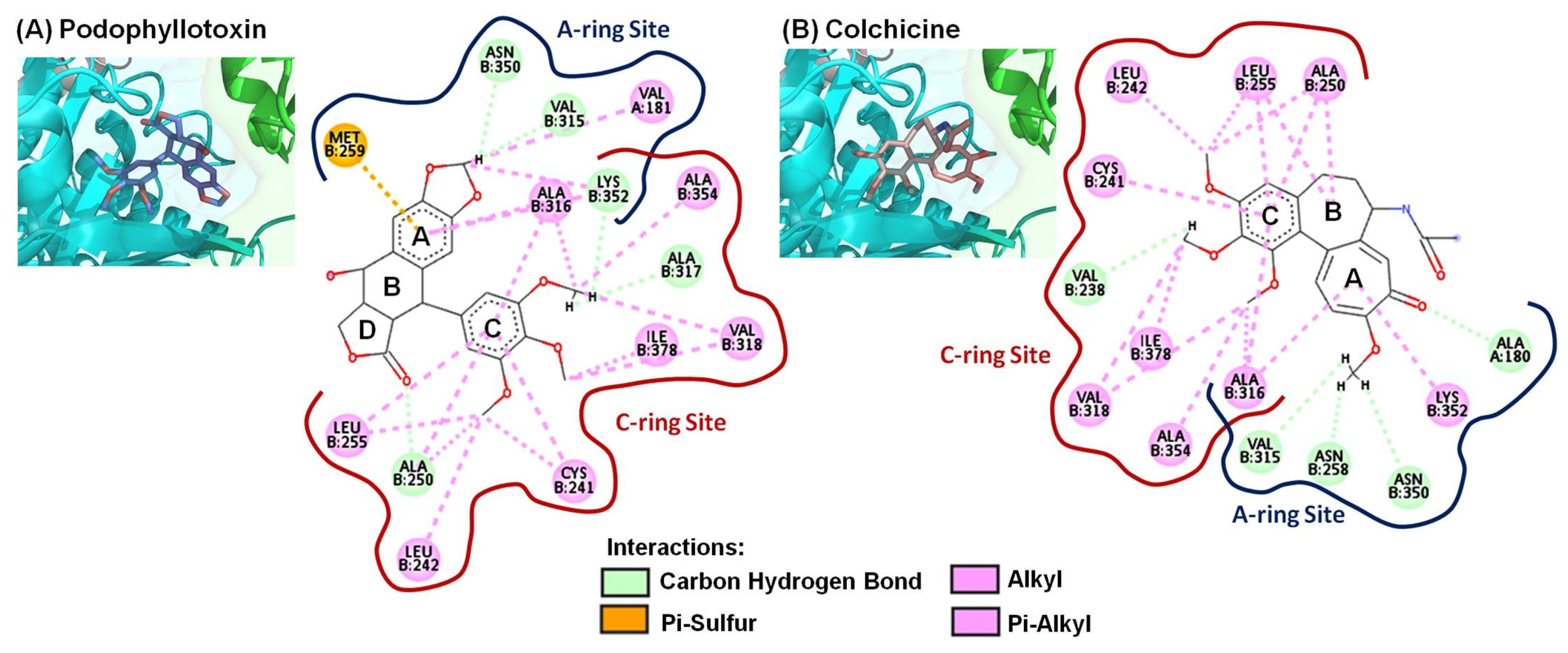
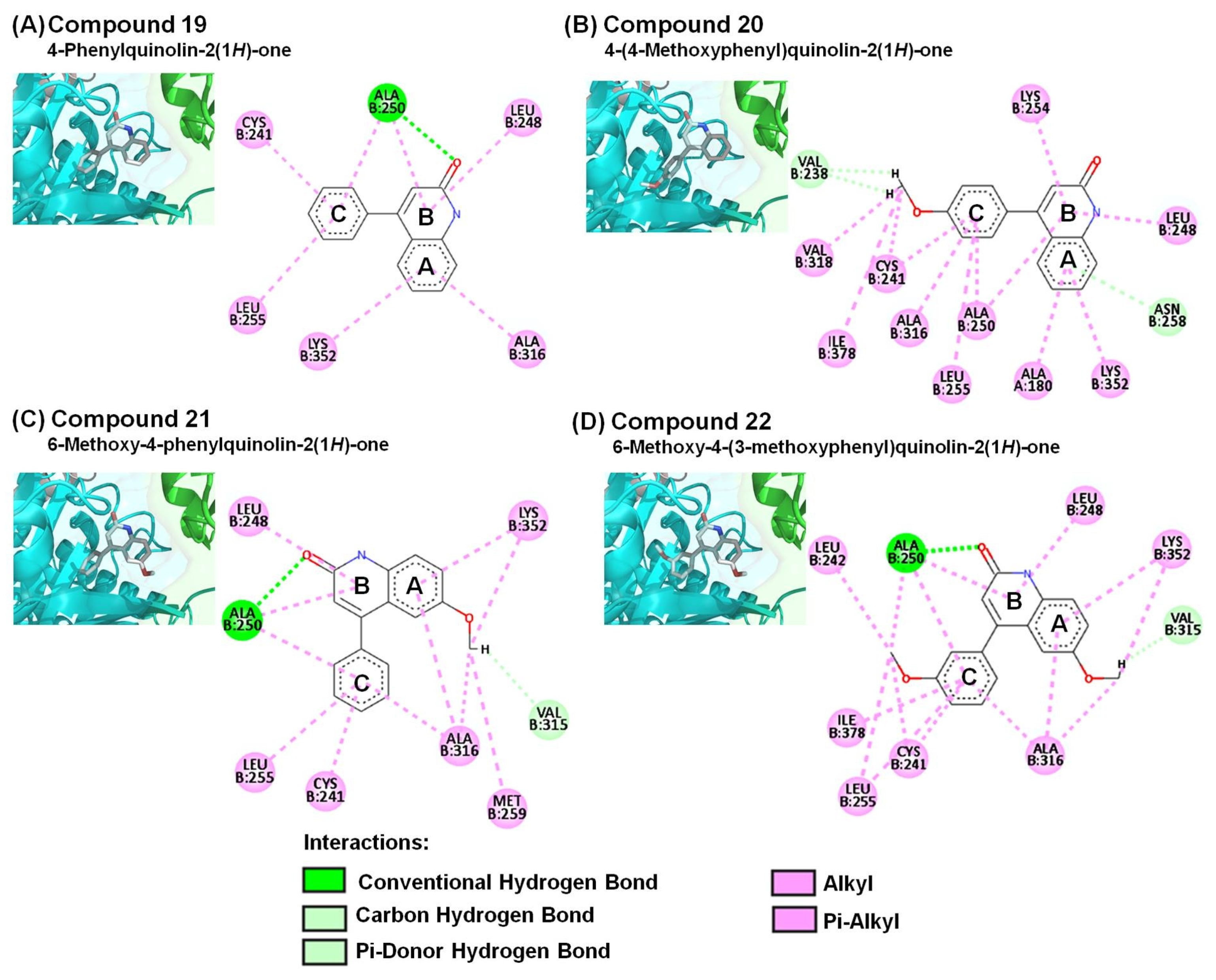
2.3. Molecular Modeling of 4-Phenyl-2-quinolone (4-PQ) Derivatives Proposes Their Docking into the Colchicine-Binding Site
2.4. In Silico Prediction of Drug-Likeness Studies
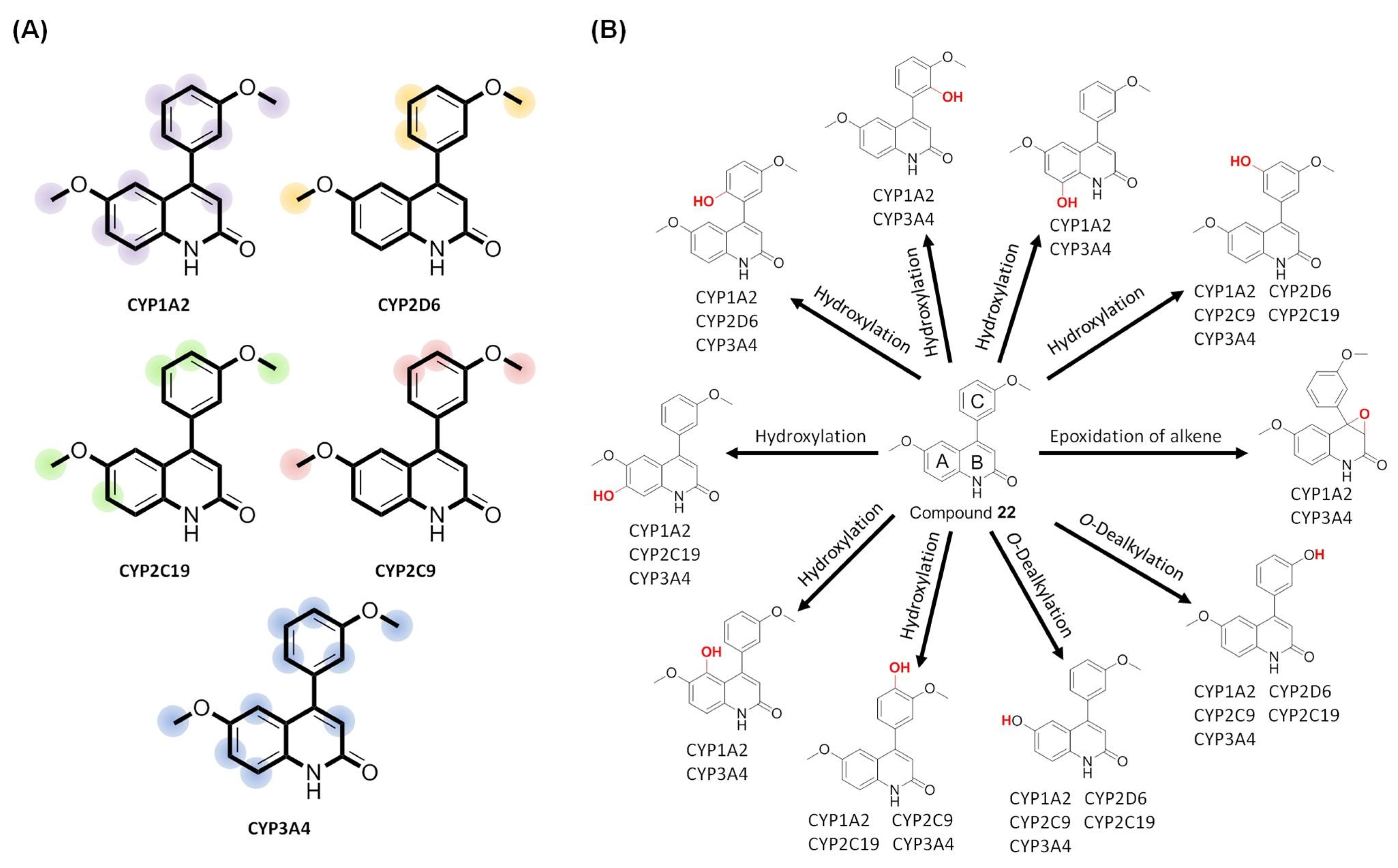
2.5. In Silico Analysis of Potential PK (ADMET) and Toxicological Properties (T)
3. Materials and Methods
3.1. Materials and Physical Measurements
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of Benzoylacetates (3a–e)
3.2.2. General Procedure for the Synthesis of Benzoylacetanilides (5–18)
3.2.3. General Procedure for the Synthesis of 5-, 6-, 7-Methoxy-substituted 4-Phenyl-2-quinolones (19–32)
3.3. MTT Assay for Antiproliferative Activity
3.4. Molecular Docking
3.5. In Silico Predictions of Physicochemical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2022, in press. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Miller, D.D.; Li, W. Molecular interactions at the colchicine binding site in tubulin: An X-ray crystallography perspective. Drug Discov. Today 2022, 27, 759–776. [Google Scholar] [CrossRef]
- Chen, Y.F.; Lin, Y.C.; Huang, P.K.; Chan, H.C.; Kuo, S.C.; Lee, K.H.; Huang, L.J. Design and synthesis of 6,7-methylenedioxy-4-substituted phenylquinolin-2(1H)-one derivatives as novel anticancer agents that induce apoptosis with cell cycle arrest at G2/M phase. Bioorganic Med. Chem. 2013, 21, 5064–5075. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Lin, Y.C.; Morris-Natschke, S.L.; Wei, C.F.; Shen, T.C.; Lin, H.Y.; Hsu, M.H.; Chou, L.C.; Zhao, Y.; Kuo, S.C.; et al. Synthesis and SAR studies of novel 6,7,8-substituted 4-substituted benzyloxyquinolin-2(1H)-one derivatives for anticancer activity. Br. J. Pharmacol. 2015, 172, 1195–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamel, E. Antimitotic natural products and their interactions with tubulin. Med. Res. Rev. 1996, 16, 207–231. [Google Scholar] [CrossRef]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; Gómez-Zurita, M.A.; García, P.A.; San Feliciano, A. Chemoinduction of cytotoxic selectivity in Podophyllotoxin-related lignans. Phytochem. Rev. 2003, 2, 219–233. [Google Scholar] [CrossRef]
- Gensler, W.J.; Murthy, C.D.; Trammell, M.H. Nonenolizable podophyllotoxin derivatives. J. Med. Chem. 1977, 20, 635–644. [Google Scholar] [CrossRef]
