

Recent Advances on Small-Molecule Antagonists Targeting TLR7
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. TLR7 Main Features
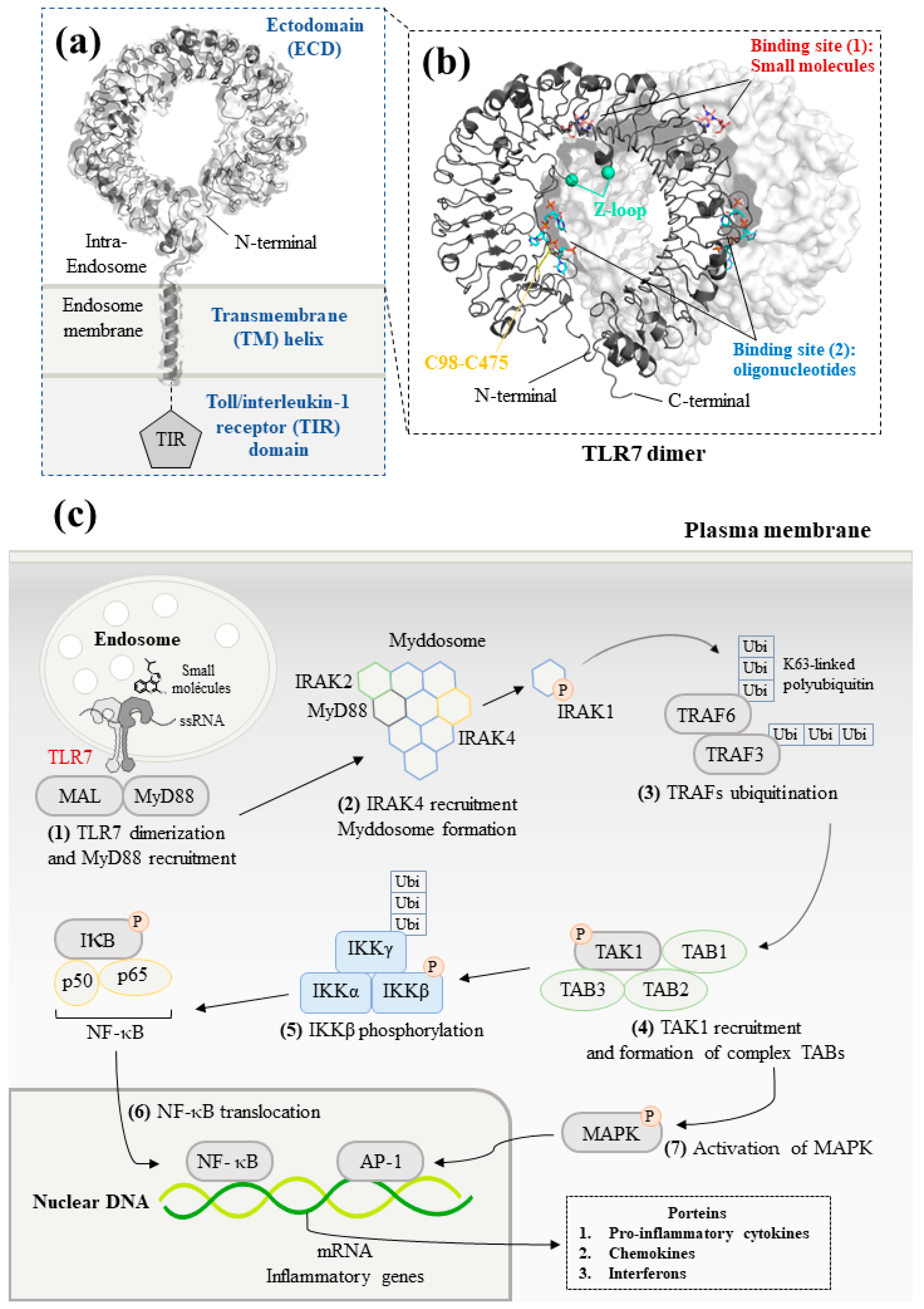
2.1. Structural Studies of TLR7
2.2. TLR7 Signaling Pathways
3. TLR7 Implication in a Variety of Clinical Diseases
3.1. Autoimmune Disorders
3.2. Immuno-Oncology
3.3. Antiviral Immunotherapy and Infection
3.4. Others
4. Small Molecule Antagonistic Ligands of TLR7
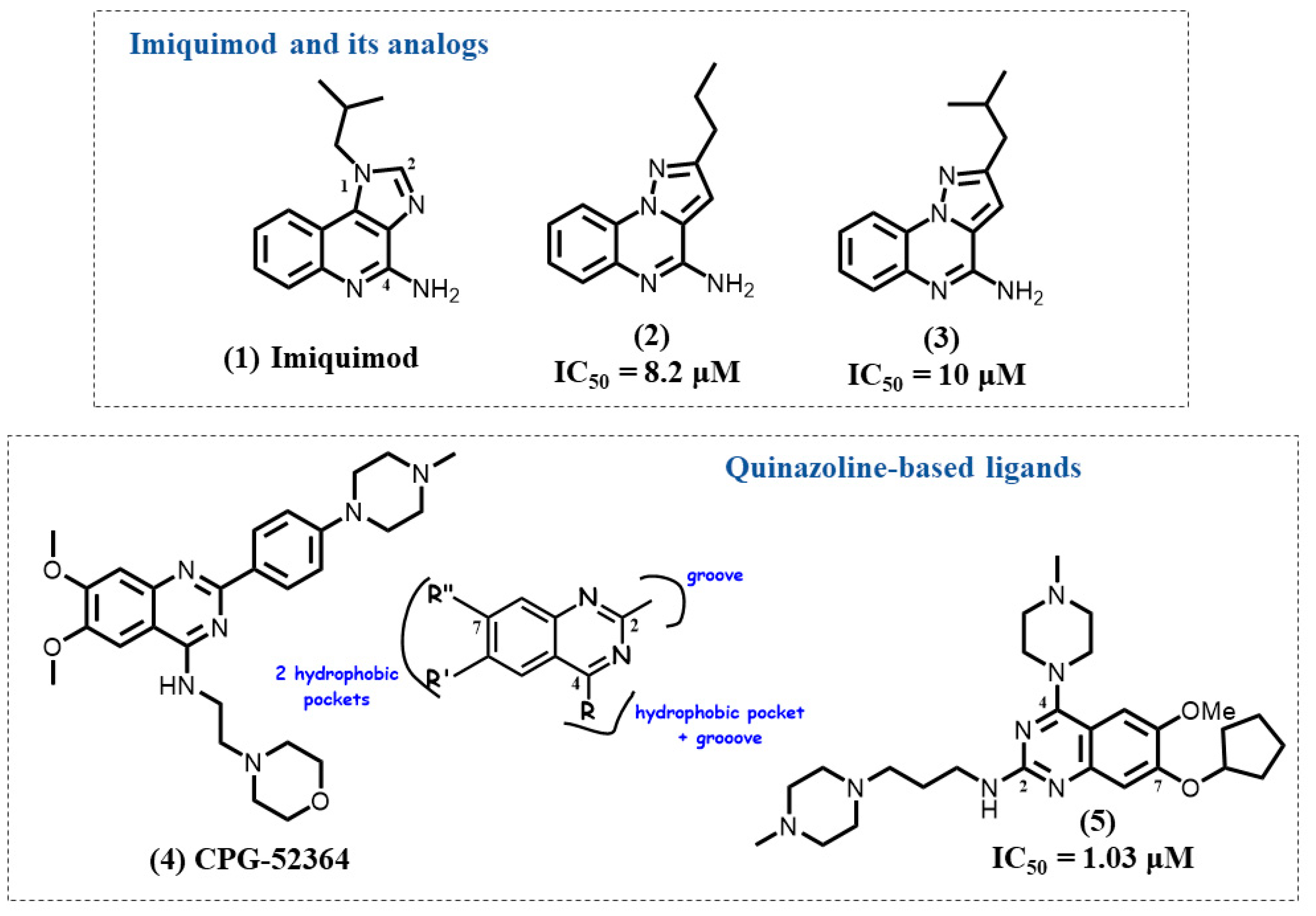
4.1. Imiquimod Analogs
4.2. Quinazoline-Based Ligands
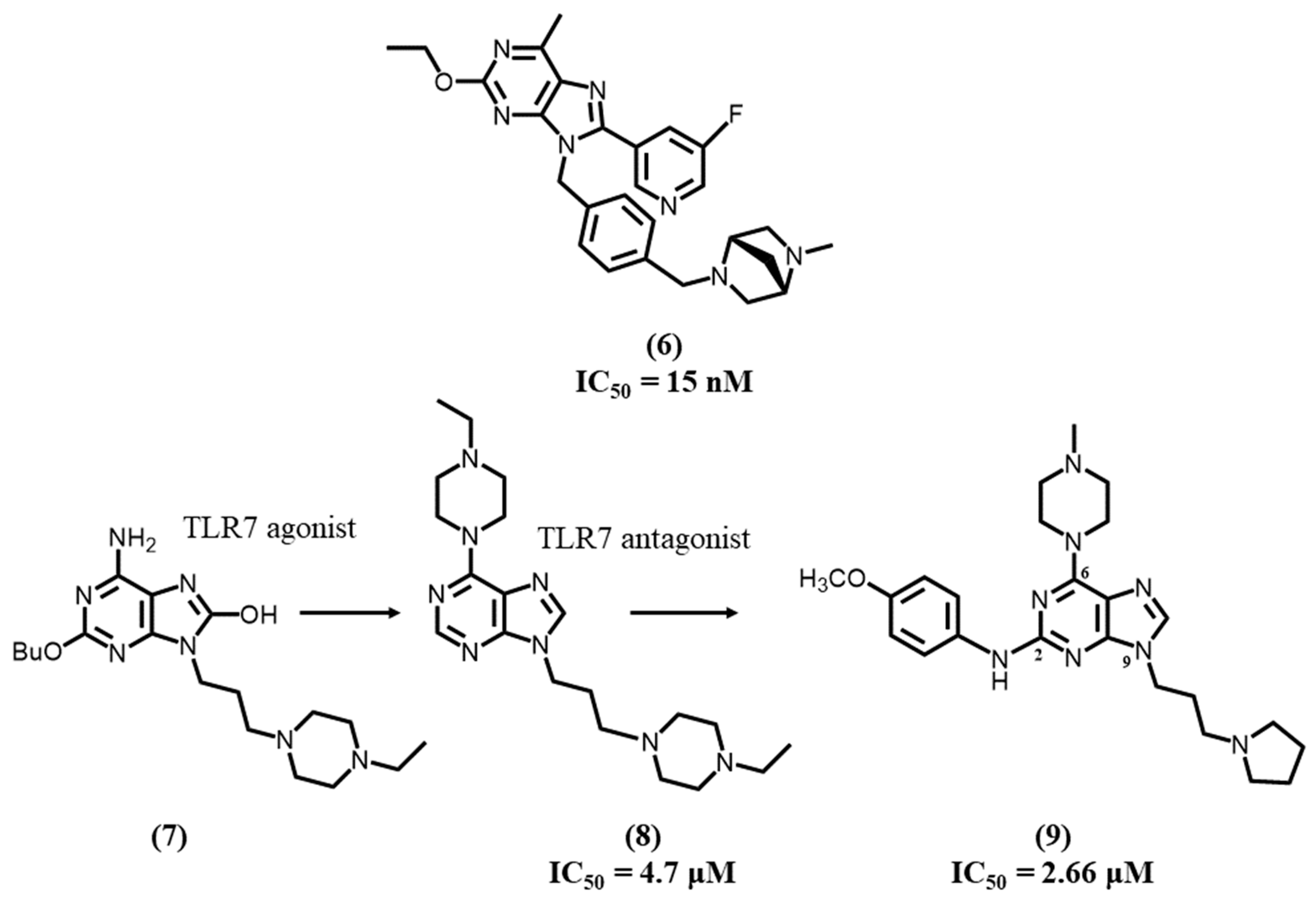
4.3. Purine-Based Ligands
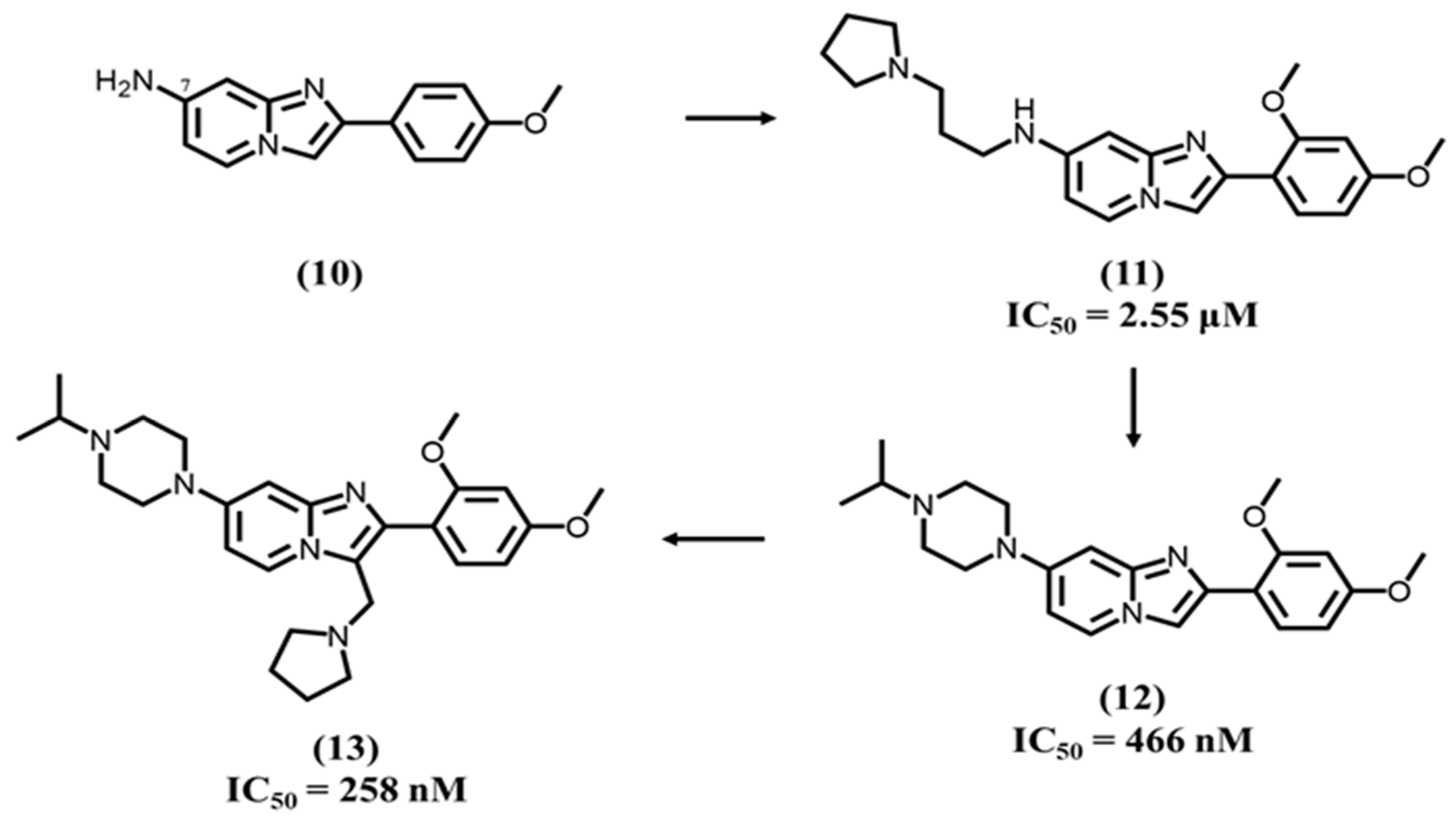
4.4. Imidazopyridine-Based Ligands
4.5. Pyridone-Based Ligands
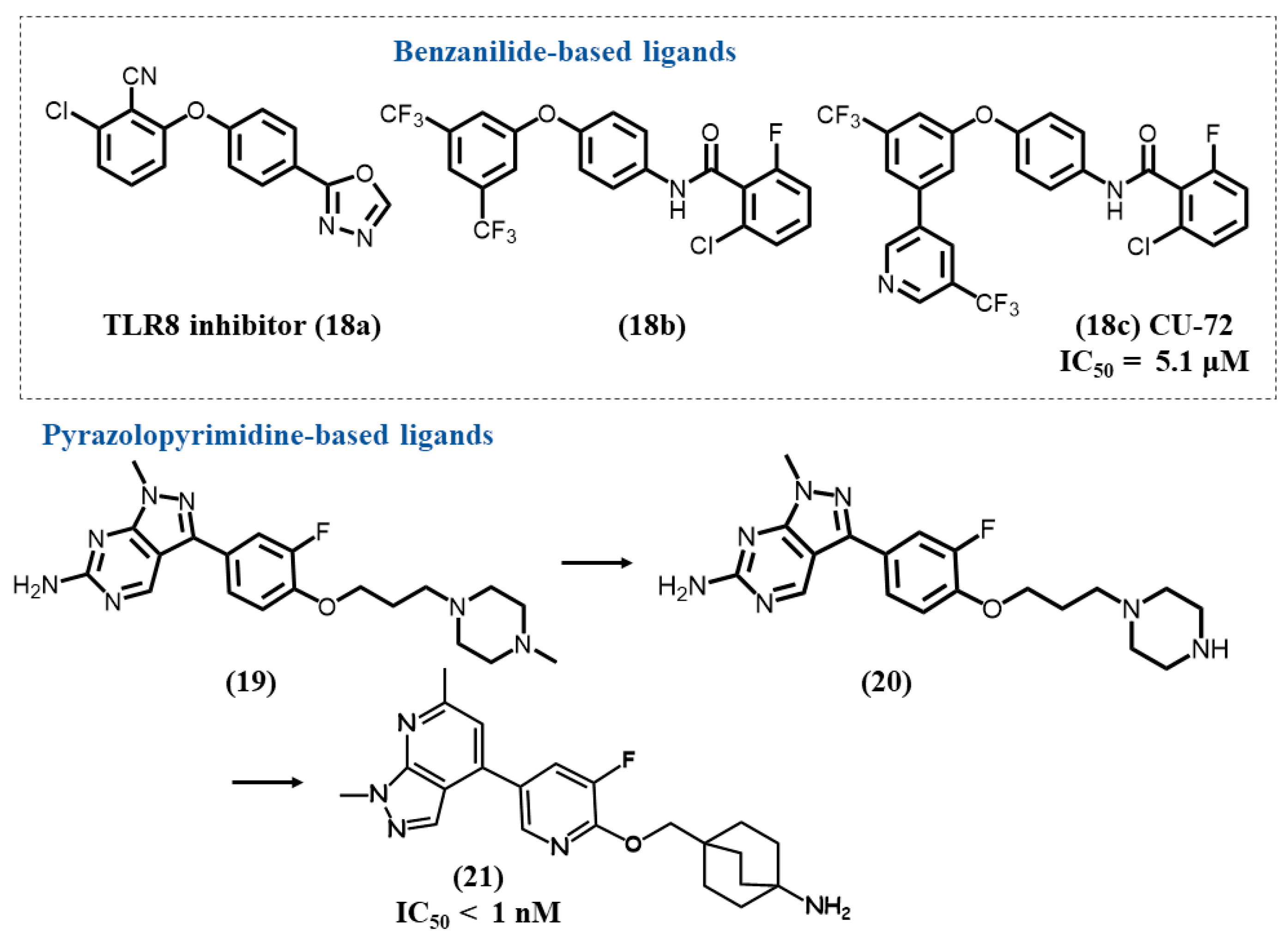
4.6. Benzanilide-Based Ligands
4.7. Pyrazolopyrimidine/Pyridine-Based Ligands
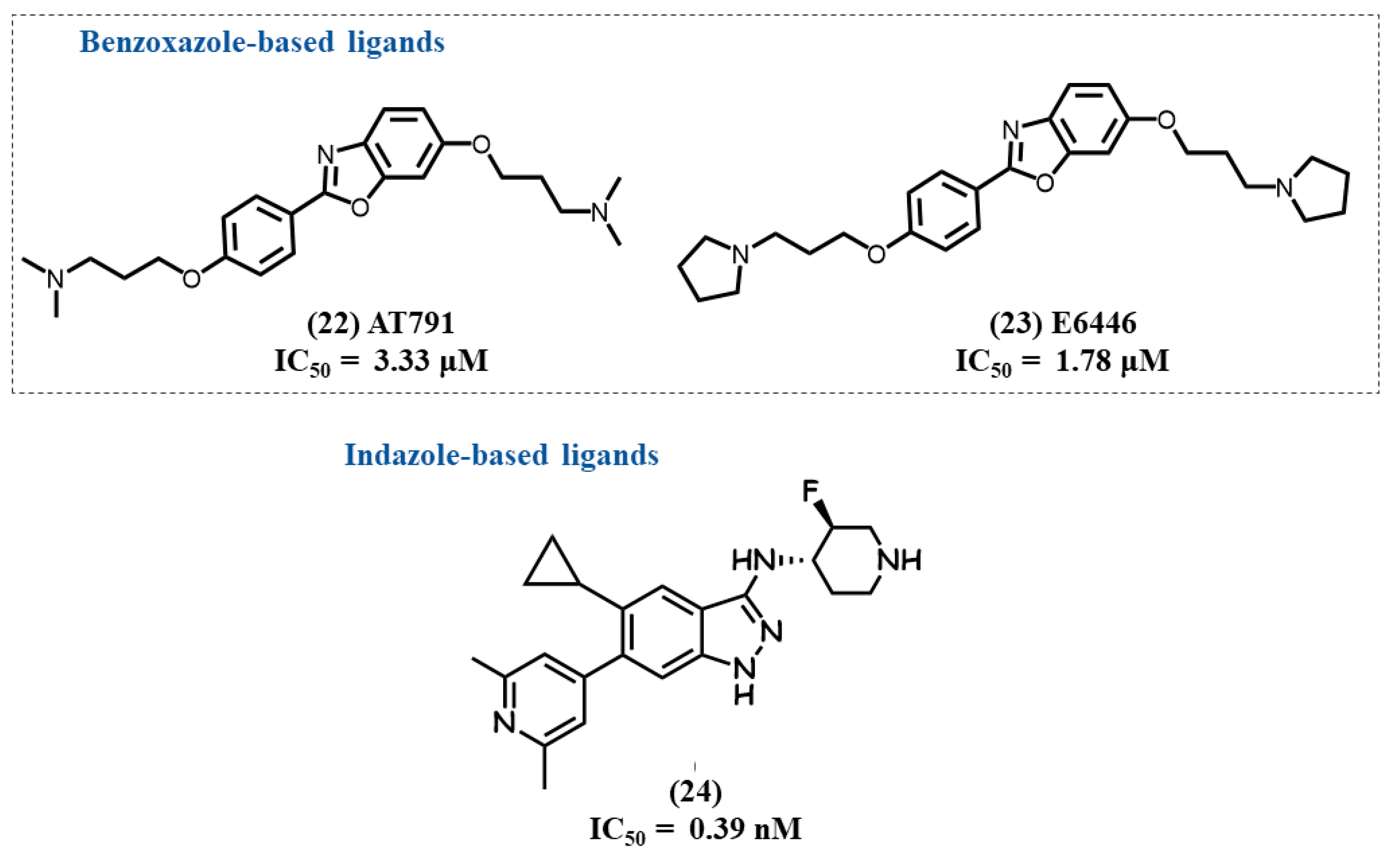
4.8. Benzoxazole-Based Ligands
4.9. Indazole-Based Ligands
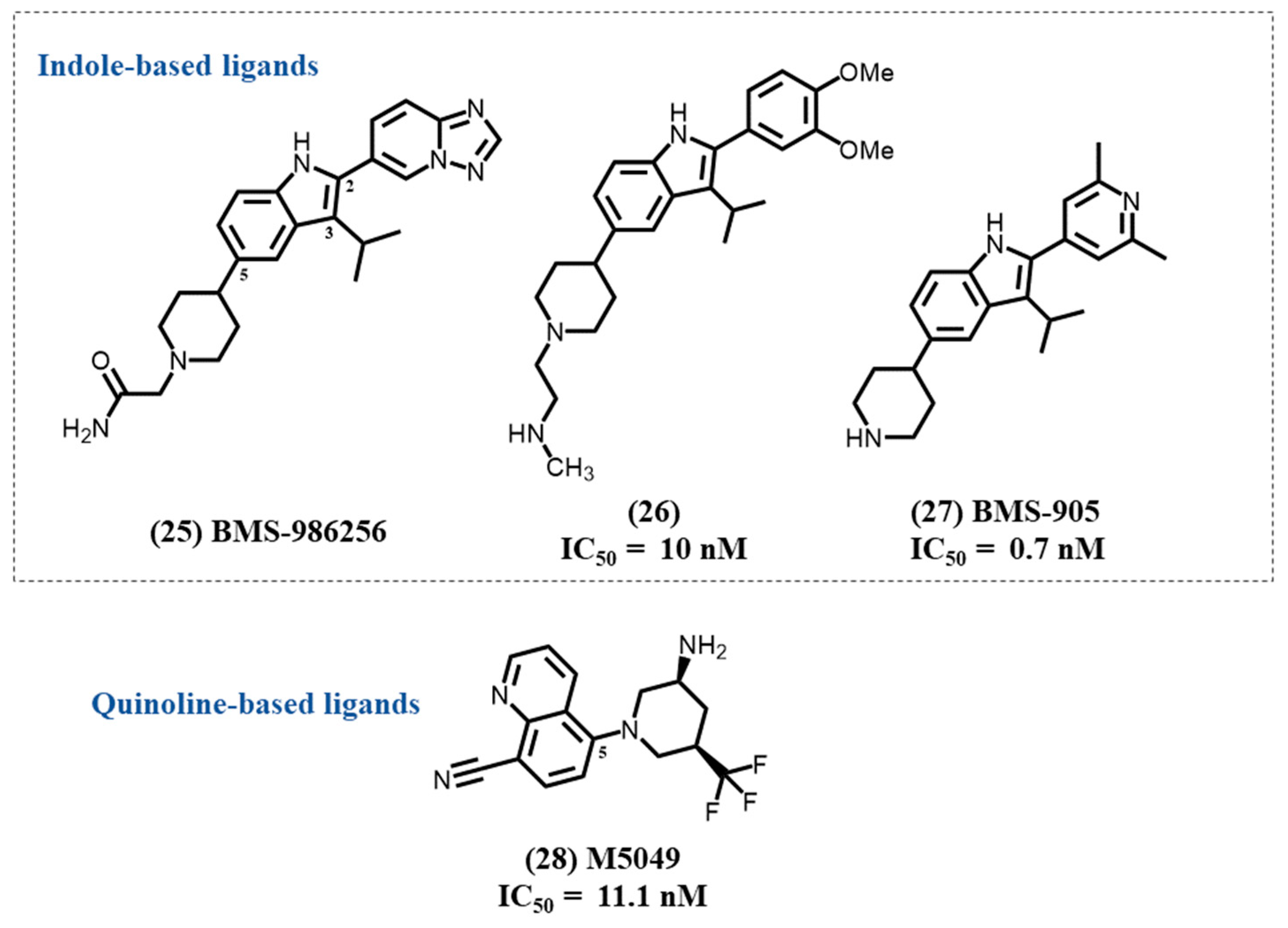
4.10. Indole-Based Ligands
4.11. Quinoline-Based Ligands
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Kawai, T.; Akira, S. Microbial Sensing by Toll-Like Receptors and Intracellular Nucleic Acid Sensors. Cold Spring Harb. Perspect. Biol. 2015, 7, a0162462015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.J.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e7. [Google Scholar] [CrossRef]
- Corzo, C.A.; Varfolomeev, E.; Setiadi, A.F.; Francis, R.; Klabunde, S.; Senger, K.; Sujatha-Bhaskar, S.; Drobnick, J.; Do, S.; Suto, E.; et al. The Kinase IRAK4 Promotes Endosomal TLR and Immune Complex Signaling in B Cells and Plasmacytoid Dendritic Cells. Sci. Signal. 2020, 13, eaaz1053. [Google Scholar] [CrossRef]
- Jarrossay, D.; Napolitani, G.; Colonna, M.; Sallusto, F.; Lanzavecchia, A. Specialization and Complementarity in Microbial Molecule Recognition by Human Myeloid and Plasmacytoid Dendritic Cells. Eur. J. Immunol. 2001, 31, 3388–3393. [Google Scholar] [CrossRef]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and Localization of Toll-like Receptor Signalling Complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- Pelka, K.; Bertheloot, D.; Reimer, E.; Phulphagar, K.; Schmidt, S.V.; Christ, A.; Stahl, R.; Watson, N.; Miyake, K.; Hacohen, N.; et al. The Chaperone UNC93B1 Regulates Toll-like Receptor Stability Independently of Endosomal TLR Transport. Immunity 2018, 48, 911–922.e7. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Junt, T.; Barchet, W. Translating Nucleic Acid-Sensing Pathways into Therapies. Nat. Rev. Immunol. 2015, 15, 529–544. [Google Scholar] [CrossRef]
- O’reilly, S.; Duffy, L. Toll-like Receptors in the Pathogenesis of Autoimmune Diseases: Recent and Emerging Translational Developments. ITT 2016, 5, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Patinote, C.; Karroum, N.B.; Moarbess, G.; Cirnat, N.; Kassab, I.; Bonnet, P.-A.; Deleuze-Masquéfa, C. Agonist and Antagonist Ligands of Toll-like Receptors 7 and 8: Ingenious Tools for Therapeutic Purposes. Eur. J. Med. Chem. 2020, 193, 112238. [Google Scholar] [CrossRef]
- Mukherjee, A.; Raychaudhuri, D.; Sinha, B.P.; Kundu, B.; Mitra, M.; Paul, B.; Bandopadhyay, P.; Ganguly, D.; Talukdar, A. A Chemical Switch for Transforming a Purine Agonist for Toll-like Receptor 7 to a Clinically Relevant Antagonist. J. Med. Chem. 2020, 63, 4776–4789. [Google Scholar] [CrossRef]
