
N-Acetyl-l-phenylalanine Racemization during TBTU Amidation: An In-Depth Study for the Synthesis of Anti-Inflammatory 2-(N-Acetyl)-l-phenylalanylamido-2-deoxy-d-glucose (NAPA)
 , , , , , and
, , , , , and
Abstract
:
1. Introduction
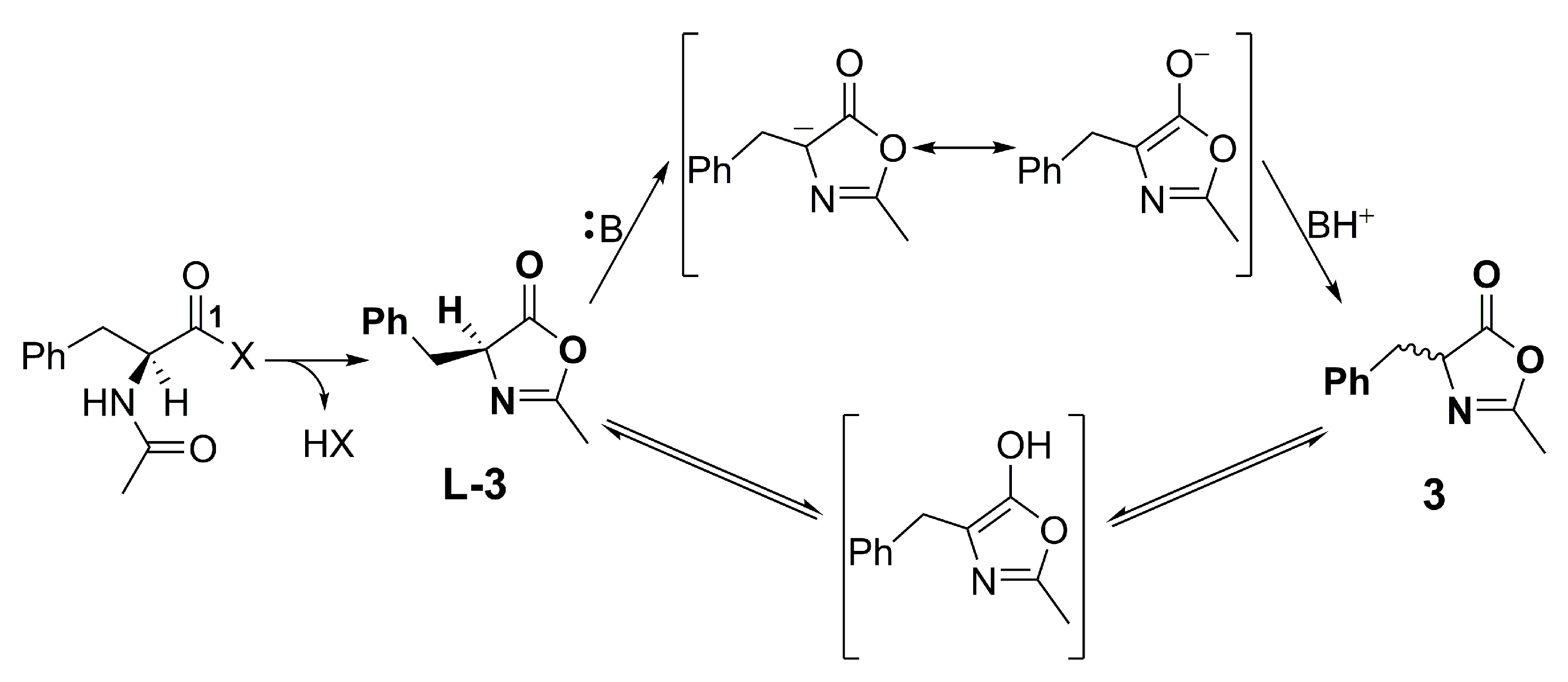
2. Results and Discussions
3. Materials and Methods
3.1. General Information
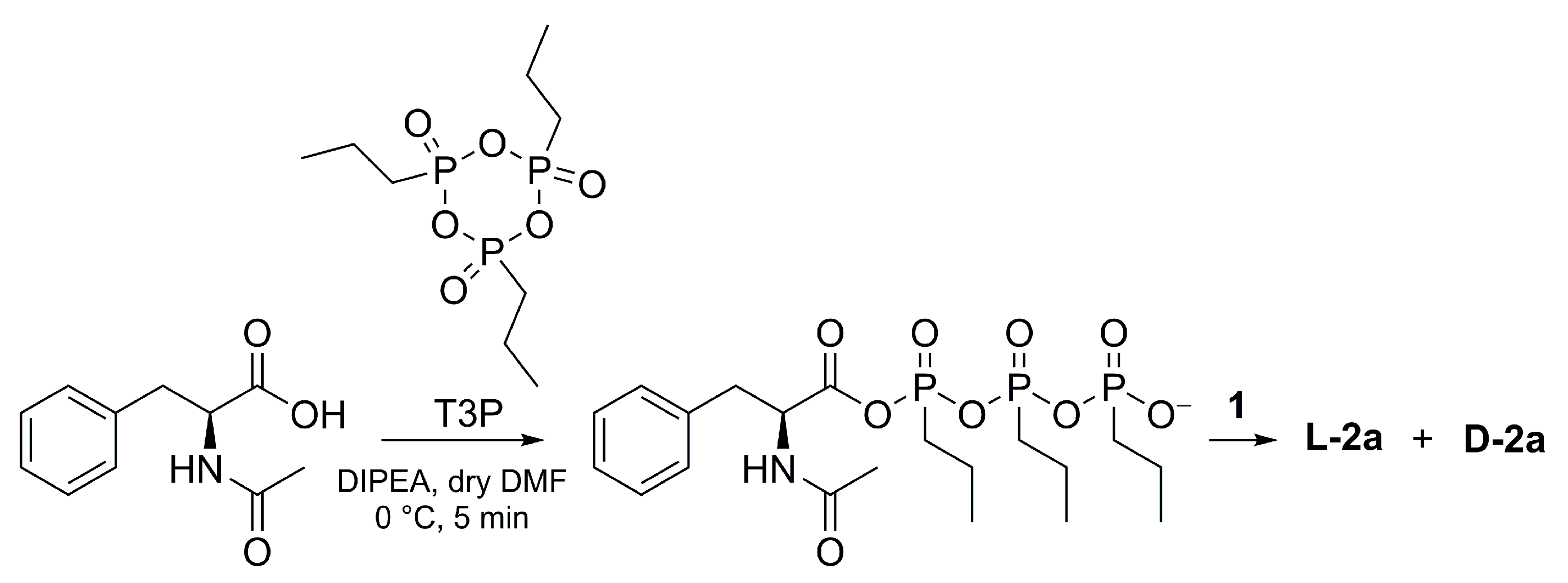
3.2. Synthesis of L-2a Using TBTU or T3P
3.3. Synthesis of D-2a
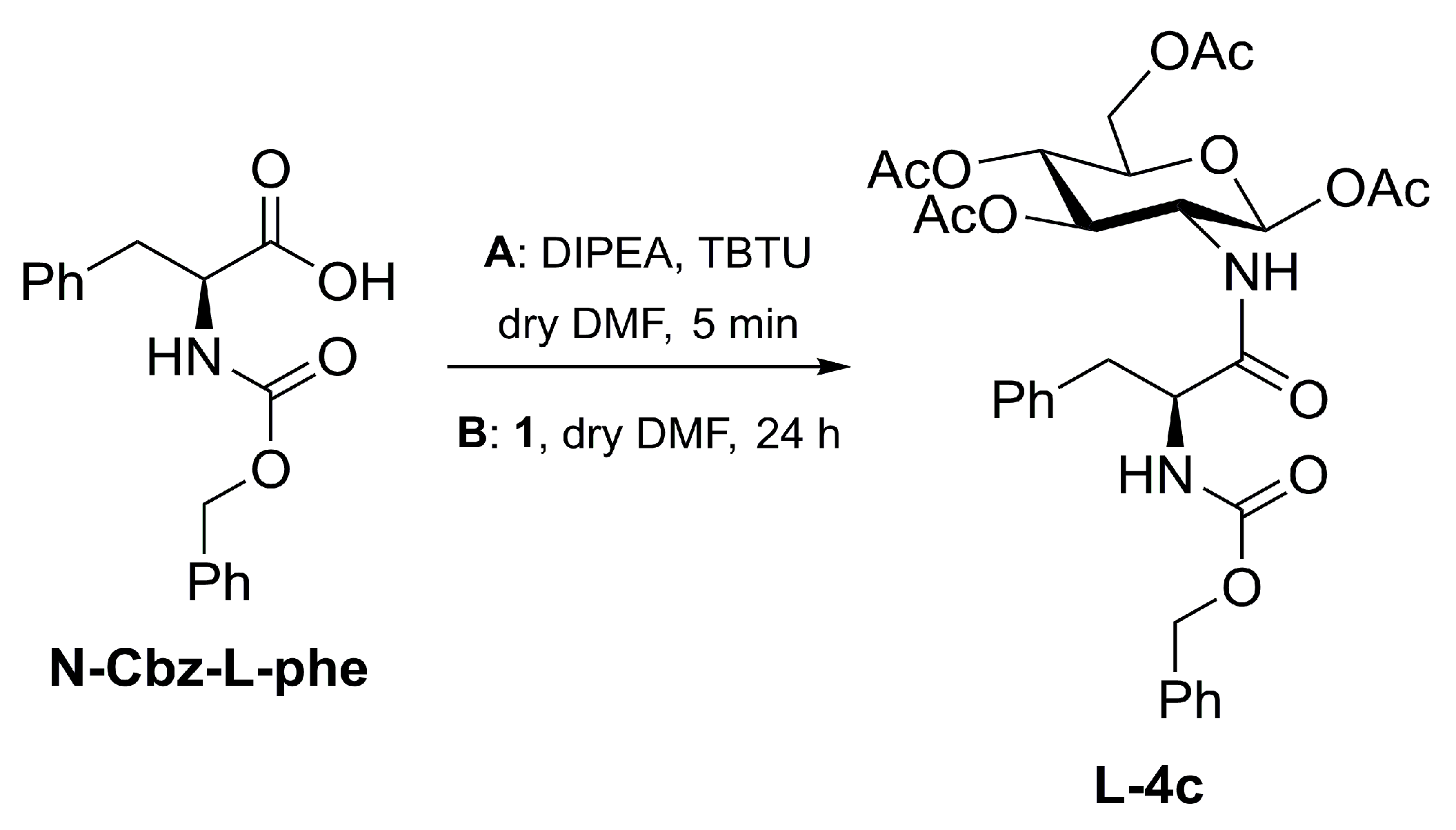
3.4. Synthesis of L-4c
3.5. Synthesis of 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Krasnov, V.P.; Zhdanova, E.A.; Solieva, N.Z.; Sadretdinova, L.S.; Bukrina, I.M.; Demin, A.M.; Levit, G.L.; Ezhikova, M.A.; Kodess, M.I. Study of the effect of the nature of the side chain in esters of α-amino acids on the diastereoselectivity of condensation with 5-(4H)-oxazolone in the synthesis of dipeptides with N-terminal N-acetylphenylalanine. Russ. Chem. Bull. 2004, 53, 1331–1334. [Google Scholar] [CrossRef]
- Chen, F.M.; Sleibioda, M.; Benoiton, N.L. Mixed carboxylic-carbonic acid anhydrides of acylamino acids and peptides as a convenient source of 2,4-dialkyl-5(4H)-oxazolones. Int. J. Pept. Protein Res. 1988, 31, 339–344. [Google Scholar] [CrossRef]
- Crisma, M.; Valle, G.; Formaggio, F.; Toniolo, C.; Bagno, A. Reactive Intermediates in Peptide Synthesis: First Crystal Structures and ab Initio Calculations of 2-Alkoxy-5 (4H)-oxazolones from Urethane-Protected Amino Acids. J. Am. Chem. Soc. 1997, 119, 4136–4142. [Google Scholar] [CrossRef]
- Yang, Y.; Hansen, L.; Baldi, A. Suppression of Simultaneous Fmoc-His(Trt)-OH Racemization and Nα-DIC-Endcapping in Solid-Phase Peptide Synthesis through Design of Experiments and Its Implication for an Amino Acid Activation Strategy in Peptide Synthesis. Org. Process. Res. Dev. 2022, 26, 2464–2474. [Google Scholar] [CrossRef]