- Magedov, I.V.; Manpadi, M.; Rozhkova, E.; Przheval’skii, N.M.; Rogelj, S.; Shors, S.T.; Steelant, W.F.; Van Slambrouck, S.; Kornienko, A. Structural simplification of bioactive natural products with multicomponent synthesis: Dihydropyridopyrazole analogues of podophyllotoxin. Bioorganic Med. Chem. Lett. 2007, 17, 1381–1385. [Google Scholar] [CrossRef]
- Magedov, I.V.; Manpadi, M.; Slambrouck, S.V.; Steelant, W.F.; Rozhkova, E.; Przheval’skii, N.M.; Rogelj, S.; Kornienko, A. Discovery and investigation of antiproliferative and apoptosis-inducing properties of new heterocyclic podophyllotoxin analogues accessible by a one-step multicomponent synthesis. J. Med. Chem. 2007, 50, 5183–5192. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Suresh, P.; Mallareddy, A.; Kumar, B.A.; Reddy, P.V.; Raju, P.; Tamboli, J.R.; Shaik, T.B.; Jain, N.; Kalivendi, S.V. Synthesis of a new 4-aza-2,3-didehydropodophyllotoxin analogues as potent cytotoxic and antimitotic agents. Bioorganic Med. Chem. 2011, 19, 2349–2358. [Google Scholar] [CrossRef]
- Semenova, M.N.; Kiselyov, A.S.; Tsyganov, D.V.; Konyushkin, L.D.; Firgang, S.I.; Semenov, R.V.; Malyshev, O.R.; Raihstat, M.M.; Fuchs, F.; Stielow, A.; et al. Polyalkoxybenzenes from plants. 5. Parsley seed extract in synthesis of azapodophyllotoxins featuring strong tubulin destabilizing activity in the sea urchin embryo and cell culture assays. J. Med. Chem. 2011, 54, 7138–7149. [Google Scholar] [CrossRef]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; García, P.A.; Gómez-Zurita, M.A.; García-Grávalos, M.D.; de la Iglesia-Vicente, J.; Gajate, C.; An, F.; Mollinedo, F.; et al. Synthesis and biological evaluation of new selective cytotoxic cyclolignans derived from podophyllotoxin. J. Med. Chem. 2004, 47, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; del Corral, J.M.; García, P.A.; Rojo, M.V.; de la Iglesia-Vicente, J.; Mollinedo, F.; Cuevas, C.; San Feliciano, A. Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities. J. Med. Chem. 2010, 53, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.C.; Lee, H.Z.; Juang, J.P.; Lin, Y.T.; Wu, T.S.; Chang, J.J.; Lednicer, D.; Paull, K.D.; Lin, C.M.; Hamel, E.; et al. Synthesis and cytotoxicity of 1,6,7,8-substituted 2-(4′-substituted phenyl)-4-quinolones and related compounds: Identification as antimitotic agents interacting with tubulin. J. Med. Chem. 1993, 36, 1146–1156. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.K.; Kuo, S.C.; Wu, T.S.; Lednicer, D.; Lin, C.M.; Hamel, E.; Lee, K.H. Antitumor agents. 150. 2’,3’,4’,5’,5,6,7-substituted 2-phenyl-4-quinolones and related compounds: Their synthesis, cytotoxicity, and inhibition of tubulin polymerization. J. Med. Chem. 1994, 37, 1126–1135. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.K.; Kuo, S.C.; Wu, T.S.; Mauger, A.; Lin, C.M.; Hamel, E.; Lee, K.H. Antitumor agents. 155. Synthesis and biological evaluation of 3’,6,7-substituted 2-phenyl-4-quinolones as antimicrotubule agents. J. Med. Chem. 1994, 37, 3400–3407. [Google Scholar] [CrossRef]
- Wang, S.W.; Pan, S.L.; Huang, Y.C.; Guh, J.H.; Chiang, P.C.; Huang, D.Y.; Kuo, S.C.; Lee, K.H.; Teng, C.M. CHM-1, a novel synthetic quinolone with potent and selective antimitotic antitumor activity against human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.W.; Lin, Y.C.; Hung, C.M.; Liu, B.L.; Kuo, S.C.; Ho, C.T.; Way, T.D.; Hung, C.H. CHM-1, a novel microtubule-destabilizing agent exhibits antitumor activity via inducing the expression of SIRT2 in human breast cancer cells. Chem. Biol. Interact. 2018, 289, 98–108. [Google Scholar] [CrossRef]
- Chan, H.C.; Liao, Y.H.; Chang, H.W.; Liu, C.H. The mechanism of anticancer activity of the new synthesized compound—6,7-Methylenedioxy-4-(2,4-dimethoxyphenyl)quinolin -2(1H)-one(12e) in human ovarian cancer cell lines. Taiwan. J. Obstet. Gynecol. 2021, 60, 266–272. [Google Scholar] [CrossRef]
- Bailly, C.; Bal, C.; Barbier, P.; Combes, S.; Finet, J.P.; Hildebrand, M.P.; Peyrot, V.; Wattez, N. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. J. Med. Chem. 2003, 46, 5437–5444. [Google Scholar] [CrossRef] [PubMed]
- Billard, C.; Menasria, F.; Quiney, C.; Faussat, A.M.; Finet, J.P.; Combes, S.; Kolb, J.P. 4-arylcoumarin analogues of combretastatins stimulate apoptosis of leukemic cells from chronic lymphocytic leukemia patients. Exp. Hematol. 2008, 36, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Ganina, O.G.; Daras, E.; Bourgarel-Rey, V.; Peyrot, V.; Andresyuk, A.N.; Finet, J.P.; Fedorov, A.Y.; Beletskaya, I.P.; Combes, S. Synthesis and biological evaluation of polymethoxylated 4-heteroarylcoumarins as tubulin assembly inhibitor. Bioorganic Med. Chem. 2008, 16, 8806–8812. [Google Scholar] [CrossRef]