- Knoepfel, T.; Nimsgern, P.; Jacquier, S.; Bourrel, M.; Vangrevelinghe, E.; Glatthar, R.; Behnke, D.; Alper, P.B.; Michellys, P.-Y.; Deane, J.; et al. Target-Based Identification and Optimization of 5-Indazol-5-Yl Pyridones as Toll-like Receptor 7 and 8 Antagonists Using a Biochemical TLR8 Antagonist Competition Assay. J. Med. Chem. 2020, 63, 8276–8295. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, T.; Jiang, S.; Yin, H. Protocol for Evaluation and Validation of TLR8 Antagonists in HEK-Blue Cells via Secreted Embryonic Alkaline Phosphatase Assay. STAR Protoc. 2022, 3, 101061. [Google Scholar] [CrossRef]
- van der Fits, L.; Mourits, S.; Voerman, J.S.A.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.-M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice Is Mediated via the IL-23/IL-17 Axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ohto, U.; Shimizu, T. Toward a Structural Understanding of Nucleic Acid-sensing Toll-like Receptors in the Innate Immune System. FEBS Lett. 2017, 591, 3167–3181. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.K.; Mullen, G.E.D.; Leifer, C.A.; Mazzoni, A.; Davies, D.R.; Segal, D.M. Leucine-Rich Repeats and Pathogen Recognition in Toll-like Receptors. Trends Immunol. 2003, 24, 528–533. [Google Scholar] [CrossRef]
- Maeda, K.; Akira, S. TLR7 Structure: Cut in Z-Loop. Immunity 2016, 45, 705–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, A.; Yamamoto, C.; Onji, M.; Fukui, R.; Saitoh, S.; Motoi, Y.; Shibata, T.; Matsumoto, F.; Muta, T.; Miyake, K. Essential Role for Toll-like Receptor 7 (TLR7)-Unique Cysteines in an Intramolecular Disulfide Bond, Proteolytic Cleavage and RNA Sensing. Int. Immunol. 2013, 25, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, H.; Asami, J.; Zhang, Z.; Nishizawa, T.; Shigematsu, H.; Ohto, U.; Shimizu, T. Cryo-EM Structures of Toll-like Receptors in Complex with UNC93B1. Nat. Struct. Mol. Biol. 2021, 28, 173–180. [Google Scholar] [CrossRef]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals That Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Ohto, U.; Nomura, S.; Kibata, K.; Motoi, Y.; Zhang, Y.; Murakami, Y.; Fukui, R.; Ishimoto, T.; Sano, S.; et al. Guanosine and Its Modified Derivatives Are Endogenous Ligands for TLR7. Int. Immunol. 2016, 28, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; Shibata, T.; Ohto, U.; Shimizu, T.; Saitoh, S.-I.; Fukui, R.; Murakami, Y. Mechanisms Controlling Nucleic Acid-Sensing Toll-like Receptors. Int. Immunol. 2018, 30, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, S.; Li, H.; Wang, H.; Zhang, T.; Hutchinson, M.R.; Yin, H.; Wang, X. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res. 2020, 53, 1046–1055. [Google Scholar] [CrossRef]
- Lushpa, V.A.; Goncharuk, M.V.; Lin, C.; Zalevsky, A.O.; Talyzina, I.A.; Luginina, A.P.; Vakhrameev, D.D.; Shevtsov, M.B.; Goncharuk, S.A.; Arseniev, A.S.; et al. Modulation of Toll-like Receptor 1 Intracellular Domain Structure and Activity by Zn2+ Ions. Commun. Biol. 2021, 4, 1003. [Google Scholar] [CrossRef]
- Nyman, T.; Stenmark, P.; Flodin, S.; Johansson, I.; Hammarström, M.; Nordlund, P. The Crystal Structure of the Human Toll-like Receptor 10 Cytoplasmic Domain Reveals a Putative Signaling Dimer. J. Biol. Chem. 2008, 283, 11861–11865. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Oh, D.S.; Jung, H.E.; Chang, J.; Lee, H.K. Plasmacytoid Dendritic Cells Contribute to the Production of IFN-β via TLR7-MyD88-Dependent Pathway and CTL Priming during Respiratory Syncytial Virus Infection. Viruses 2019, 11, 730. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Sig Transduct Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- AbdAllah, N.B.; Toraih, E.A.; Al Ageeli, E.; Elhagrasy, H.; Gouda, N.S.; Fawzy, M.S.; Helal, G.M. MYD88, NFKB1, and IL6 Transcripts Overexpression Are Associated with Poor Outcomes and Short Survival in Neonatal Sepsis. Sci. Rep. 2021, 11, 13374. [Google Scholar] [CrossRef]