- Albericio, F.; Bofill, J.M.; El-Faham, A.; Kates, S.A. Use of onium salt-based coupling reagents in peptide synthesis. J. Org. Chem. 1998, 63, 9678–9683. [Google Scholar] [CrossRef]
- Twibanire, J.D.A.K.; Grindley, T.B. Efficient and controllably selective preparation of esters using uronium-based coupling agents. Org. Lett. 2011, 13, 2988–2991. [Google Scholar] [CrossRef]
- Fields, C.G.; Lloyd, D.H.; Macdonald, R.L.; Otteson, K.M.; Noble, R.L. HBTU activation for automated Fmoc solid-phase peptide synthesis. Pept. Res. 1991, 4, 95–101. [Google Scholar]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Mali, S.M.; Ganesh Kumar, M.; Katariya, M.M.; Gopi, H.N. HBTU mediated 1-hydroxybenzotriazole (HOBt) conjugate addition: Synthesis and stereochemical analysis of β-benzotriazole N-oxide substituted γ-amino acids and hybrid peptides. Org. Biomol. Chem. 2014, 12, 8462–8472. [Google Scholar] [CrossRef]
- Brufani, M.; Ceccacci, F.; Filocamo, L.; Garofalo, L.; Joudioux, R.; La Bella, A.; Leonelli, F.; Migneco, M.L.; Marini Bettolo, R.; Farina, P.M.; et al. Novel Locally Active Estrogens Accelerate Cutaneous Wound Healing. A Preliminary Study. Mol. Pharm. 2009, 6, 543–556. [Google Scholar] [CrossRef]
- Furlotti, G.; Alisi, M.A.; Cazzolla, N.; Ceccacci, F.; Garrone, B.; Gasperi, T.; La Bella, A.; Leonelli, F.; Loreto, M.A.; Magaro, G.; et al. Targeting Serotonin 2A and Adrenergic alpha1 Receptors for Ocular Antihypertensive Agents: Discovery of 3,4-Dihydropyrazino[1,2-b]indazol-1(2H)-one Derivatives. Chem. Med. Chem. 2018, 13, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Petaccia, M.; Giansanti, L.; Leonelli, F.; Bella, A.; Gradella Villalva, D.; Mancini, G. Synthesis, characterization and inclusion into liposomes of a new cationic pyrenyl amphiphile. Chem. Phys. Lipids 2016, 200, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Gallina, C.; Consalvi, V.; Scandurra, R. Synthesis and properties of D-glucosamine N-peptidyl derivatives as substrate analog inhibitors of papain and cathepsin B. Eur. J. Med. Chem. 1991, 26, 153–162. [Google Scholar] [CrossRef]