- Combes, S.; Barbier, P.; Douillard, S.; McLeer-Florin, A.; Bourgarel-Rey, V.; Pierson, J.T.; Fedorov, A.Y.; Finet, J.P.; Boutonnat, J.; Peyrot, V. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. Part 2. J. Med. Chem. 2011, 54, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Chai, H.-B.; Shin, Y.G.; García, R.; Mejía, M.; Gao, Q.; Fairchild, C.R.; Lane, K.E.; Menendez, A.T.; Farnsworth, N.R.; et al. Cytotoxic Constituents of the Roots of Exostema acuminatum. Tetrahedron 2000, 56, 6401–6405. [Google Scholar] [CrossRef]
- Rivero-Cruz, I.; Cristians, S.; Ovalle-Magallanes, B.; Mata, R. Mexican copalchis of the Rubiaceae family: More than a century of pharmacological and chemical investigations. Phytochem. Rev. 2019, 18, 1435–1455. [Google Scholar] [CrossRef]
- Garazd, M.M.; Garazd, Y.L.; Khilya, V.P. Neoflavones. 1. Natural Distribution and Spectral and Biological Properties. Chem. Nat. Compd. 2003, 39, 54–121. [Google Scholar] [CrossRef]
- Rappl, C.; Barbier, P.; Bourgarel-Rey, V.; Grégoire, C.; Gilli, R.; Carre, M.; Combes, S.; Finet, J.P.; Peyrot, V. Interaction of 4-arylcoumarin analogues of combretastatins with microtubule network of HBL100 cells and binding to tubulin. Biochemistry 2006, 45, 9210–9218. [Google Scholar] [CrossRef]
- Mutai, P.; Breuzard, G.; Pagano, A.; Allegro, D.; Peyrot, V.; Chibale, K. Synthesis and biological evaluation of 4 arylcoumarin analogues as tubulin-targeting antitumor agents. Bioorganic Med. Chem. 2017, 25, 1652–1665. [Google Scholar] [CrossRef]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Sai, K.K.; Gilbert, T.M.; Klumpp, D.A. Knorr cyclizations and distonic superelectrophiles. J. Org. Chem. 2007, 72, 9761–9764. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, N.; Simenel, C.; Delepierre, M.; Barale, J.-C.; Janin, Y.L. On the Knorr Synthesis of 6-Bromo-4-methylquinolin-2(1H)-one. Synthesis 2011, 2011, 934–942. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Wang, J.; Li, C.