
Interrupted Nef and Meyer Reactions: A Growing Point for Diversity-Oriented Synthesis Based on Nitro Compounds

Abstract
:
1. Introduction
2. Interruption of the Nef and Meyer Reactions by Halide Anions
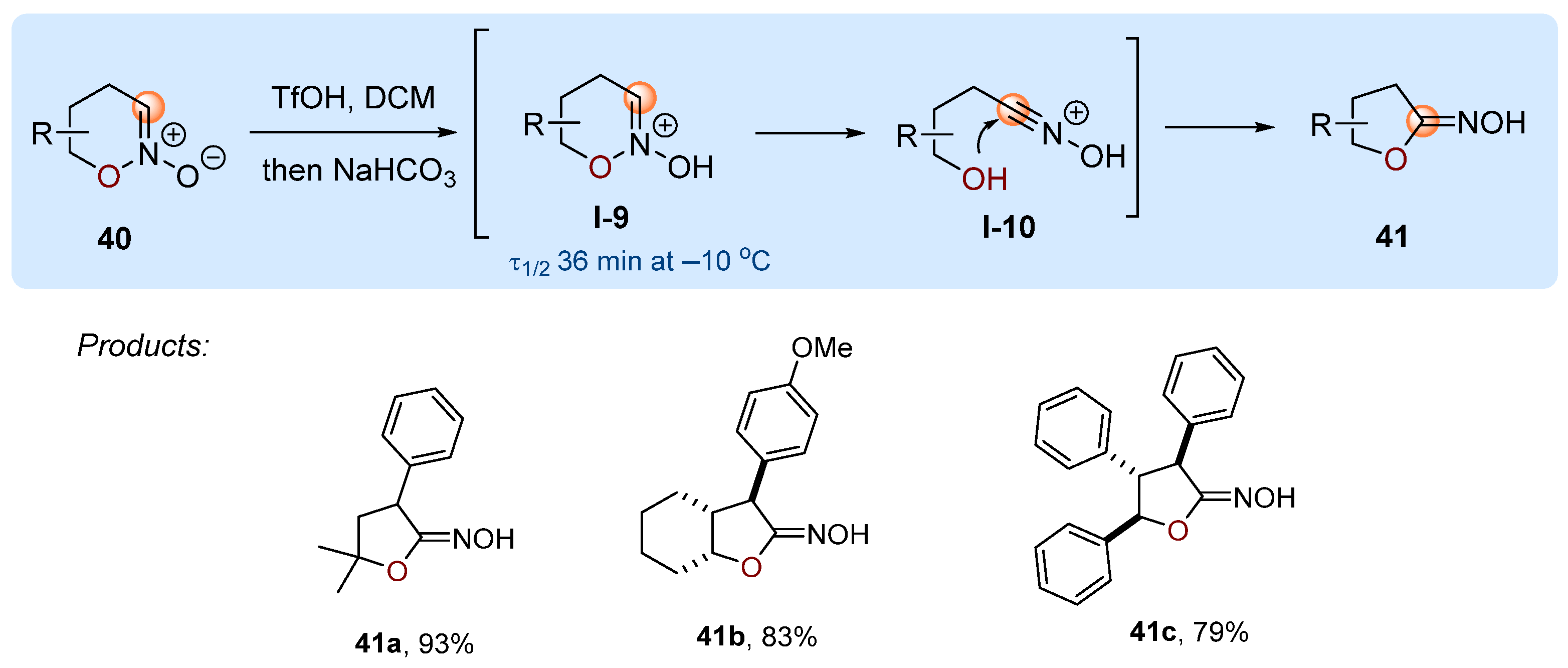
3. Interruption of the Nef and Meyer Reactions by O- and S-Nucleophiles
4. Interruption of the Nef and Meyer Reactions by N-Nucleophiles
5. Interruption of the Nef and Meyer Reactions by C-Nucleophiles
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meyer, V.; Wurster, C. Ueber die nitroverbindungen der fettreihe. Sechste mittheilung. Chem. Ber. 1873, 6, 1168–1172. [Google Scholar] [CrossRef] [Green Version]
- Konovalov, M. Nitrating action of nitric acid on saturated hydrocarbons. J. Russ. Phys. Chem. Soc. 1893, 25, 509. [Google Scholar]
- Nef, J.U. Ueber die constitution der salze der nitroparaffine. Liebigs Ann. Chem. 1894, 280, 263–291. [Google Scholar] [CrossRef]
- Bamberger, E.; Rust, E. Transformation of nitroparaffins. Chem. Ber. 1902, 35, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Nametkin, S.S. Structure of isonitro compounds. J. Russ. Phys. Chem. Soc. 1914, 45, 1414–1420. [Google Scholar]
- Sowden, J.C. The nitromethane and 2-nitroethanol syntheses. In Advances in Carbohydrate Chemistry; Hudso, C.S., Canto, S.M., Eds.; Academic Press: Cambridge, MA, USA, 1951; Volume 6, pp. 291–318. [Google Scholar]
- Tamelen, E.E.V.; Thiede, R.J. The synthetic application and mechanism of the Nef reaction. J. Am. Chem. Soc. 1952, 74, 2615–2618. [Google Scholar] [CrossRef]
- Noland, W.E. The Nef reaction. Chem. Rev. 1955, 55, 137–155. [Google Scholar] [CrossRef]
- Hawthorne, M.F. aci-Nitroalkanes. II. The mechanism of the Nef reaction. J. Am. Chem. Soc. 1957, 79, 2510–2515. [Google Scholar] [CrossRef]
- Kornblum, N.; Brown, R.A. The action of acids on nitronic esters and nitroparaffin salts. Concerning the mechanisms of the Nef and the hydroxamic acid forming reactions of nitroparaffins. J. Am. Chem. Soc. 1965, 87, 1742–1747. [Google Scholar] [CrossRef]
- Sun, S.; Folliard, J. The participation of water in the Nef reaction of aci-nitro compounds. Tetrahedron 1971, 27, 323–330. [Google Scholar] [CrossRef]
- Edward, J.T.; Tremaine, P.H. The Meyer reaction of phenylnitromethane in acid. III. The tautomerization to the aci-Form. Can. J. Chem. 1971, 49, 3493–3501. [Google Scholar] [CrossRef] [Green Version]
- McMurry, J.E.; Melton, J. New method for the conversion of nitro groups into carbonyls. J. Org. Chem. 1973, 38, 4367–4373. [Google Scholar] [CrossRef]
- Seebach, D.; Colvin, E.V.; Lehr, F.; Weller, T. Nitroaliphatic compounds-ideal intermediates in organic synthesis. Chimia 1979, 33, 1. [Google Scholar]
- Kornblum, N.; Erickson, A.S.; Kelly, W.J.; Henggeler, B. Conversion of nitro paraffins into aldehydes and ketones. J. Org. Chem. 1982, 47, 4534–4538. [Google Scholar] [CrossRef]