- Scotto d’Abusco, A.; Politi, L.; Giordano, C.; Scandurra, R. A peptidyl-glucosamine derivative affects IKKα kinase activity in human chondrocytes. Arthritis Res. Ther. 2010, 12, R18. [Google Scholar] [CrossRef] [Green Version]
- Towheed, T.; Maxwell, L.; Anastassiades, T.P.; Shea, B.; Houpt, J.B.; Welch, V.; Hochberg, M.C.; Wells, G.A. Glucosamine therapy for treating osteoarthritis. Cochrane Database Syst. Rev. 2005, 2, CD002946. [Google Scholar] [CrossRef]
- Uitterlinden, E.J.; Koevoet, J.L.; Verkoelen, C.F.; Bierma-Zeinstra, S.M.; Jahr, H.; Weinans, H.; Verhaar, J.A.; van Osch, G.J. Glucosamine increases hyaluronic acid production in human osteoarthritic synovium explants. BMC Musculoskelet. Disord. 2008, 9, 120. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, M.; Kaga, I.; Takamori, Y.; Sakamoto, K.; Miyazawa, K.; Nagaoka, I. Effects of glucosamine derivatives and uronic acids on the production of glycosaminoglycans by human synovial cells and chondrocytes. Int. J. Mol. Med. 2011, 27, 821–827. [Google Scholar]
- Schulz, R.M.; Bader, A. Cartilage tissue engineering and bioreactor systems for the cultivation and stimulation of chondrocytes. Eur. Biophys. J. 2007, 36, 539–568. [Google Scholar] [CrossRef]
- Scotto d’Abusco, A.; Corsi, A.; Grillo, M.G.; Cicione, C.; Calamia, V.; Panzini, G.; Sansone, A.; Giordano, C.; Politi, L.; Scandurra, R. Effects of intra-articular administration of glucosamine and a peptidyl-glucosamine derivative in a rabbit model of experimental osteoarthritis: A pilot study. Rheumatol. Int. 2008, 28, 437–443. [Google Scholar] [CrossRef]
