Abstract
A novel hydrogen bond surrogate-based (HBS) α-helix mimetic was designed by the combination of covalent H-bond replacement and the use of an ether linkage to substitute an amide bond within a short peptide sequence. The new helix template could be placed in position other than the N-terminus of a short peptide, and the CD studies demonstrate that the template adopts stable conformations in aqueous buffer at exceptionally high temperatures.
1. Introduction
The α-helix is the most abundant secondary structural element in proteins and is found frequently at the interfaces of protein–protein interactions [1]. For example, transcriptional activator p53 contain a short α-helical sequence that mediates function by direct interaction with a receptor [2]. Stable isolated peptides with a defined short α-helical segment would be ideal inhibitors of macromolecular interactions. However, those with less than ~15 amino acid residues rarely adopt a defined conformation in isolation [3,4,5,6], and often lack the ability to fold into their bioactive conformation due to an entropic penalty for folding. New techniques for stabilizing short peptide helices may aid the design of inhibitors or mimics of protein function [7,8,9,10,11,12]. The head-to-backbone cross-linking strategy for helix stabilization includes salt bridges, metal chelates, and covalent cyclization methods such as disulfide and lactam bridges [13,14], hydrocarbon stapling [15,16], and hydrogen-bond surrogate (HBS) methods [17,18,19]. P53 can act as a tumor suppressor and induce cancer cell death, and the levels of p53 can be increased by blocking the p53–MDM2 interaction and reactivate the p53 function [20]. Thus, it is a promising therapeutic strategy for the treatment of cancers by designing and developing small-molecule inhibitors of the MDM2–p53 interaction [21,22]. We discovered that the CUL4A-DDB1-ROC1-L2DTL-PCNA ubiquitin E3 ligase complex interacts with MDM2 and p53 and regulates p53 polyubiquitination and proteolysis through MDM2. We also found that both p53 and MDM2 bind to PCNA directly through a conserved PIP box in p53 and MDM2 [23]. Based on these findings and our experience obtained from medicinal chemistry [24] and total synthesis of marine natural products [25,26,27,28,29,30,31,32], we initiated a chemical biology program aiming to develop inhibitors to block p53 degradation based on the PCNA binding motif, a helix-containing PIP-box of PCNA interaction proteins.
Strategically placed covalent linkages of the type C = X − Y − N to replace the weak (i, i + 4) hydrogen bond, hydrogen-bond surrogates (HBS), have been shown to stabilize the helical conformations in short peptide sequences [17,18,19]. Modifications of the peptide backbone by the replacement of the hydrogen bond with hydrazine [17], carbon–carbon links [33], and thioether linkage [34] have been reported. This strategy provides peptides with increased target affinity and allows a stabilization of the α-helical conformation [9]. However, this approach seems limited to the N-terminal position of a short peptide, and could not be extended to our targeted system. In order to introduce the covalent H-bond replacement at an internal helical turn, we strategically employed an ether linkage to substitute an amide bond as well as replaced the corresponding (i, i + 4) hydrogen bond with a covalent ethylane bridge to afford a novel hydrogen bond surrogate-based (HBS) α-helix 1 (see Scheme 1). Notably, the new helix template (1) could be placed in position other than N-terminus of a short peptide. In connection with the previously mentioned chemical biology program, we selected the helix mimetic template 1 as a model system to explore the ability of the newly designed cyclopeptide to promote an α-helical conformation in aqueous solution.
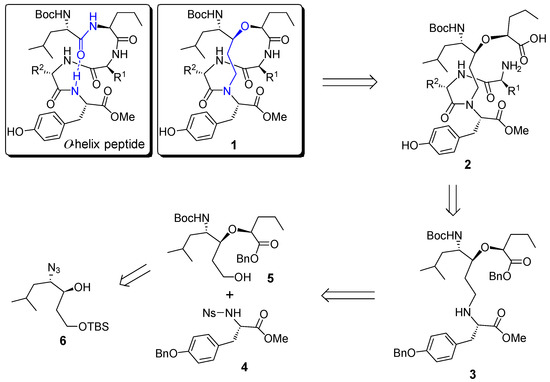
Scheme 1.
Structure of α-helix peptide and retrosynthetic analysis of helix template 1.