-M.; Ahn, S.; Barrett, C.M.; Dalton, J.T.; Li, W.; Miller, D.D. Design, Synthesis, and Biological Evaluation of Stable Colchicine Binding Site Tubulin Inhibitors as Potential Anticancer Agents. J. Med. Chem. 2014, 57, 7355–7366. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, R.; Aramburu, L.; Puebla, P.; Caballero, E.; González, M.; Vicente, A.; Medarde, M.; Peláez, R. Pyridine Based Antitumour Compounds Acting at the Colchicine Site. Curr. Med. Chem. 2016, 23, 1100–1130. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Pang, Y.; Chen, J.; Sheng, J.; Hu, J.; Huang, L.; Li, X. Synthesis, biological evaluation and mechanism study of a class of cyclic combretastatin A-4 analogues as novel antitumour agents. RSC Adv. 2015, 5, 98527–98537. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.N.H.; Huy, N.T.; Bay, M.V.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model. 2020, 60, 204–211. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Eddershaw, P.J.; Beresford, A.P.; Bayliss, M.K. ADME/PK as part of a rational approach to drug discovery. Drug Discov. Today 2000, 5, 409–414. [Google Scholar] [CrossRef]
- Yu, H.; Adedoyin, A. ADME-Tox in drug discovery: Integration of experimental and computational technologies. Drug Discov. Today 2003, 8, 852–861. [Google Scholar] [CrossRef]
- Wan, H. What ADME tests should be conducted for preclinical studies? ADMET DMPK 2013, 1, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Habet, S.A.; Baweja, R.K.; Burckart, G.J.; et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Lynch, T.; Price, A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am. Fam. Physician 2007, 76, 391–396. [Google Scholar] [PubMed]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef]
- Zhou, P.-z.; Babcock, J.; Liu, L.-q.; Li, M.; Gao, Z.-b. Activation of human ether-a-go-go related gene (hERG) potassium channels by small molecules. Acta Pharmacol. Sin. 2011, 32, 781–788. [Google Scholar] [CrossRef]
- Danker, T.; Möller, C. Early identification of hERG liability in drug discovery programs by automated patch clamp. Front. Pharmacol. 2014, 5, 203. [Google Scholar] [CrossRef] [Green Version]
- Environmental Protection Agency U.S. Label Review Manual Chapter 7: Precautionary Statements. 2018. Available online: https://www.epa.gov/pesticide-registration/label-review-manual (accessed on 23 December 2022).