- Veronesi, F.; Giavaresi, G.; Maglio, M.; Scotto d’Abusco, A.; Politi, L.; Scandurra, R.; Olivotto, E.; Grigolo, B.; Borzì, R.M.; Fini, M. Chondroprotective activity of N-acetyl phenylalanine glucosamine derivative on knee joint structure and inflammation in a murine model of osteoarthritis. Osteoarthr. Cartil. 2017, 25, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Scotto d’Abusco, A.; Cicione, C.; Calamia, V.; Negri, R.; Giordano, C.; Grigolo, B.; Politi, L.; Scandurra, R. Glucosamine and its N-acetyl-phenylalanine derivative prevent TNF-alpha-induced transcriptional activation in human chondrocytes. Clin. Exp. Rheumatol. 2007, 25, 847. [Google Scholar] [PubMed]
- Biswas, N.N.; Yu, T.T.; Kimyon, O.; Nizalapur, S.; Gardner, C.R.; Manefield, M.; Griffith, R.; Black, D.S.; Kumar, N. Synthesis of antimicrobial glucosamides as bacterial quorum sensing mechanism inhibitors. Bioorg. Med. Chem. 2017, 25, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.S.; Zhalnina, A.A.; Shishkov, S.V. A convergent approach to synthesis of bortezomib: The use of TBTU suppresses racemization in the fragment condensation. Tetrahedron 2009, 65, 7105–7108. [Google Scholar] [CrossRef]
- Novelli, F.; De Santis, S.; Punzi, P.; Giordano, C.; Scipioni, A.; Masci, G. Self-assembly and drug release study of linear L,D-oligopeptide-poly(ethylene glycol) conjugates. New Biotechnol. 2017, 37, 99–107. [Google Scholar] [CrossRef] [PubMed]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef]
- Windridge, G.C.; Jorgensen, E.C. 1-Hydroxybenzotriazole as a Racemization-Suppressing Reagent for the Incorporation of im-Benzyl-l-histidine into Peptides. J. Am. Chem. Soc. 1971, 93, 6318–6319. [Google Scholar] [CrossRef]
- Chen, F.M.F.; Benoiton, N.L. H n.m.r. long-range coupling in 2,4-disubstituted-5(4H)-oxazolones and 4-alkyl-5(2H)-oxazolones generated therefrom by the action of triethylamine. Int. J. Pept. Protein Res. 1987, 30, 683–688. [Google Scholar] [CrossRef]