The retrosynthetic analysis of the designed helix template (1) was shown in Scheme 1. The target molecule 1 could be obtained from a macrocyclization of the corresponding linear precursor 2, which should be readily available from a coupling reaction of the advanced intermediate 3 with a suitable dipeptide. Further disconnection of 3 gave rise to the protected tyrosine derivative 4 and alcohol 5, which is readily available from β-hydroxy azide 6.
2. Results and Discussion
2.1. Synthesis of Helix Mimetic Template
The synthesis commenced with the substrate controlled allylation of the known α-homochiral aldehyde 7 [35] (Scheme 2). Thus, the treatment of aldehyde 7 with allyltributyltin in the presence of SnCl4 at −78 °C afforded the syn- (8) and anti-homoallylic alcohols in a diastereomeric ratio of 9:1 [36,37]. Homoallylic alcohol 8 was converted into acetonide 9 in 76% overall yield via a three-step sequence including (i) oxazolidine formation with 2,2-dimethoxypropane/PTSA, (ii) the dihydroxylation of the terminal alkene with osmium tetroxide and 4-methylmorpholine N-oxide as co-oxidant, followed by sodium periodate cleavage of the diol, resulted in the corresponding aldehyde, and (iii) reduction in the resulting aldehyde with NaBH4 in methanol. The simultaneous removal of N-Boc and isopropylidene protective groups of 9 was realized under acidic conditions (6 M HCl in THF) to afford the corresponding amino diol, which was then converted into azide diol 10 in 74% yield via a diazo-transfer reaction with triflyl azide. The regioselective protection of the primary alcohol as its TBS ether afforded the key intermediate 6 in 87% yield.
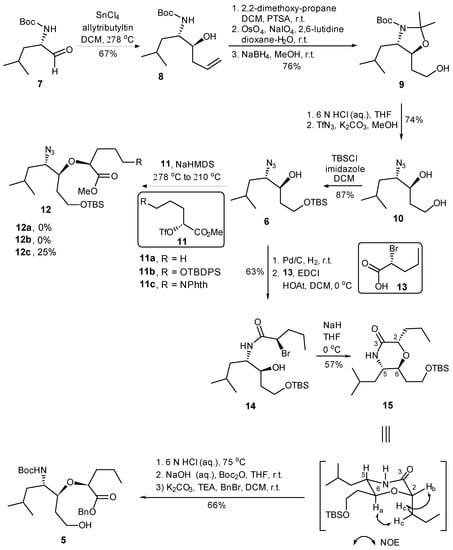
Scheme 2.
Synthesis of fragment 5.
We then addressed the formation of ether linkage of 12 so as to generate the required precursor 5 for further coupling reaction. Considering the nature of chiral triflate, also supported by literature precedents [38], we anticipated that the displacement of the triflate 11 by alkoxy anion derived from 6 could be achieved. Unfortunately, the attempted synthesis of the requisite ether 12 via the displacement reaction proved to be frustrating. All attempts to convert 6 to 12 by the reaction of the anion derived from the former with triflate 11(a–c) [38], under a variety of reaction conditions provided no product or resulted in a much lower conversion (Scheme 2). In order to avoid more side-reaction derived from the slow intermolecular displacement process, we elected to construct the ether linkage via an intramolecular SN2 reaction, which would also avoid any ambiguity of the stereochemistry. Thus, the hydrogenation of the azide alcohol 6 over Pd/C gave the corresponding amine, which was condensed with (R)-2-bromopentanoic acid (13) employing EDCI to provide amide 14 in 63% yield over two steps. To our delight, upon the treatment of 14 with sodium hydride in THF, the intramolecular SN2 reaction proceeded smoothly to furnish the desired morphorlinone 15 as a single diastereomer in 57% yield. The inversion of the C-2 stereogenic center of 13 leading to the structure of 15 with the all-S stereochemical configurations was ascertained by NMR correlation and NOESY experiments. Morphorlinone 15 was then converted into the key intermediate 5 in 66% overall yield by a three-step sequence of straightforward transformations: (i) acid cleavage of TBS ether and the hydrolysis of amide to the corresponding amino acid; (ii) protection of the resultant amino group as its Boc carbamate; and (iii) protection of the carboxyl functionality as its benzyl ester.