- Barton, D.H.; Motherwell, W.B.; Zard, S.Z. A mild oxidative Nef reaction. Tetrahedron Lett. 1983, 24, 5227–5230. [Google Scholar] [CrossRef]
- Petruš, L.; Petrušová, M.; Pham-Huu, D.-P.; Lattová, E.; Pribulová, B.; Turjan, J. Timely Research Perspectives in Carbohydrate Chemistry; Schmid, W., Stütz, A.E., Eds.; Springer: Vienna, Austria, 2002; pp. 33–42. [Google Scholar]
- Ballini, R.; Petrini, M. Recent synthetic developments in the nitro to carbonyl conversion (Nef reaction). Tetrahedron 2004, 60, 1017–1047. [Google Scholar] [CrossRef]
- Pinnick, H.W. The Nef reaction. In Organic Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2004; Volume 38, pp. 655–792. [Google Scholar]
- Ballini, R.; Petrini, M.; Rosini, G. Nitroalkanes as central reagents in the synthesis of spiroketals. Molecules 2008, 13, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durchschein, K.; Ferreira-da Silva, B.; Wallner, S.; Macheroux, P.; Kroutil, W.; Glueck, S.M.; Faber, K. The flavoprotein-catalyzed reduction of aliphatic nitro-compounds represents a biocatalytic equivalent to the Nef-reaction. Green Chem. 2010, 12, 616–619. [Google Scholar] [CrossRef]
- Bhat, C.; Tilve, S.G. Synthesis of (−)-hygrine, (−)-norhygrine, (−)-pseudohygroline and (−)-hygroline via Nef reaction. Tetrahedron Lett. 2011, 52, 6566–6568. [Google Scholar] [CrossRef]
- Zard, S.Z. Some aspects of the chemistry of nitro compounds. Helv. Chim. Acta 2012, 95, 1730–1757. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. The nitro to carbonyl conversion (Nef reaction): New perspectives for a classical transformation. Adv. Synth. Catal. 2015, 357, 2371–2402. [Google Scholar] [CrossRef]
- Kawamoto, Y.; Ozone, D.; Kobayashi, T.; Ito, H. Total synthesis of (±)-chondrosterin I using a desymmetric aldol reaction. Org. Biomol. Chem. 2018, 16, 8477–8480. [Google Scholar] [CrossRef]
- Lupidi, G.; Palmieri, A.; Petrini, M. Synthesis of nitro alcohols by riboflavin promoted tandem Nef-Henry reactions on nitroalkanes. Adv. Synth. Catal. 2021, 363, 742–746. [Google Scholar] [CrossRef]
- Tran, V.-P.; Matsumoto, N.; Nalaoh, P.; Jing, H.; Chen, C.-Y.; Lindsey, J.S. Dihydrooxazine byproduct of a McMurry-Melton reaction en route to a synthetic bacteriochlorin. Organics 2022, 3, 262–274. [Google Scholar] [CrossRef]
- Qu, Z.-W.; Zhu, H.; Grimme, S. Mechanistic insights for nitromethane activation into reactive nitrogenating reagents. ChemCatChem 2021, 13, 2132–2137. [Google Scholar] [CrossRef]
- Smirnov, V.O.; Khomutova, Y.A.; Tartakovsky, V.A.; Ioffe, S.L. C–C coupling of acyclic nitronates with silyl ketene acetals under silyl triflate catalysis: Reactivity umpolung of aliphatic nitro compounds. Eur. J. Org. Chem. 2012, 2012, 3377–3384. [Google Scholar] [CrossRef]