- Katritzky, A.R. Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 1997; Volume 70. [Google Scholar]
- De Castro, P.P.; Batista, G.M.F.; Pinheiro, D.L.J.; Dos Santos, H.F.; Amarante, G.W. Old drawback on azlactone formation revealed by a combination of theoretical and experimental studies. J. Braz. Chem. Soc. 2018, 29, 2213–2219. [Google Scholar] [CrossRef]
- De Castro, P.P.; Carpanez, A.G.; Amarante, G.W. Azlactone Reaction Developments. Chem.—Eur. J. 2016, 22, 10294–10318. [Google Scholar] [CrossRef]
- De Castro, P.P.; Batista, G.M.F.; Amarante, G.W. Theoretical Study on the Epimerization of Azlactone Rings: Keto−Enol Tautomerism or Base-Mediated Racemization? ACS Omega 2018, 3, 3507–3512. [Google Scholar] [CrossRef]
- Zhdanova, E.A.; Solieva, N.Z.; Sadretdinova, L.S.; Ezhikova, M.A.; Kodess, M.I.; Krasnov, V.P. Study of the influence of the alkyl ester group in (S)-valinates on diastereoselectivity of their condensation with N-acetylphenylalanine by the mixed anhydride method. Russ. Chem. Bull. 2006, 55, 925–927. [Google Scholar] [CrossRef]
- Waring, A.; Joullie, M.M.; Lassen, K.M. Evolution of amide bond formation. Arkivoc 2010, 2010, 189–250. [Google Scholar]
- Dunet, J.R.; Xiang, Y.; Baldwin, A.; Ringling, J. General and Scalable Amide Bond Formation with Epimerization-Prone Substrates Using T3P and Pyridine. Org. Lett. 2011, 13, 5048–5051. [Google Scholar] [CrossRef] [PubMed]
- Waghmare, A.A.; Hindupur, R.M.; Pati, H.N. Propylphosphonic anhydride (T3P®): An expedient reagent for organic synthesis. Rev. J. Chem. 2014, 4, 53–131. [Google Scholar] [CrossRef]
- Pinzón Martín, S.M.; Medina, R.F.; Iregui Castro, C.A.; Rivera Monroy, Z.J.; García Castañeda, J.E. Novel Synthesis of N-Glycosyl Amino Acids Using T3P®: Propylphosphonic Acid Cyclic Anhydride as Coupling Reagent. Int. J. Pept. Res. 2017, 24, 291–298. [Google Scholar] [CrossRef]