With alcohol 5 at hand, attention was turned to the installation of the tyrosine units via the Fukuyama–Mitsunobu protocol and later-stage macrocyclization (Scheme 3). Thus, the treatment of sulfonamide-protected amine 4 [39,40,41] and alcohol 5 in the presence of diethylazodicarboxylate and triphenylphosphine afforded the corresponding alkylation adduct 16 in 78% yield. The nosyl group (Ns-) of 16 was then cleaved with Fukuyama’s thiophenol/K2CO3/CH3CN conditions [42], giving rise to the corresponding free secondary amine 3 in 85% yield. It is known that the coupling of a secondary amine and carboxylic acid using standard peptide coupling techniques is often a low-yielding process with certain difficulties. Gratifyingly, the HATU/HOAt-promoted coupling reaction between secondary amine 3 and dipeptide acid 17a provided 2a in 85% yield. The simultaneous removal of the benzyl ester, O-Bn ether and Cbz-protecting group was achieved by the hydrogenolysis of 2a with Pd(OH)2 on carbon to produce the desired amino acid which was immediately activated by HATU/HOAT in the presence of N-methylmorpholine to afford cyclodepsipeptide 1a in 62% yield. The steric hinderance of amine 3 significantly deterioate the reaction rate and yield of this peptide coupling step. Among all the tested coupling reagents such as PyAOP, DEPBT. HATU, BOPCl etc., HATU/HOAT condition was found with best results. The removal of the Boc- protecting group of 1a with trifluoroacetic acid in CH2Cl2 produced the more hydrophilic template 18 in 78% yield (See Supplementary Materials).
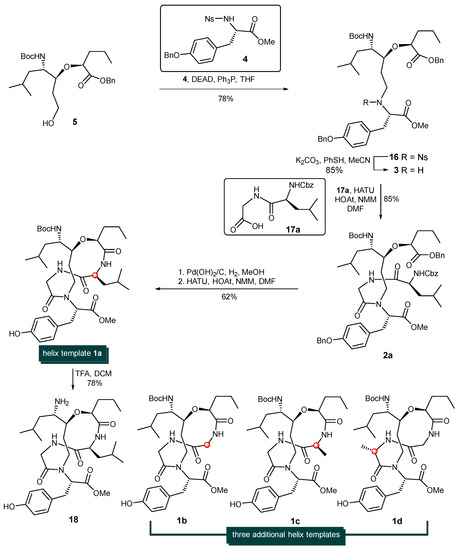
Scheme 3.
Synthesis of helix templates 1a, 1b, 1c, 1d.
By employing three additional dipeptides (Cbz-Gly-Gly-OH (17b), Cbz-L-Ala-Gly-OH (17c), and Cbz-Gly-L-Ala-OH (17d)) as coupling partners, helix-templates 1b, 1c, and 1d were constructed via the identical procedure for the synthesis of 1a. The structures of 1b, 1c, and 1d are shown in Scheme 3.
2.2. CD Spectra of Helix Templates
The solution conformation of the helix-template 1a–d was investigated by circular dichroism spectroscopy (Figure 1). All samples were measured at 10 mM concentration in pH 7.4 phosphate buffer. The CD spectra obtained for 1a and 1c were typical for the α-helical structure, showing the characteristic two molar ellipticity minima (λ = 222, 208 nm) and an ellipticity maximum (λ = 195 nm) [43,44]. However, the CD spectra of 1b and 1d showed almost no α-helical secondary structure (Figure 1, left panel). The loss of helicity in 1b and 1d relative to 1a and 1c indicated the stereogenic center and the substituent R1 of 1 (Scheme 1) are vital for the overall stability of helical conformation. Similar to 1a, the CD spectra of 18 also showed two negative peaks at 208 and 222 nm wave trough and a positive value at 195 nm, which almost did not change spectral intensities by adding 2,2,2-trifluoroethanol (Figure 1, right panel). Previous thermal stability studies of HBS helices have shown that the conformations of these peptides remain remarkably consistent at high temperatures [19,33,45]. The thermal stabilities of 18 were investigated by monitoring the temperature-dependent change in the intensity of the 220 nm bands in the CD spectra (Figure 2). To our delight, we observed that the spectral lineshapes or intensities almost did not change along with a gradual increase in the temperature, even at 95 °C. Overall, the CD studies demonstrated that the template 18 adopts stable conformations.