- Tabolin, A.A.; Sukhorukov, A.Y.; Ioffe, S.L.; Dilman, A.D. Recent advances in the synthesis and chemistry of nitronates. Synthesis 2017, 49, 3255–3268. [Google Scholar] [CrossRef]
- Das, S.; Mitschke, B.; De, C.K.; Harden, I.; Bistoni, G.; List, B. Harnessing the ambiphilicity of silyl nitronates in a catalytic asymmetric approach to aliphatic β3-amino acids. Nat. Catal. 2021, 4, 1043–1049. [Google Scholar] [CrossRef]
- Trudel, V.; Tien, C.-H.; Trofimova, A.; Yudin, A.K. Interrupted reactions in chemical synthesis. Nat. Rev. Chem. 2021, 5, 604–623. [Google Scholar] [CrossRef]
- Tabolin, A.A.; Sukhorukov, A.Y.; Ioffe, S.L. α-Electrophilic reactivity of nitronates. Chem. Rec. 2018, 18, 1489–1500. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Aksenov, A.V.; Ovcharov, S.N.; Aksenov, D.A.; Rubin, M. Electrophilically activated nitroalkanes in reactions with carbon based nucleophiles. Front. Chem. 2020, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioffe, S.L. Nitronates. In Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; Feuer, H., Ed.; Wiley: Hoboken, NJ, USA, 2007; p. 625. [Google Scholar]
- Laali, K.K. Nitro and nitroso transformations in superacids. Coord. Chem. Rev. 2000, 210, 47–71. [Google Scholar] [CrossRef]
- Steinkopf, W.; Jürgens, B. Zur kenntnis aliphatischer nitrokörper. XII. Mitteilung: Über die konstitution der aci-nitrokörper. J. Prakt. Chem. 1911, 84, 686–713. [Google Scholar] [CrossRef]
- Gil, V.; MacLeod, A. Synthesis of glucosinolates. Tetrahedron 1980, 36, 779–783. [Google Scholar] [CrossRef]
- Zhang, Q.; Lebl, T.; Kulczynska, A.; Botting, N.P. The synthesis of novel hexa-13C-labelled glucosinolates from [13C6]-D-glucose. Tetrahedron 2009, 65, 4871–4876. [Google Scholar] [CrossRef]
- Nenitzescu, C.D.; Isacescu, D.A. aci-Nitro compounds. IV. The mechanism of the conversion of the nitro derivative into hydroxamic acid. Bull. Soc. Chim. Rom. 1932, 14, 53–61. [Google Scholar]
- Arndt, F.; Rose, J.D. Relations between acidity and tautomerism. Part III. The nitro-group and the nitronic esters. J. Chem. Soc. 1935, 1–10. [Google Scholar] [CrossRef]
- Benn, M.; Ettlinger, M. The synthesis of sinigrin. Chem. Commun. 1965, 445–447. [Google Scholar] [CrossRef]
- Abramski, W.; Chmielewski, M. Practical synthesis of sinigrin. J. Carbohydr. Chem. 1996, 15, 109–113. [Google Scholar] [CrossRef]
- Hurd, C.D.; Nilson, M.E. Aliphatic nitro ketones. J. Org. Chem. 1955, 20, 927–936. [Google Scholar] [CrossRef]
- Yao, C.-F.; Kao, K.-H.; Liu, J.-T.; Chu, C.-M.; Wang, Y.; Chen, W.-C.; Lin, Y.-M.; Lin, W.-W.; Yan, M.-C.; Liu, J.-Y.; et al. Generation of nitroalkanes, hydroximoyl halides and nitrile oxides from the reactions of β-nitrostyrenes with Grignard or organolithium reagents. Tetrahedron 1998, 54, 791–822. [Google Scholar] [CrossRef]