- De Castro, P.P.; Rimulo, I.M.R.; De Almeida, A.M.; Diniz, R.; Amarante, G.W. Bronsted Acid-Catalyzed Epimerization-Free Preparation of Dual-Protected Amino Acid Derivatives. ACS Omega 2017, 2, 2967–2976. [Google Scholar] [CrossRef]
- Hu, L.; Xu, S.; Zhao, Z.; Yang, Y.; Peng, Z.; Yang, M.; Wang, C.; Zhao, J. Ynamides as Racemization-Free Coupling Reagents for Amide and Peptide Synthesis. J. Am. Chem. Soc. 2016, 138, 13135–13138. [Google Scholar] [CrossRef]
- Perrin, D.D.; Dempsey, B.; Serjeant, E.P. pKa Prediction for Organic Acids and Bases; Chapman and Hall: London, UK, 1981; Volume 1. [Google Scholar]
- Han, K.J.; Tae, B.S.; Kim, M. A simple procedure for the conversion of carboxylic acids to the corresponding amides. Org. Prep. Proced. Int. 2005, 37, 198–203. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Amino Acid Configuration | DIPEA (eq) | TR (°C) | tR (h) | Yield (%) | RD (L-2a:D-2a) |
|---|---|---|---|---|---|---|
| 1 | L | 2 | r.t. | 24 | 87 | 31:69 |
| 2 | 2 | 3 | 82 | 34:66 | ||
| 3 | 1 | 24 | 78 | 24:76 | ||
| 4 | 1 | 3 | 71 | 29:71 | ||
| 5 | 1 | −10 | 3 | 49 | 65:35 | |
| 6 | D | 2 | r.t. | 24 | 93 | 35:65 |
| 7 | 1 | 24 | 93 | 24:76 | ||
| 8 | 1 | 3 | 82 | 26:78 | ||
| 9 | 1 | −10 | 3 | 88 | 8:92 |
| Entry | DIPEA (eq.) | TR (°C) | tR (h) | Yield (%) | RD (L-4c:D-4c) |
|---|---|---|---|---|---|
| 1-Z | 1 | r.t. | 24 | 80 | 100:0 |
| 2-Z | 2 | r.t. | 24 | 85 | 100:0 |
| Entry | Amino Acid | DIPEA (eq.) | TR (°C) | tR (h) | Yield (%) | RD (L:D) |
|---|---|---|---|---|---|---|
| 1-T3P | N-Ac-l-phe | 2 | r.t. | 24 | 84 | 34:66 |
| 2-T3P | N-Cbz-l-phe | 2 | r.t. | 24 | 87 | 100:0 |
| Entry | Base 2 | Eq. | TR (°C) | tR (h) | Yield (%) | RD (L-2a:D-2a) | L (%) | D (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | - | - | r.t. | 3 | - | - | - | - |
| 2 | - | - | r.t. | 24 | 38 | 80:20 | 30 | 8 |