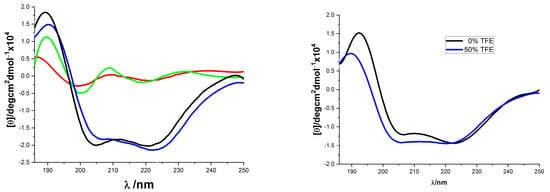
Figure 1.
CD spectra of compounds 1 and 18. ((Left) panel: 1a (blue), 1b (red), 1c (black), and 1d (green), all were recorded in 10 mM sodium phosphate buffer (pH 7.4); (Right) panel: 18 in 10 mM phosphate buffer pH 7.4 (black) and in 50% TFE (blue)).
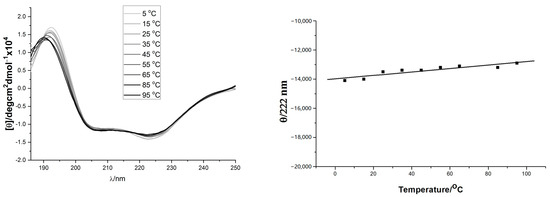
Figure 2.
(Left) panel: Thermo-stability of 18 measured by CD spectra (from slim to thick line with 10 °C interval increase); (Right) panel: Temperature dependence of 18’s mean peptide ellipticity at 222 nm (by linear fit).
3. Materials and Methods
3.1. General Experimental Details
All non-aqueous reactions were performed under an atmosphere of nitrogen or argon using oven-dried (120 °C) or flame-dried glassware under a N2 atmosphere. Commercially available reagents were used without further purification. All solvents were distilled prior to use: tetrahydrofuran (THF) from Na/benzophenone, dichloromethane (DCM), triethylamine (TEA) and dimethylformamide (DMF) were distilled from CaH2. Methanol (MeOH) was distilled under a N2 atmosphere from Mg/I2. 1H NMR and 13C NMR spectra were recorded in CDCl3 or MeOH-d4 on a Bruker Avance AV500 or Bruker Avance AV400 at 500 MHz (125 MHz) or 400 MHz (100 MHz), respectively. Chemical shifts are reported as δ values (ppm) referenced to either a tetramethylsilane (TMS) internal standard or the signals due to the solvent residual. Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, br = broad, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant (Hz), integration. Mass spectra were measured on ABI Q-star Elite. Optical rotations were measured on a Perkin-Elmer 351 polarimeter at 589 nm with a 100 mm path length cell at 20 °C (reported as follows: concentration (c (in 1 g/100 mL), solvent). The reaction progress was checked on TLC plates. TLC was carried out using pre-coated sheets (Qingdao silica gel 60-F250, 0.2 mm) which, after development, were visualized under UV light at 254 nm, and/or staining in p-anisole, ninhydrin or phosphomolybdic acid solution followed by heating. Flash column chromatography was performed using the indicated solvents on Qingdao silica gel 60 (230–400 mesh ASTM). Yields refer to chromatographically purified compounds, unless otherwise stated.
CD spectroscopy studies: CD spectra were recorded on an Applied Photophysics Chirascan CD spectrometer equipped with a temperature controller using 0.1 cm path length cells. Experiments were performed at 20 °C using a 0.1 cm cell, at a scan speed of 100 nm/min from 250 to 186 nm. Spectral baselines were obtained under analogous conditions as that for the samples. All spectra were baseline subtracted, converted to a uniform scale of molar ellipticity, and smoothed using OriginPro 8.0 software. The samples were dissolved in 10 mM phosphate buffer, measured as pH 7.4. The raw CD data of 1a–1d and 18 were recorded in raw ellipticity units. The concentrations of 1a–1d and 18 were determined by quantitative RP-HPLC against a standard of known concentrations. All samples were between 20 and 70 μM, and the CD spectra were recorded in a range of concentrations to confirm that the sample aggregation did not occur. CD data were converted to mean the residue ellipticity, [θ] (deg∙cm2∙dmol−1), using the equation [θ] = θ/(10 × c × l × n), where c is the sample concentration (M) and l is the cell path length (cm), n is number of amino acid residues in the peptides (n = 4).