- Kao, K.-H.; Yang, C.-S.; Liu, J.-T.; Lin, W.-W.; Fang, H.-Y.; Yao, C.-F.; Chen, K. One-pot synthesis of the hydroximoyl chlorides and [3.3.0] bicyclic compounds from the reactions of β-nitrostyrenes with stabilized nucleophiles. Tetrahedron 1998, 54, 13997–14014. [Google Scholar] [CrossRef]
- Fusco, R.; Rossi, S. β-Nitroketones. Chem. Ind. 1957, 1650. [Google Scholar]
- Carr, J.B.; Durham, H.G.; Hass, D.K. Isoxazole anthelmintics. J. Med. Chem. 1977, 20, 934–939. [Google Scholar] [CrossRef]
- Malykhin, R.S.; Boyko, Y.D.; Nelyubina, Y.V.; Ioffe, S.L.; Sukhorukov, A.Y. Interrupted Nef reaction of cyclic nitronates: Diastereoselective access to densely substituted α-chloronitroso compounds. J. Org. Chem. 2022, 87, 16617–16631. [Google Scholar] [CrossRef]
- Adams, M.A.; Duggan, A.J.; Smolanoff, J.; Meinwald, J. Total synthesis of (±)-pederamide. J. Am. Chem. Soc. 1979, 101, 5364–5370. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G.; Uselli, A. A simple, efficient, two-step synthesis of symmetric 2,7-dialkyl-1,6-dioxaspiro[4.4]nonanes. J. Heterocycl. Chem. 1994, 31, 259–260. [Google Scholar] [CrossRef]
- Occhiato, E.G.; Guarna, A.; De Sarlo, F.; Scarpi, D. Baker’s yeast reduction of prochiral γ-nitroketones. II. straightforward enantioselective synthesis of 2,7-dimethyl-1,6-dioxaspiro[4.4]nonanes. Tetrahedron Asymmetry 1995, 6, 2971–2976. [Google Scholar] [CrossRef]
- Occhiato, E.G.; Scarpi, D.; Menchi, G.; Guarna, A. Synthesis of enantiopure 2,7-diaryl-1,6-dioxaspiro[4.4]nonanes via enantioselective reduction of prochiral γ-nitroketones by diisopinocampheylchloroborane (DIP-C1™). Tetrahedron Asymmetry 1996, 7, 1929–1942. [Google Scholar] [CrossRef]
- Rosinidy, G.; Ballini, R.; Marotta, E. Functionalized nitroalkanes in synthesis of 1,6-dioxaspiro[4.5]decane components of paravespula vulgaris pheromone. Tetrahedron 1989, 45, 5935–5942. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M.; Rosini, G. New and efficient synthesis of ω-nitroalcohols and spiroketals by chemio- and regioselective reductive cleavage of 2-nitrocycloalkanones. Tetrahedron 1990, 46, 7531–7538. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. Hydroxy-functionalized conjugated nitroolefins as immediate precursors of spiroketals. A new synthesis of 1,7-dioxaspiro[5.5]undecane and (E)-2-methyl-1,7-dioxaspiro[5.6]dodecane. J. Chem. Soc. Perkin Trans. 1 1992, 3159–3160. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G.; Schaafstra, R. Nitro ketones in organic synthesis: A new, short synthesis of racemic trans-2-methyl-1,7-dioxaspiro[5.5]undecane, trans, trans- and trans, cis-2,8-dimethyl-1,7-dioxaspiro[5.5]undecane by Henry reaction. Liebigs Ann. Chem. 1994, 1994, 1235–1237. [Google Scholar] [CrossRef]