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. SOMP: Web server for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics 2015, 31, 2046–2048. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tian, S.; Allen, D.; Oler, E.; Peters, H.; Lui, V.W.; Gautam, V.; Djoumbou-Feunang, Y.; Greiner, R.; Metz, T.O. BioTransformer 3.0—A web server for accurately predicting metabolic transformation products. Nucleic Acids Res. 2022, 50, W115–W123. [Google Scholar] [CrossRef] [PubMed]
- Allinger, N.L.; Yuh, Y.H.; Lii, J.H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
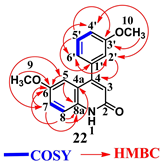 | ||||
|---|---|---|---|---|
| Position | δH, mult. (J in Hz) | δc | COSY | HMBC a (H→C) |
| 1 | 11.76, br. s | – | – | – |
| 2 | – | 159.94, C, overlapped | – | – |
| 3 | 6.38, s | 106.52, CH | – | – |
| 4 | – | 149.47, C | – | H-2′ (3J) |
| 4a | – | 126.20, C | – | H-8 (3J) |
| 5 | 7.51, d (2.4) | 104.27, CH | H-7 b | H-7 (3J) |
| 6 | – | 156.08, C, overlapped | – | H-5 (2J), H-7 (2J), H-8 (3J), H-9 (3J) |
| 7 | 7.32, dd (8.8, 2.4) | 122.63, CH | H-5 b, H-8 | H-5 (3J) |
| 8 | 7.76, d (8.8) | 121.18, CH | H-7 | – |
| 8a | – | 156.08, C, overlapped | – | H-5 (3J), H-7 (3J), H-8 (2J) |
| 9 | 3.84, s | 55.73, -OCH3 | – | – |
| 10 | 3.86, s | 55.77,-OCH3 | – | – |
| 1′ | – | 136.16, C | – | H-5′ (3J) |
| 2′ | 7.36–7.39, m overlapped | 113.11, CH | – | H-4′ (3J), H-6′ (3J) |
| 3′ | – | 159.94, C, overlapped | – | H-5′ (3J), H-10 (3J) |
| 4′ | 7.36–7.39, m overlapped | 119.92, CH | H-5′ | H-2′ (3J), H-6′ (3J) |
| 5′ | 7.48, t (8.0) | 130.57, CH | H-4′, H-6′ | – |
| 6′ | 7.12, d (7.6) | 116.31, CH | H-5′ | H-2′ (3J), H-4′ (3J) |
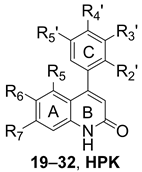 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Ring A | Ring C | IC50 (μM) a Against Cell Lines b | |||||||||
| R5 | R6 | R7 | R2’ | R3’ | R4’ | R5’ | COLO205 | A498 | H460 | Hep3B | Detroit-551 | |
| 19 | H | H | H | H | H | H | H | 32.88 | >50 | 36.49 | >50 | >50 |
| 20 | H | H | H | H | H | OCH3 | H | 23.33 | >50 | 47.1 | >50 | >50 |
| 21 | H | OCH3 | H | H | H | H | H | >50 | >50 | 45.7 | >50 | >50 |
| 22 | H | OCH3 | H | H | OCH3 | H | H | 0.32 | >50 | 0.89 | >50 | >50 |
| 23 | H | OCH3 | H | H | H | OCH3 | H | >50 | >50 | >50 | >50 | >50 |
| 24 | OCH3 | H | OCH3 | H | H | H | H | >50 | >50 | >50 | >50 | >50 |
| 25 | OCH3 | H | OCH3 | H | OCH3 | H | H | 37.6 | >50 | 46.25 | >50 | >50 |
| 26 | OCH3 | H | OCH3 | H | H | OCH3 | H | >50 | >50 | >50 | >50 | >50 |
| 27 | OCH3 | H | OCH3 | OCH3 | H | OCH3 | H | 7.85 | >50 | >50 | >50 | >50 |
| 28 | OCH3 | H | OCH3 | OCH3 | H | H | OCH3 | 38.55 | >50 | 32.27 | ND c | ND c |
| 29 | OCH3 | OCH3 | OCH3 | H | H | H | H | >50 | >50 | >50 | >50 | >50 |
| 30 | OCH3 | OCH3 | OCH3 | H | H | OCH3 | H | >50 | >50 | >50 | >50 | >50 |
| 31 | OCH3 | OCH3 | OCH3 | OCH3 | H | OCH3 | H | >50 | >50 | >50 | >50 | >50 |
| 32 | OCH3 | OCH3 | OCH3 | OCH3 | H | H | OCH3 | >50 | >50 | >50 | >50 | >50 |