- Balka, K.R.; Nardo, D. Understanding Early TLR Signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef]
- Chen, Z.J. Ubiquitination in Signaling to and Activation of IKK: Ubiquitin-Mediated Activation of IKK. Immunol. Rev. 2012, 246, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.-R.; Michaelis, K.A.; Nozik-Grayck, E.; Seedorf, G.J.; Hartman-Filson, M.; Abman, S.H.; Wright, C.J. The NF-ΚB Inhibitory Proteins IκBα and IκBβ Mediate Disparate Responses to Inflammation in Fetal Pulmonary Endothelial Cells. J. Immunol. 2013, 190, 2913–2923. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Sig. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, M.; Baron, B. The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. Int. J. Inflam. 2017, 2017, 8391230. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Marken, J.; Chen, J.; Tran, V.B.; Li, Q.-Z.; Li, M.; Cerosaletti, K.; Elkon, K.B.; Zeng, X.; Giltiay, N.V. High TLR7 Expression Drives the Expansion of CD19+CD24hiCD38hi Transitional B Cells and Autoantibody Production in SLE Patients. Front. Immunol. 2019, 10, 1243. [Google Scholar] [CrossRef] [Green Version]
- Illescas-Montes, R.; Corona-Castro, C.C.; Melguizo-Rodríguez, L.; Ruiz, C.; Costela-Ruiz, V.J. Infectious Processes and Systemic Lupus Erythematosus. Immunology 2019, 158, 153–160. [Google Scholar] [CrossRef]
- Zhou, Y.; Yuan, J.; Pan, Y.; Fei, Y.; Qiu, X.; Hu, N.; Luo, Y.; Lei, W.; Li, Y.; Long, H.; et al. T Cell CD40LG Gene Expression and the Production of IgG by Autologous B Cells in Systemic Lupus Erythematosus. Clin. Immunol. 2009, 132, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshak-Rothstein, A.; Rifkin, I.R. Immunologically Active Autoantigens: The Role of Toll-Like Receptors in the Development of Chronic Inflammatory Disease. Annu. Rev. Immunol. 2007, 25, 419–441. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Role of Type I Interferons in the Activation of Autoreactive B Cells. Immunol. Cell Biol 2012, 90, 498–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, J.R.; Molano, I.; Ruiz, P.; Coutermarsh-Ott, S.; Cunningham, M.A. TLR7 Agonism Accelerates Disease and Causes a Fatal Myeloproliferative Disorder in NZM 2410 Lupus Mice. Front. Immunol. 2020, 10, 3054. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Lee, H.; Yamamoto, M.; Jones, L.A.; Dayalan, J.; Hopkins, R.; Zhou, X.J.; Yarovinsky, F.; Connolly, J.E.; Curotto de Lafaille, M.A.; et al. B Cell TLR7 Expression Drives Anti-RNA Autoantibody Production and Exacerbates Disease in Systemic Lupus Erythematosus–Prone Mice. J. Immunol. 2012, 189, 5786–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, E.R.; Pisitkun, P.; Voynova, E.; Deane, J.A.; Scott, B.L.; Caspi, R.R.; Bolland, S. Dual Signaling by Innate and Adaptive Immune Receptors Is Required for TLR7-Induced B-Cell–Mediated Autoimmunity. Proc. Natl. Acad. Sci. USA 2012, 109, 16276–16281. [Google Scholar] [CrossRef] [Green Version]
- Deane, J.A.; Pisitkun, P.; Barrett, R.S.; Feigenbaum, L.; Town, T.; Ward, J.M.; Flavell, R.A.; Bolland, S. Control of Toll-like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity 2007, 27, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Sakata, K.; Nakayamada, S.; Miyazaki, Y.; Kubo, S.; Ishii, A.; Nakano, K.; Tanaka, Y. Up-Regulation of TLR7-Mediated IFN-α Production by Plasmacytoid Dendritic Cells in Patients with Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1957. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.J.; Cañete, P.F.; Wang, H.; Medhavy, A.; Bones, J.; Roco, J.A.; He, Y.; Qin, Y.; Cappello, J.; Ellyard, J.I.; et al. TLR7 Gain-of-Function Genetic Variation Causes Human Lupus. Nature 2022, 605, 349–356. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Hoffmann, J.A.; Galluzzi, L. Pharmacological Modulation of Nucleic Acid Sensors—Therapeutic Potential and Persisting Obstacles. Nat. Rev. Drug Discov. 2019, 18, 845–867. [Google Scholar] [CrossRef]