- Huang, W.-L.; Raja, A.; Hong, B.-C.; Lee, G.-H. Organocatalytic enantioselective Michael–acetalization–reduction–Nef reaction for a one-pot entry to the functionalized aflatoxin system. Total synthesis of (−)-dihydroaflatoxin D2 and (−)- and (+)-microminutinin. Org. Lett. 2017, 19, 3494–3497. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-Y.; Raja, A.; Hong, B.-C.; Kotame, P.; Chang, W.-C.; Lee, G.-H. Organocatalytic enantioselective Michael–acetalization–Henry reaction cascade of 2-hydroxynitrostyrene and 5-oxohexanal for the entry to the hexahydro-6H-benzo[c]chromenones with four consecutive stereogenic centers and an approach to aflatoxin analogues. J. Org. Chem. 2017, 82, 12840–12848. [Google Scholar] [CrossRef]
- Wilson, H.; Lewis, E.S. Neighboring group participation in proton transfers. J. Am. Chem. Soc. 1972, 94, 2283–2285. [Google Scholar] [CrossRef]
- Krawczyk, H.; Wolf, W.M.; Śliwiński, M. Nitroalkanes as nucleophiles in a self-catalytic Michael reaction. J. Chem. Soc. Perkin Trans. 1 2002, 2794–2798. [Google Scholar] [CrossRef]
- Keumi, T.; Morita, T.; Mitzui, T.; Jōka, T.; Kitajima, H. A convenient synthesis of polysubstituted phthalic acid derivatives via side-chain nitration of polymethylbenzoic acids. Synthesis 1985, 1985, 223–224. [Google Scholar] [CrossRef]
- Krawczyk, H.; Albrecht, Ł.; Wojciechowski, J.; Wolf, W.M. Spontaneous Nef reaction of 3-aryl-2-(diethoxyphosphoryl)-4-nitroalkanoic acids. Tetrahedron 2006, 62, 9135–9145. [Google Scholar] [CrossRef]
- Weber, M.; Frey, W.; Peters, R. Asymmetric palladium(II)-catalyzed cascade reaction giving quaternary amino succinimides by 1,4-addition and a Nef-type reaction. Angew. Chem. Int. Ed. 2013, 52, 13223–13227. [Google Scholar] [CrossRef]
- Mendler, B.; Kazmaier, U.; Huch, V.; Veith, M. Straightforward approach to iminoxazines and azetidinimines via 1,4-additions of chelated enolates toward nitroalkenes. Org. Lett. 2005, 7, 2643–2646. [Google Scholar] [CrossRef] [PubMed]
- Ansell, G.B.; Moore, D.W.; Nielsen, A.T. Formation and crystal structure of 3-(4-bromophenyl)-2-hydroxyimino-6,7-dihydro-4 (5H)-benzofuranone. Michael addition of cyclohexane-1,3-dione to 4-bromo-ω-nitrostyrene. Chem. Commun. 1970, 23, 1602–1603. [Google Scholar] [CrossRef]
- Ansell, G.B.; Moore, D.W.; Nielsen, A.T. Intramolecular reactions of nitro-olefin–cyclohexane-1,3-dione Michael adducts. Crystal structure of 3-(4-bromophenyl)-6,7-dihydro-2-hydroxyiminobenzofuran-4(5H)-one. J. Chem. Soc. B: Phys. Org. 1971, 2376–2382. [Google Scholar] [CrossRef]
- Hrnčiar, P.; Čulák, I. Michael addition of 1,3-cyclopentanedione, 1,3-cyclohexanedione and 1,3-cycloheptanedione to 1-(X-phenyl)-2-nitroethylenes. Collect. Czech. Chem. Commun. 1984, 49, 1421–1431. [Google Scholar] [CrossRef]