| HPKd | H | -O-CH2-O- | OCH3 | H | OCH3 | H | 7.4 d | 48 d | 0.9 d | 1.0 d | >25 d | |
| PPT e | 0.01 e | 0.01 e | 0.01 e | ND c | ND c | |||||||
| Colchicine e | 0.025 e | 0.03 e | 0.02 e | ND c | ND c | |||||||
| Compound | Cell Lines | |||
|---|---|---|---|---|
| COLO205 | H460 | |||
| IC50 (μM) | Selectivity Index b | IC50 (μM) | Selectivity Index b | |
| 19 | 32.88 | 0.64 | 36.49 | 0.82 |
| 20 | 23.33 | 0.91 | 47.1 | 0.64 |
| 21 | >50 | ND c | 45.7 | 0.66 |
| 22 | 0.32 | 66.04 | 0.89 | 33.64 |
| 25 | 37.6 | 0.56 | 46.25 | 0.65 |
| 27 | 7.85 | 2.69 | >50 | ND c |
| 28 | 38.55 | 0.55 | 32.27 | 0.93 |
| HPK | 7.4 | 2.86 | 0.9 | 33.27 |
| Total Mean a | 21.13 | ND c | 29.94 | ND c |
| Compound | ΔG (kcal/mol) a | ΔG (kcal/mol) b |
|---|---|---|
| 19 | −7.57 | −7.5 |
| 20 | −7.46 | −7.4 |
| 21 | −7.59 | −8.1 |
| 22 | −7.97 | −8.1 |
| 25 | −7.78 | −6.1 |
| 27 | −7.62 | −5.9 |
| 28 | −7.51 | −5.2 |
| HPK | −7.90 | −8.3 |
| PPT | −9.91 | −9.0 |
| Colchicine | −8.17 | −7.6 |
| Compound | Interacting Amino Acid Residues |
|---|---|
| 19 | a, b, d (ALA250), d (CYS241, LEU248, LEU255, ALA316, LYS352) |
| 20 | b (VAL238), c (ASN258), d (ALA180, CYS241, LEU248, ALA250, LYS254, LEU255, ALA316, VAL318, LYS352, ILE378) |
| 21 | a, b, d (ALA250), b (VAL315), d (CYS241, LEU248, LEU255, MET259, ALA316, LYS352) |
| 22 | a, b, d (ALA250), b (VAL315), d (CYS241, LEU242, LEU248, LEU255, ALA316, LYS352, ILE378) |
| 25 | b, d (ALA250), b (VAL238), d (VAL181, CYS241, LEU242, LEU248, LEU255, ALA316, LYS352), e (LYS254) |
| 27 | d (VAL181, CYS241, LEU242, LEU248, ALA250, LEU255, MET259, ALA316, VAL318, LYS352, ALA354) |
| 28 | a (THR179), d (VAL181, CYS241, LEU248, ALA250, LYS254, LEU255, MET259, ALA316, VAL318, LYS352), e (ALA250, MET259) |
| HPK | b (VAL238, ASN258, VAL315), d (VAL181, CYS241, LEU242, LEU248, ALA250, LEU255, MET259, ALA316, VAL318, LYS352, ALA354) |
| PPT | b (ALA250, VAL315, ALA317, LYS352, ASN350), d (VAL181, CYS241, LEU242, ALA250, LEU255, ALA316, VAL318, LYS352, ALA354, ILE378), e (MET259) |
| Colchicine | b (ALA180, VAL238, ASN258, VAL315, ASN350), d (CYS241, LEU242, ALA250, LEU255, ALA316, VAL318, LYS352, ALA354, ILE378) |
| Compound | M.W a (Da) | HBA b | HBD c | Log P d | Log S e | TPSA f | PAINS g | Drug Likeliness | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipinski’s Rules | Veber’s Rules | Ghose’s Rules | Egan’s Rules | Muegge’s Rules | ||||||||
| 19 | 221.25 | 1 | 1 | 3.05 | −3.42 | 32.86 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| 20 | 251.28 | 2 | 1 | 3.03 | −3.45 | 42.09 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| 21 | 251.28 | 2 | 1 | 3.03 | −3.45 | 42.09 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| 22 | 281.31 | 3 | 1 | 3.01 | −3.49 | 51.32 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| 25 | 311.33 | 4 | 1 | 2.98 | −3.55 | 60.55 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| 27 | 341.36 | 5 | 1 | 2.97 | −3.61 | 69.78 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| 28 | 341.36 | 5 | 1 | 2.96 | −3.61 | 69.78 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| HPK | 325.32 | 5 | 1 | 2.85 | −3.58 | 69.78 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| PPT | 414.41 | 8 | 1 | 2.25 | −3.71 | 92.68 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| Colchicine | 399.44 | 6 | 1 | 2.36 | −2.9 | 83.09 Å2 | 0 alert | Yes | Yes | Yes | Yes | Yes |