- Akilesh, H.M.; Buechler, M.B.; Duggan, J.M.; Hahn, W.O.; Matta, B.; Sun, X.; Gessay, G.; Whalen, E.; Mason, M.; Presnell, S.R.; et al. Chronic TLR7 and TLR9 Signaling Drives Anemia via Differentiation of Specialized Hemophagocytes. Science 2019, 363, eaao5213. [Google Scholar] [CrossRef]
- Rimbach, K.; Kaiser, S.; Helm, M.; Dalpke, A.H.; Eigenbrod, T. 2’-O-Methylation within Bacterial RNA Acts as Suppressor of TLR7/TLR8 Activation in Human Innate Immune Cells. J. Innate Immun. 2015, 7, 482–493. [Google Scholar] [CrossRef]
- Schmitt, F.C.F.; Freund, I.; Weigand, M.A.; Helm, M.; Dalpke, A.H.; Eigenbrod, T. Identification of an Optimized 2′- O -Methylated Trinucleotide RNA Motif Inhibiting Toll-like Receptors 7 and 8. RNA 2017, 23, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.-Y.; Su, Y.-W.; Lin, K.-I.; Hsu, L.-C.; Chuang, T.-H. Natural Modulators of Endosomal Toll-Like Receptor-Mediated Psoriatic Skin Inflammation. J. Immunol. Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Fariñas, M.; Arbeit, R.; Jiang, W.; Ortenzio, F.S.; Sullivan, T.; Krueger, J.G. Suppression of Molecular Inflammatory Pathways by Toll-Like Receptor 7, 8, and 9 Antagonists in a Model of IL-23-Induced Skin Inflammation. PLoS ONE 2013, 8, e84634. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Wang, D.; Jiang, W.; Bhagat, L.; Agrawal, S. Sa1757 Targeting Innate Immune Receptors to Treat Inflammatory Bowel Disease: Preclinical Activity of IMO-9200, an Antagonist of TLRS 7, 8, and 9 in Mouse Models of Colitis. Gastroenterology 2015, 148, S-324. [Google Scholar] [CrossRef]
- Pearson, J.A.; Tai, N.; Ekanayake-Alper, D.K.; Peng, J.; Hu, Y.; Hager, K.; Compton, S.; Wong, F.S.; Smith, P.C.; Wen, L. Norovirus Changes Susceptibility to Type 1 Diabetes by Altering Intestinal Microbiota and Immune Cell Functions. Front. Immunol. 2019, 10, 2654. [Google Scholar] [CrossRef]
- Huang, J.; Peng, J.; Pearson, J.A.; Efthimiou, G.; Hu, Y.; Tai, N.; Xing, Y.; Zhang, L.; Gu, J.; Jiang, J.; et al. Toll-like Receptor 7 Deficiency Suppresses Type 1 Diabetes Development by Modulating B-Cell Differentiation and Function. Cell Mol. Immunol. 2021, 18, 328–338. [Google Scholar] [CrossRef]
- Lee, A.S.; Ghoreishi, M.; Cheng, W.K.; Chang, T.-Y.E.; Zhang, Y.Q.; Dutz, J.P. Toll-like Receptor 7 Stimulation Promotes Autoimmune Diabetes in the NOD Mouse. Diabetologia 2011, 54, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Tomita, T. Apoptosis of Pancreatic β-Cells in Type 1 Diabetes. Bosn J. Basic Med. Sci. 2017, 17, 183–193. [Google Scholar] [CrossRef]
- Eizirik, D.L.; Mandrup-Poulsen, T. A Choice of Death—The Signal-Transduction of Immune-Mediated Beta-Cell Apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Roussel-Queval, A.; Chasson, L.; Hanna Kazazian, N.; Marcadet, L.; Nezos, A.; Sieweke, M.H.; Mavragani, C.; Alexopoulou, L. TLR7 Signaling Drives the Development of Sjögren’s Syndrome. Front. Immunol. 2021, 12, 676010. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L. Nucleic Acid-Sensing Toll-like Receptors: Important Players in Sjögren’s Syndrome. Front. Immunol. 2022, 13, 980400. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, M.; Jonsson, R.; Brun, J.G.; Appel, S.; Hansen, T. TLR-7 and -9 Stimulation of Peripheral Blood B Cells Indicate Altered TLR Signalling in Primary Sjögren’s Syndrome Patients by Increased Secretion of Cytokines. Scand. J. Immunol. 2015, 82, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like Receptors and Cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Choi, S. Toll-like Receptors from the Perspective of Cancer Treatment. Cancers 2020, 12, 297. [Google Scholar] [CrossRef] [Green Version]
- Urban-Wojciuk, Z.; Khan, M.M.; Oyler, B.L.; Fåhraeus, R.; Marek-Trzonkowska, N.; Nita-Lazar, A.; Hupp, T.R.; Goodlett, D.R. The Role of TLRs in Anti-Cancer Immunity and Tumor Rejection. Front. Immunol. 2019, 10, 2388. [Google Scholar] [CrossRef]
- Grimmig, T.; Matthes, N.; Hoeland, K.; Tripathi, S.; Chandraker, A.; Grimm, M.; Moench, R.; Moll, E.-M.; Friess, H.; Tsaur, I.; et al. TLR7 and TLR8 Expression Increases Tumor Cell Proliferation and Promotes Chemoresistance in Human Pancreatic Cancer. Int. J. Oncol. 2015, 47, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Fukata, M.; Abreu, M.T. Role of Toll-like Receptors in Gastrointestinal Malignancies. Oncogene 2008, 27, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yan, Z.; Wang, J.; Yao, X. Toll-like Receptors 7 and 8 Expression Correlates with the Expression of Immune Biomarkers and Positively Predicts the Clinical Outcome of Patients with Melanoma. OTT 2017, 10, 4339–4346. [Google Scholar] [CrossRef]
- Sheyhidin, I. Overexpression of TLR3, TLR4, TLR7 and TLR9 in Esophageal Squamous Cell Carcinoma. WJG 2011, 17, 3745. [Google Scholar] [CrossRef]
- Keshavarz, A.; Pourbagheri-Sigaroodi, A.; Zafari, P.; Bagheri, N.; Ghaffari, S.H.; Bashash, D. Toll-like Receptors (TLRs) in Cancer; with an Extensive Focus on TLR Agonists and Antagonists. IUBMB Life 2021, 73, 10–25. [Google Scholar] [CrossRef]
- Chatterjee, S.; Crozet, L.; Damotte, D.; Iribarren, K.; Schramm, C.; Alifano, M.; Lupo, A.; Cherfils-Vicini, J.; Goc, J.; Katsahian, S.; et al. TLR7 Promotes Tumor Progression, Chemotherapy Resistance, and Poor Clinical Outcomes in Non–Small Cell Lung Cancer. Cancer Res. 2014, 74, 5008–5018. [Google Scholar] [CrossRef] [Green Version]
- Caron, G.; Duluc, D.; Frémaux, I.; Jeannin, P.; David, C.; Gascan, H.; Delneste, Y. Direct Stimulation of Human T Cells via TLR5 and TLR7/8: Flagellin and R-848 Up-Regulate Proliferation and IFN-γ Production by Memory CD4+ T Cells. J. Immunol. 2005, 175, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 Promotes Tumor Growth and Suppresses Tumor Immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [Green Version]
- Meås, H.Z.; Haug, M.; Beckwith, M.S.; Louet, C.; Ryan, L.; Hu, Z.; Landskron, J.; Nordbø, S.A.; Taskén, K.; Yin, H.; et al. Sensing of HIV-1 by TLR8 Activates Human T Cells and Reverses Latency. Nat. Commun. 2020, 11, 147. [Google Scholar] [CrossRef] [Green Version]
- Fugger, L.; Jensen, L.T.; Rossjohn, J. Challenges, Progress, and Prospects of Developing Therapies to Treat Autoimmune Diseases. Cell 2020, 181, 63–80. [Google Scholar] [CrossRef]
- Patamawenu, A.A.; Wright, N.E.; Shofner, T.; Evans, S.; Manion, M.M.; Doria-Rose, N.; Migueles, S.A.; Mendoza, D.; Peterson, B.; Wilhelm, C.; et al. Toll-like Receptor 7-Adapter Complex Modulates Interferon-α Production in HIV-Stimulated Plasmacytoid Dendritic Cells. PLoS ONE 2019, 14, e0225806. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Villar, M.; Gautron, A.-S.; de Marcken, M.; Keller, M.J.; Hafler, D.A. TLR7 Induces Anergy in Human CD4+ T Cells. Nat. Immunol. 2015, 16, 118–128. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Mueller, Y.M.; Yang, G.; Boesteanu, A.C.; Gracias, D.T.; Do, D.H.; Hope, J.L.; Kathuria, N.; McGettigan, S.E.; Lewis, M.G.; et al. Type I Interferon Upregulates Bak and Contributes to T Cell Loss during Human Immunodeficiency Virus (HIV) Infection. PLoS Pathog. 2013, 9, e1003658. [Google Scholar] [CrossRef]