3.2. Procedures and Analytical Description of Compounds
tert-Butyl ((4S,5S)-5-hydroxy-2-methyloct-7-en-4-yl)carbamate 8: SnCl4 (163 mL, 163 mmol, 1.0 M in CH2Cl2) was dropwise added to a solution of N-Boc-leucinal 7 (17.60 g, 81.6 mmol) in CH2Cl2 (380 mL) at −78 °C, 10 min later, allyltributyltin (35.20 g, 106 mmol) was dropwise added. The reaction mixture was stirred at −78 °C for 2 h and then quenched with saturated aqueous NH4Cl (50 mL). The organic layer was separated and dried with Na2SO4 and concentrated in vacuo. The residue was then dissolved in MeOH (400 mL) at 0 °C, after NaBH4 (5.40 g, 143 mmol) was added in three portions, the resulting mixture was vigorously stirred for 15 min and quenched with a saturated aqueous solution of NaHCO3 (100 mL). The reaction mixture was concentrated, and the residue was extracted with CH2Cl2 (100 mL × 3). The combined organic phase was dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel to give compound 8 (14.10 g, 67%) as a colorless oil. [α]D23 = −33.8 (c 3.7, MeOH). 1H NMR (500 MHz, CDCl3) δ 5.84–5.77 (m, 1H), 5.10–5.06 (m, 2H), 4.80 (d, J = 9.5 Hz, 1H), 3.60–3.54 (m, 2H), 2.69 (brs, 1H), 2.28–2.23 (m, 1H), 2.19–2.13 (m, 1H), 1.65–1.59 (m, 1H), 1.49–1.36 (m, 11H), 0.90–0.86 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ 156.1, 134.7, 117.8, 78.9, 72.8, 51.8, 41.7, 38.9, 28.2, 24.7, 23.0, 22.0 ppm. HRMS (ESI, m/z) calculated for C14H28NO3+ [M+H]+: 258.2064, found 258.2058.
tert-Butyl (4S,5S)-5-(2-hydroxyethyl)-4-isobutyl-2,2-dimethyloxazolidine-3-carbo-xylate 9: To a solution of 8 (5.00 g, 19.4 mmol) and 2,2-dimethoxypropane (3.6 mL, 29.3 mmol) in CH2Cl2 (150 mL) was added TsOH·H2O (85.0 mg, 0.45 mmol) at room temperature. The reaction mixture was stirred for 3 h at room temperature and then quenched with saturated aqueous NaHCO3 (50 mL) and extracted with additional CH2Cl2 (50 mL × 3). The combined organic layer was dried with Na2SO4. The residue was purified by column chromatography on silica gel to give the desired acetonide 8a (5.25 g, 91%) as a colorless oil. [α]D23 = +10.7 (c 2.0, CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.78–5.69 (m, 1H), 5.08–5.04 (m, 2H), 3.82–3.84 (m, H), 3.79–3.55 (m, 1H), 2.36–2.33 (m, 1H), 2.28–2.24 (m, 1H), 1.60–1.35 (m, 18H), 0.88 (s, 3H), 0.87 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 151.7, 134.1, 117.6, 94.0, 80.3, 79.5, 60.0, 43.4, 40.3, 28.4, 25.4, 23.9, 21.2 ppm. HRMS (ESI, mz) calculated for C17H32NO3+ [M+H]+: 298.2377, found 298.2374.
The above acetonide 8a (3.50 g, 11.7 mmol) was dissolved in dioxane–water (100 mL, 3:1), 2,6-lutidine (1.5 mL, 13.0 mmol), OsO4 (2.9 mL, 0.23 mmol, 0.079 M in tert-butanol) and NaIO4 (9.90 g, 46.0 mmol) were sequentially added to the solution at room temperature. The reaction mixture was stirred at room temperature and monitored by TLC. Upon all the starting acetonide being consumed, Na2S2O3 (30 mL, 60.0 mmol, 2 M in water) was added and the mixture was stirred for an additional 3 h to quench the reaction. The reaction mixture was extracted with ethyl acetate (50 mL × 4). The combined organic phase was washed with saturated aqueous NH4Cl (40 mL × 3), and brine (40 mL), dried over Na2SO4 and concentrated in vacuo. The residue was then dissolved in methanol (20 mL) at 0 °C, after NaBH4 (1.32 g, 35.0 mmol) was added, the reaction mixture was stirred at 0 °C for 15 min and then quenched by addition of a saturated aqueous solution of NH4Cl (50 mL). Volatiles were removed in vacuo, and the residue was extracted with ethyl acetate (50 mL × 3). The combined organic phase was washed with water (50 mL), brine (50 mL), dried over Na2SO4 and concentrated in vacuo. The purification of the residue by chromatography yielded 9 (2.97 g, 84%) as a colorless oil. [α]D23 = +4.96 (c 3.1, CHCl3). 1H NMR (500 MHz, CDCl3) δ 4.04–4.00 (m, 1H), 3.79–3.70 (m, 3H), 2.40 (s, 1H), 1.88–1.84 (m, 1H), 1.77–1.74 (m, 1H), 1.53–1.35 (m, 18H), 0.90 (s, 3H), 0.88 (s, 3H) ppm.13C NMR (125 MHz, CDCl3) δ 151.8, 94.0, 79.7, 79.2, 60.9, 60.2, 37.9, 43.5, 42.3, 37.9, 28.4, 25.5, 23.9, 21.4 ppm. HRMS (ESI, m/z) calculated for C16H32NO4+ [M+H]+: 302.2326, found: 302.2329.