| Compound | HIA a | BBB b | P-gp c Substrate | Cytochrome P450 Inhibition | ||||
|---|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | ||||
| 19 | High | Yes | No | Yes | No | No | No | No |
| 20 | High | Yes | No | Yes | Yes | No | No | No |
| 21 | High | Yes | No | Yes | Yes | No | No | No |
| 22 | High | Yes | No | Yes | No | Yes | Yes | Yes |
| 25 | High | Yes | No | Yes | No | Yes | Yes | Yes |
| 27 | High | Yes | No | Yes | No | Yes | Yes | Yes |
| 28 | High | Yes | No | Yes | No | Yes | Yes | Yes |
| HPK | High | Yes | Yes | Yes | No | Yes | Yes | Yes |
| PPT | High | No | No | No | No | No | Yes | Yes |
| Colchicine | High | No | Yes | No | No | No | Yes | Yes |
| Compound | hERG Inhibition a | AMES Toxicity | Carcinogenicity | Rate Acute Toxicity (LD50, mol/kg) b | Acute Oral Toxicity c |
|---|---|---|---|---|---|
| 19 | None | None | None | 2.1994 | Class III |
| 20 | None | Toxic | None | 2.2696 | Class III |
| 21 | None | Toxic | None | 2.2696 | Class III |
| 22 | None | Toxic | None | 2.4292 | Class III |
| 25 | None | Toxic | None | 2.5121 | Class III |
| 27 | None | None | None | 2.7135 | Class III |
| 28 | None | None | None | 2.7135 | Class III |
| HPK | None | Toxic | None | 2.6129 | Class III |
| PPT | None | None | None | 3.0013 | Class III |
| Colchicine | None | None | None | 2.3748 | Class III |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-F.; Lawal, B.; Huang, L.-J.; Kuo, S.-C.; Sumitra, M.R.; Mokgautsi, N.; Lin, H.-Y.; Huang, H.-S. In Vitro and In Silico Biological Studies of 4-Phenyl-2-quinolone (4-PQ) Derivatives as Anticancer Agents. Molecules 2023, 28, 555. https://doi.org/10.3390/molecules28020555
Chen Y-F, Lawal B, Huang L-J, Kuo S-C, Sumitra MR, Mokgautsi N, Lin H-Y, Huang H-S. In Vitro and In Silico Biological Studies of 4-Phenyl-2-quinolone (4-PQ) Derivatives as Anticancer Agents. Molecules. 2023; 28(2):555. https://doi.org/10.3390/molecules28020555
Chicago/Turabian StyleChen, Yi-Fong, Bashir Lawal, Li-Jiau Huang, Sheng-Chu Kuo, Maryam Rachmawati Sumitra, Ntlotlang Mokgautsi, Hung-Yun Lin, and Hsu-Shan Huang. 2023. "In Vitro and In Silico Biological Studies of 4-Phenyl-2-quinolone (4-PQ) Derivatives as Anticancer Agents" Molecules 28, no. 2: 555. https://doi.org/10.3390/molecules28020555
APA StyleChen, Y.-F., Lawal, B., Huang, L.-J., Kuo, S.-C., Sumitra, M. R., Mokgautsi, N., Lin, H.-Y., & Huang, H.-S. (2023). In Vitro and In Silico Biological Studies of 4-Phenyl-2-quinolone (4-PQ) Derivatives as Anticancer Agents. Molecules, 28(2), 555. https://doi.org/10.3390/molecules28020555