- Simmons, R.P.; Scully, E.P.; Groden, E.E.; Arnold, K.B.; Chang, J.J.; Lane, K.; Lifson, J.; Rosenberg, E.; Lauffenburger, D.A.; Altfeld, M. HIV-1 Infection Induces Strong Production of IP-10 through TLR7/9-Dependent Pathways. AIDS 2013, 27, 2505–2517. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zuo, X.; Zhang, S.; Ouyang, Z.; Jiang, S.; Wang, F.; Wang, G. The Mechanism behind Influenza Virus Cytokine Storm. Viruses 2021, 13, 1362. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.; Yang, Z. The Cytokine Storm of Severe Influenza and Development of Immunomodulatory Therapy. Cell Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappe, J.C.F.; Finsterbusch, K.; Crotta, S.; Mack, M.; Priestnall, S.L.; Wack, A. A TLR7 Antagonist Restricts Interferon-Dependent and -Independent Immunopathology in a Mouse Model of Severe Influenza. J. Exp. Med. 2021, 218, e20201631. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Huang, L.; Luo, Q.; Tu, Q.; Liu, J.; Yu, R.; Huang, J.; Chen, T.; Yin, Y.; Cao, J. Absence of Toll-like receptor 7 protects mice against Pseudomonas aeruginosa pneumonia. Int. Immunopharmacol. 2021, 96, 107739. [Google Scholar] [CrossRef]
- Mancuso, G.; Gambuzza, M.; Midiri, A.; Biondo, C.; Papasergi, S.; Akira, S.; Teti, G.; Beninati, C. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat. Immunol. 2009, 10, 587–594. [Google Scholar] [CrossRef]
- Lee, C.Y.Q.; Chan, Y.T.; Cheok, Y.Y.; Tan, G.M.Y.; Tang, T.F.; Cheong, H.C.; Vadivelu, J.; Abdullah, S.; Looi, C.Y.; Wong, W.F. Helicobacter pylori Infection Elicits Type I Interferon Response in Human Monocytes via Toll-Like Receptor 8 Signaling. J. Immunol. Res. 2022, 2022, 3861518. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.-Y. MicroRNAs Are Ligands of Toll-like Receptors. RNA 2013, 19, 737–739. [Google Scholar] [CrossRef] [Green Version]
- Hoshikawa, N.; Sakai, A.; Takai, S.; Suzuki, H. Targeting Extracellular MiR-21-TLR7 Signaling Provides Long-Lasting Analgesia in Osteoarthritis. Mol. Ther. Nucleic Acids 2020, 19, 199–207. [Google Scholar] [CrossRef]
- Liu, C.-L.; Santos, M.M.; Fernandes, C.; Liao, M.; Iamarene, K.; Zhang, J.-Y.; Sukhova, G.K.; Shi, G.-P. Toll-like Receptor 7 Deficiency Protects Apolipoprotein E-Deficient Mice from Diet-Induced Atherosclerosis. Sci. Rep. 2017, 7, 847. [Google Scholar] [CrossRef]
- Salagianni, M.; Galani, I.E.; Lundberg, A.M.; Davos, C.H.; Varela, A.; Gavriil, A.; Lyytikäinen, L.-P.; Lehtimäki, T.; Sigala, F.; Folkersen, L.; et al. Toll-Like Receptor 7 Protects From Atherosclerosis by Constraining “Inflammatory” Macrophage Activation. Circulation 2012, 126, 952–962. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small Anti-Viral Compounds Activate Immune Cells via the TLR7 MyD88–Dependent Signaling Pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef]
- Sauder, D.N. Imiquimod: Modes of Action. Br. J. Derm. 2003, 149, 5–8. [Google Scholar] [CrossRef]
- Buck, H.W. Imiquimod (Aldara? Cream). Infect. Dis. Obstet. Gynecol. 1998, 6, 49–51. [Google Scholar] [CrossRef]