(3S,4S)-4-Azido-6-methylheptane-1,3-diol 10: A solution of HCl (5 mL, 6 M in water) was added to a solution of 9 (4.15 g, 13.7 mmol) in THF (20 mL). The reaction mixture was stirred at room temperature overnight. Volatiles were removed in vacuo, the residue was dissolved in MeOH (40 mL) and cooled to −15 °C. After K2CO3 (7.56 g, 54.8 mmol) and TfN3 (3.80 g, 22.0 mmol in CH2Cl2) were added, the reaction mixture was stirred at −15 °C for 1 h and allowed to warm to room temperature within 45 min. The reaction was then quenched with saturated aqueous NH4Cl (50 mL). Layers were separated, and the aqueous layer was extracted with CH2Cl2 (50 mL × 3). The combined organic layer was washed with brine (50 mL), dried over Na2SO4, and concentrated in vacuo. The residue was subjected to chromatographic purification, yielding compound 10 (1.89 g, 74%, two steps) as colorless oil. [α]D23 = −36.4 (c 1.6, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.93–3.89 (m, 1H), 3.85–3.79 (m, 2H), 3.60 (s, 1H), 3.32 (s, 1H), 3.20 (dt, J = 9.6, 4.1 Hz, 1H), 1.80–1.74 (m, 2H), 1.71–1.67 (m, 1H), 1.55 (ddd, J = 14.3, 10.0, 5.1 Hz, 1H), 1.33 (ddd, J = 13.5, 9.1, 4.1 Hz, 1H), 0.94–0.90 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ 73.3, 65.1, 60.5, 39.3, 35.5, 25.0, 23.1, 21.6 ppm. HRMS (ESI, m/z) calculated for C8H18N3O2+ [M+H]+: 188.1394, found: 188.1497.
(3S,4S)-4-Azido-1-((tert-butyldimethylsilyl)oxy)-6-methylheptan-3-ol 6: tert-Butyl-dimethylsilyl chloride (2.80 g, 15.0 mmol) and imidazole (1.22 g, 18.0 mmol) were added to a solution of 10 (2.1 g, 11.2mmol) in DMF (10 mL) at room temperature. The reaction mixture was then stirred for 16 h before it was quenched by saturated sodium bicarbonate (20 mL) and extracted with ethyl acetate (50 mL × 3). The combined organic layers were washed with water (50 mL) and brine (50 mL), and dried over Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography to give compound 6 (3.93 g, 87%) as a colorless oil. [α]D23 = −6.5 (c 1.7, CHCl3). 1H NMR (500 MHz, CDCl3) δ 3.93–3.89 (m, 1H), 3.85–3.79 (m, 2H), 3.40 (d, J = 3.0 Hz, 1H), 3.18 (dt, J = 9.8, 4.0 Hz, 1H), 1.86–1.79 (m, 2H), 1.68–1.58 (m, 2H), 1.33 (ddd, J = 13.4, 9.0, 4.1 Hz, 1H), 0.96–0.93 (m, 6H), 0.89 (s, 9H), 0.08 (s, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ 74.3, 64.5, 62.2, 39.1, 35.5, 25.8, 25.0, 23.2, 21.7, 18.1, −5.6, −5.6 ppm. HRMS (ESI, m/z) calculated for C8H18N3O2+ [M+H]+: 302.2258, found: 302.2267.