| 3 | DMA | 1 | r.t. | 3 | 30 | 97:3 | 29 | 1 |
| 4 | Py | 1 | r.t. | 3 | 43 | 93:7 | 40 | 3 |
| 5 | 1 | r.t. | 5 | 50 | 87:13 | 44 | 6 | |
| 6 | 1 | 0 | 5 | 16 | 100:00 | 16 | 0 | |
| 7 | 1 | 5 | 15 | 43 | 87:13 | 37 | 6 | |
| 8 | 2 | r.t. | 3 | 40 | 93:7 | 37 | 3 | |
| 9 | Lut | 1 | r.t. | 3 | 55 | 83:17 | 46 | 9 |
| 10 | DMAP | 1 | r.t. | 3 | 71 | 35:65 | 25 | 46 |
| 11 | DIPEA | 1 | r.t. | 3 | 71 | 29:71 | 21 | 50 |
| 12 | DBU | 1 | r.t. | 3 | 64 | 40:60 | 26 | 38 |
| Entry | Amino Acid Conf. | Base | Eq. | tR (h) | Yield (%) | RE (L-5:D-5) |
|---|---|---|---|---|---|---|
| 1 | L | DIPEA | 1 | 3 | 60 | 56:44 |
| 2 | D | 1 | 59 | 52:48 | ||
| 3 | L:D = 1:1 | 1 | 66 | 51:49 | ||
| 4 | L | Py | 1 | 15 | 98:2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sturabotti, E.; Vetica, F.; Toscano, G.; Calcaterra, A.; Martinelli, A.; Migneco, L.M.; Leonelli, F. N-Acetyl-l-phenylalanine Racemization during TBTU Amidation: An In-Depth Study for the Synthesis of Anti-Inflammatory 2-(N-Acetyl)-l-phenylalanylamido-2-deoxy-d-glucose (NAPA). Molecules 2023, 28, 581. https://doi.org/10.3390/molecules28020581
Sturabotti E, Vetica F, Toscano G, Calcaterra A, Martinelli A, Migneco LM, Leonelli F. N-Acetyl-l-phenylalanine Racemization during TBTU Amidation: An In-Depth Study for the Synthesis of Anti-Inflammatory 2-(N-Acetyl)-l-phenylalanylamido-2-deoxy-d-glucose (NAPA). Molecules. 2023; 28(2):581. https://doi.org/10.3390/molecules28020581
Chicago/Turabian StyleSturabotti, Elisa, Fabrizio Vetica, Giorgia Toscano, Andrea Calcaterra, Andrea Martinelli, Luisa Maria Migneco, and Francesca Leonelli. 2023. "N-Acetyl-l-phenylalanine Racemization during TBTU Amidation: An In-Depth Study for the Synthesis of Anti-Inflammatory 2-(N-Acetyl)-l-phenylalanylamido-2-deoxy-d-glucose (NAPA)" Molecules 28, no. 2: 581. https://doi.org/10.3390/molecules28020581