- Marks, R.; Gebauer, K.; Shumack, S.; Amies, M.; Bryden, J.; Fox, T.L.; Owens, M.L. Imiquimod 5% Cream in the Treatment of Superficial Basal Cell Carcinoma: Results of a Multicenter 6-Week Dose-Response Trial. J. Am. Acad Derm. 2001, 44, 807–813. [Google Scholar] [CrossRef]
- Bou Karroum, N.; Moarbess, G.; Guichou, J.-F.; Bonnet, P.-A.; Patinote, C.; Bouharoun-Tayoun, H.; Chamat, S.; Cuq, P.; Diab-Assaf, M.; Kassab, I.; et al. Novel and Selective TLR7 Antagonists among the Imidazo [1,2- a ]Pyrazines, Imidazo [1,5- a ]Quinoxalines, and Pyrazolo [1,5- a ]Quinoxalines Series. J. Med. Chem. 2019, 62, 7015–7031. [Google Scholar] [CrossRef]
- Yang, M.; Larson, P.G.; Brown, L.; Schultz, J.R.; Kucaba, T.A.; Griffith, T.S.; Ferguson, D.M. Toll-like Receptor 7 and 8 Imidazoquinoline-Based Agonist/Antagonist Pairs. Bioorg. Med. Chem. Lett. 2022, 59, 128548. [Google Scholar] [CrossRef]
- Pal, S.; Paul, B.; Bandopadhyay, P.; Preethy, N.; Sarkar, D.; Rahaman, O.; Goon, S.; Roy, S.; Ganguly, D.; Talukdar, A. Synthesis and Characterization of New Potent TLR7 Antagonists Based on Analysis of the Binding Mode Using Biomolecular Simulations. Eur. J. Med. Chem. 2021, 210, 112978. [Google Scholar] [CrossRef]
- Dharmapuri, S.; Aurisicchio, L.; Neuner, P.; Verdirame, M.; Ciliberto, G.; La Monica, N. An Oral TLR7 Agonist Is a Potent Adjuvant of DNA Vaccination in Transgenic Mouse Tumor Models. Cancer Gene Ther. 2009, 16, 462–472. [Google Scholar] [CrossRef]
- Hirota, K.; Kazaoka, K.; Niimoto, I.; Kumihara, H.; Sajiki, H.; Isobe, Y.; Takaku, H.; Tobe, M.; Ogita, H.; Ogino, T.; et al. Discovery of 8-Hydroxyadenines as a Novel Type of Interferon Inducer. J. Med. Chem. 2002, 45, 5419–5422. [Google Scholar] [CrossRef]
- Bazin, H.G.; Li, Y.; Khalaf, J.K.; Mwakwari, S.; Livesay, M.T.; Evans, J.T.; Johnson, D.A. Structural Requirements for TLR7-Selective Signaling by 9-(4-Piperidinylalkyl)-8-Oxoadenine Derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 1318–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Wada, H.; Kurebayashi, H.; McInally, T.; Bonnert, R.; Isobe, Y. Synthesis and Evaluation of 8-Oxoadenine Derivatives as Potent Toll-like Receptor 7 Agonists with High Water Solubility. Bioorg. Med. Chem. Lett. 2013, 23, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Tojo, S.; Zhang, Z.; Matsui, H.; Tahara, M.; Ikeguchi, M.; Kochi, M.; Kamada, M.; Shigematsu, H.; Tsutsumi, A.; Adachi, N.; et al. Structural Analysis Reveals TLR7 Dynamics Underlying Antagonism. Nat. Commun. 2020, 11, 5204. [Google Scholar] [CrossRef] [PubMed]
- Kundu, B.; Raychaudhuri, D.; Mukherjee, A.; Sinha, B.P.; Sarkar, D.; Bandopadhyay, P.; Pal, S.; Das, N.; Dey, D.; Ramarao, K.; et al. Systematic Optimization of Potent and Orally Bioavailable Purine Scaffold as a Dual Inhibitor of Toll-Like Receptors 7 and 9. J. Med. Chem. 2021, 64, 9279–9301. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Bandopadhyay, P.; Roy, S.; Sinha, B.P.; Dastidar, U.G.; Rahaman, O.; Pal, S.; Ganguly, D.; Talukdar, A. Development, Optimization, and In Vivo Validation of New Imidazopyridine Chemotypes as Dual TLR7/TLR9 Antagonists through Activity-Directed Sequential Incorporation of Relevant Structural Subunits. J. Med. Chem. 2022, 65, 11607–11632. [Google Scholar] [CrossRef]
- Padilla-Salinas, R.; Anderson, R.; Sakaniwa, K.; Zhang, S.; Nordeen, P.; Lu, C.; Shimizu, T.; Yin, H. Discovery of Novel Small Molecule Dual Inhibitors Targeting Toll-Like Receptors 7 and 8. J. Med. Chem. 2019, 62, 10221–10244. [Google Scholar] [CrossRef]
- Alper, P.B.; Deane, J.; Betschart, C.; Buffet, D.; Collignon Zipfel, G.; Gordon, P.; Hampton, J.; Hawtin, S.; Ibanez, M.; Jiang, T.; et al. Discovery of Potent, Orally Bioavailable in Vivo Efficacious Antagonists of the TLR7/8 Pathway. Bioorg. Med. Chem. Lett. 2020, 30, 127366. [Google Scholar] [CrossRef]