- Nielsen, A.T.; Archibald, T.G. Intramolecular reactions of nitroolefin-β-diketone Michael adducts: Formation of 3-oxo-2,3-dihydro-4H-1,2-benzoxazine and 4(5H)-benzofuranone derivatives. Tetrahedron 1969, 25, 2393–2400. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Hassanin, H.M.; Gabr, Y.A.; Alnamer, Y.A. Studies on the chemical behavior of 3-(nitroacetyl)-1-ethyl-4-hydroxyquinolin-2(1H)-one towards some electrophilic and nucleophilic reagents. J. Braz. Chem. Soc. 2012, 23, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Lai, X.; Zha, G.; Xu, Y.; Sun, P.; Xia, T.; Shen, Y. The squaramide-catalyzed asymmetric Michael/cyclization tandem reaction for the synthesis of chiral trifluoromethylated hydroxyimino tetrahydrobenzofuranones. Org. Biomol. Chem. 2016, 14, 3603–3607. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Li, K.; Wang, N.; He, T.; Yu, X.-Q. A Novel Catalyst-Free Tandem Reaction for the Synthesis of 5-Hydroxy-1,5-dihydro-2H-pyrrol-2-ones in Water Medium. Synthesis 2011, 2011, 1831–1839. [Google Scholar] [CrossRef]
- Smirnov, V.O.; Khomutova, Y.A.; Ioffe, S.L. Six-membered cyclic nitronates as Brönsted bases: Protonation and rearrangement into butyrolactone oximes. Mendeleev Commun. 2008, 5, 255–257. [Google Scholar] [CrossRef]
- Kiprianov, A.I.; Verbovskaya, T.M. Condensation of ethyl nitroacetate with o-aminophenyl mercaptan. Zh. Obshch. Khim. 1961, 31, 531–537. [Google Scholar]
- Kiprianov, A.I.; Verbovskaya, T.M. Condensation of nitroacetone, nitroacetophenone and nitroacetonitrile with o-aminophenyl mercaptan. Zh. Obshch. Khim. 1962, 32, 3703–3707. [Google Scholar]
- Dighe, S.U.; Mukhopadhyay, S.; Priyanka, K.; Batra, S. Metal-free oxidative nitration of α-carbon of carbonyls leads to one-pot synthesis of thiohydroximic acids from acetophenones. Org. Lett. 2016, 18, 4190–4193. [Google Scholar] [CrossRef]
- Bachman, G.B.; Goldmacher, J.E. Conversion of carboxylic acids to amines and their derivatives. J. Org. Chem. 1964, 29, 2576–2579. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Smirnov, A.N.; Aksenov, N.A.; Bijieva, A.S.; Aksenova, I.V.; Rubin, M. Benzimidazoles and benzoxazoles via the nucleophilic addition of anilines to nitroalkanes. Org. Biomol. Chem. 2015, 13, 4289–4295. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aksenov, N.A.; Kirilov, N.K.; Skomorokhov, A.A.; Aksenov, D.A.; Kurenkov, I.A.; Sorokina, E.A.; Nobi, M.A.; Rubin, M. Does electrophilic activation of nitroalkanes in polyphosphoric acid involve formation of nitrile oxides? RSC Adv. 2021, 11, 35937–35945. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Rubio, O.; Awartani, R. A novel preparation of the 1,3-benzoxazepine ring system. Heterocycles 1984, 22, 1155–1159. [Google Scholar] [CrossRef]
- Hirao, S.; Kobiro, K.; Sawayama, J.; Saigo, K.; Nishiwaki, N. Ring construction via pseudo-intramolecular hydrazonation using bifunctional δ-keto nitrile. Tetrahedron Lett. 2012, 53, 82–85. [Google Scholar] [CrossRef]