- Lamphier, M.; Zheng, W.; Latz, E.; Spyvee, M.; Hansen, H.; Rose, J.; Genest, M.; Yang, H.; Shaffer, C.; Zhao, Y.; et al. Novel Small Molecule Inhibitors of TLR7 and TLR9: Mechanism of Action and Efficacy In Vivo. Mol. Pharm. 2014, 85, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Betschart, C.; Faller, M.; Zink, F.; Hemmig, R.; Blank, J.; Vangrevelinghe, E.; Bourrel, M.; Glatthar, R.; Behnke, D.; Barker, K.; et al. Structure-Based Optimization of a Fragment-like TLR8 Binding Screening Hit to an In Vivo Efficacious TLR7/8 Antagonist. ACS Med. Chem. Lett. 2022, 13, 658–664. [Google Scholar] [CrossRef]
- Mussari, C.P.; Dodd, D.S.; Sreekantha, R.K.; Pasunoori, L.; Wan, H.; Posy, S.L.; Critton, D.; Ruepp, S.; Subramanian, M.; Watson, A.; et al. Discovery of Potent and Orally Bioavailable Small Molecule Antagonists of Toll-like Receptors 7/8/9 (TLR7/8/9). ACS Med. Chem. Lett. 2020, 11, 1751–1758. [Google Scholar] [CrossRef]
- Sreekantha, R.K.; Mussari, C.P.; Dodd, D.S.; Pasunoori, L.; Hegde, S.; Posy, S.L.; Critton, D.; Ruepp, S.; Subramanian, M.; Salter-Cid, L.M.; et al. Identification of 2-Pyridinylindole-Based Dual Antagonists of Toll-like Receptors 7 and 8 (TLR7/8). ACS Med. Chem. Lett. 2022, 13, 812–818. [Google Scholar] [CrossRef]
- Vlach, J.; Bender, A.T.; Przetak, M.; Pereira, A.; Deshpande, A.; Johnson, T.L.; Reissig, S.; Tzvetkov, E.; Musil, D.; Morse, N.T.; et al. Discovery of M5049: A Novel Selective Toll-Like Receptor 7/8 Inhibitor for Treatment of Autoimmunity. J. Pharm. Exp. Ther. 2021, 376, 397–409. [Google Scholar] [CrossRef]
- Port, A.; Shaw, J.V.; Klopp-Schulze, L.; Bytyqi, A.; Vetter, C.; Hussey, E.; Mammasse, N.; Ona, V.; Bachmann, A.; Strugala, D.; et al. Phase 1 Study in Healthy Participants of the Safety, Pharmacokinetics, and Pharmacodynamics of Enpatoran (M5049), a Dual Antagonist of Toll-like Receptors 7 and 8. Pharm. Res. Perspect. 2021, 9, e00842. [Google Scholar] [CrossRef]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like Receptors in the Pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef]
- Dyavar, S.R.; Singh, R.; Emani, R.; Pawar, G.P.; Chaudhari, V.D.; Podany, A.T.; Avedissian, S.N.; Fletcher, C.V.; Salunke, D.B. Role of Toll-like Receptor 7/8 Pathways in Regulation of Interferon Response and Inflammatory Mediators during SARS-CoV2 Infection and Potential Therapeutic Options. Biomed. Pharm. 2021, 141, 111794. [Google Scholar] [CrossRef]
- Klopp-Schulze, L.; Shaw, J.V.; Dong, J.Q.; Khandelwal, A.; Vazquez-Mateo, C.; Goteti, K. Applying Modeling and Simulations for Rational Dose Selection of Novel Toll-Like Receptor 7/8 Inhibitor Enpatoran for Indications of High Medical Need. Clin. Pharma. Ther. 2022, 112, 297–306. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, H.; Wu, P.; Bonnet, P.-A. Recent Advances on Small-Molecule Antagonists Targeting TLR7. Molecules 2023, 28, 634. https://doi.org/10.3390/molecules28020634
Zheng H, Wu P, Bonnet P-A. Recent Advances on Small-Molecule Antagonists Targeting TLR7. Molecules. 2023; 28(2):634. https://doi.org/10.3390/molecules28020634
Chicago/Turabian StyleZheng, Haoyang, Peiyang Wu, and Pierre-Antoine Bonnet. 2023. "Recent Advances on Small-Molecule Antagonists Targeting TLR7" Molecules 28, no. 2: 634. https://doi.org/10.3390/molecules28020634
APA StyleZheng, H., Wu, P., & Bonnet, P.-A. (2023). Recent Advances on Small-Molecule Antagonists Targeting TLR7. Molecules, 28(2), 634. https://doi.org/10.3390/molecules28020634