- Dauzonne, D.; Demerseman, P.; Royer, R. Sur la reactionentre le chlorure de pyridinium et les nitroalcanes primaires. Bull. Soc. Chim. Fr. 1980, 11–12, 601–608. [Google Scholar]
- Berrier, C.; Brahmi, R.; Carreyre, H.; Coustard, J.M.; Jacquesy, J.C. Nitronate anions as precursors of hydroxynitrilium ion equivalents in electrophilic aromatic substitution—A novel route to oximes. Tetrahedron Lett. 1989, 30, 5763–5766. [Google Scholar] [CrossRef]
- Ohwada, T.; Yamagata, N.; Shudo, K. Friedel-Crafts-type reactions involving di- and tricationic species. Onium-allyl dications and O,O-diprotonated aci-nitro species bearing a protonated carbonyl group. J. Am. Chem. Soc. 1991, 113, 1364–1373. [Google Scholar] [CrossRef]
- Coustard, J.-M.; Jacquesy, J.-C.; Violeau, B. Direct carbohydroximoylation of aromatics with primary nitroalkanes in triflic acid (TFSA). Tetrahedron Lett. 1992, 33, 8085–8086. [Google Scholar] [CrossRef]
- Coustard, J.-M.; Jacquesy, J.C.; Violeau, B. Hydroxynitrilium ions as intermediates in the reaction of nitroderivatives with aromatics. Tetrahedron Lett. 1991, 32, 3075–3078. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aksenov, N.A.; Nadein, O.N.; Aksenova, I. V, Nitromethane in polyphosphoric acid—A new reagent for carboxyamidation and carboxylation of activated aromatic compounds. Synth. Commun. 2012, 42, 541–547. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aksenov, N.A.; Nadein, O.N.; Aksenova, I.V. Nitroethane in polyphosphoric acid: A new reagent for acetamidation and amination of aromatic compounds. Synlett 2010, 2010, 2628–2630. [Google Scholar] [CrossRef]
- Nakamura, S.; Sugimoto, H.; Ohwada, T. Formation of 4H-1,2-benzoxazines by intramolecular cyclization of nitroalkanes. scope of aromatic oxygen-functionalization reaction involving a nitro oxygen atom and mechanistic insights. J. Am. Chem. Soc. 2007, 129, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kaji, E.; Zen, S. A novel ring transformation of 3, 5-bis(methoxycarbonyl)-4-phenyl-2-isoxazoline-2-oxides into 2-methoxycarbonyl-1-oxido-3H-indole-3-acetates. Chem. Pharm. Bull. 1985, 33, 8–15. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhorukov, A.Y. Interrupted Nef and Meyer Reactions: A Growing Point for Diversity-Oriented Synthesis Based on Nitro Compounds. Molecules 2023, 28, 686. https://doi.org/10.3390/molecules28020686
Sukhorukov AY. Interrupted Nef and Meyer Reactions: A Growing Point for Diversity-Oriented Synthesis Based on Nitro Compounds. Molecules. 2023; 28(2):686. https://doi.org/10.3390/molecules28020686
Chicago/Turabian StyleSukhorukov, Alexey Yu. 2023. "Interrupted Nef and Meyer Reactions: A Growing Point for Diversity-Oriented Synthesis Based on Nitro Compounds" Molecules 28, no. 2: 686. https://doi.org/10.3390/molecules28020686