(R)-2-Bromo-N-((3S,4S)-1-((tert-butyldimethylsilyl)oxy)-3-hydroxy-6-methylheptan-4-yl)pentanamide 14: Compound 6 (2.75 g, 9.1 mmol) was dissolved in MeOH (50 mL). After Pd(OH)2/C (0.23 g, 10% wt) was added, the reaction vessel was sealed and changed to a hydrogen atmosphere and stirred for 3 h. The catalyst was filtered off and the filtrate was dried over Na2SO4 and concentrated in vacuo to afford the crude amine, which was directly used in the next step coupling reaction. (2R)-2-Bromo-pentanoic acid 13 (2.7 g, 15.0 mmol) was dissolved in CH2Cl2 (15 mL), and pre-activated by EDCI (2.91 g, 15.1 mmol) and HOAt (2.28 g, 15.0 mmol) at room temperature for 0.5 h. A solution of the above crude amine in CH2Cl2 (20 mL) was added to the reaction mixture at 0 °C. The reaction mixture was then stirred overnight before it was quenched by saturated sodium bicarbonate (20 mL) and extracted with ethyl acetate (60 mL × 2). The combined organic layers were washed with water (20 mL), brine (20 mL), and evaporated in vacuo. The residue was purified by flash chromatography to give compound 14 (2.51 g, 63% two steps) as a colorless oil. [α]D23 = +0.5 (c 2.3, CHCl3). 1H NMR (500 MHz, CDCl3) δ 6.56 (d, J = 9.5 Hz, 1H), 4.28 (dd, J = 8.0, 5.7 Hz, 1H), 3.96–3.78 (m, 4H), 2.13–2.04 (m, 1H), 2.01–1.92 (m, 1H), 1.78–1.69 (m, 1H), 1.62–1.31 (m, 6H), 0.94–0.86 (m, 18H), 0.06 (s, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ 168.8, 74.1, 62.8, 51.8, 51.1, 41.4, 37.7, 35.7, 25.7, 25.7, 24.7, 23.1, 22.1, 20.6, 18.0, 13.2, −5.6 (2C) ppm. HRMS (ESI, m/z) calculated for C19H41BrNO3Si+ [M+H]+: 438.2034, found: 438.2053.
(2S,5S,6S)-6-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-5-isobutyl-2-propylmorpholin-3-one 15: A solution of compound 14 (2.50 g, 5.70 mmol) in THF (10 mL) was dropwise added to a suspension of NaH (410 mg, 6.84 mmol, 60% dispensed in mineral oil) in THF (10 mL) at −20 °C. The reaction mixture was stirred for 1 h at −20 °C, then it was allowed to warm up slowly to 0 °C within 3 h and kept at 0 °C till TLC indicated the complete consumption of compound 14. The reaction was quenched by the addition of a saturated aqueous solution of NH4Cl (20 mL) and extracted with ethyl acetate (50 mL × 3). The combined organic layer was washed with water (15 mL × 2) and brine (15 mL), dried over Na2SO4 and evaporated in vacuo. The residue was purified by flash chromatography to give compound 15 (1.17 g, 57%) as a colorless oil. [α]D23 = −25.5 (c 2.1, CHCl3). 1H NMR (500 MHz, CDCl3) δ 6.50 (s, 1H), 4.15–4.11 (m, 1H), 3.77–3.68 (m, 3H), 3.32–3.26 (m, 1H), 1.83–1.74 (m, 3H), 1.75–1.66 (m, 2H), 1.58–1.28 (m, 4H), 0.96–0.90 (m, 9H), 0.89 (s, 9H), 0.05 (s, 6H) ppm; 13CNMR (125 MHz, CDCl3) δ 172.1, 73.6, 68.8, 59.0, 54.0, 42.4, 34.2, 33.1, 25.8, 23.9, 23.6, 21.5, 18.9, 18.1, 13.6, −5.5 (2C) ppm. HRMS (ESI, m/z) calculated for C19H40NO3Si+ [M+H]+: 358.2772, found: 358.2761.
Benzyl (S)-2-(((3S,4S)-4-((tert-butoxycarbonyl)amino)-1-hydroxy-6-methylheptan-3-yl)oxy)pentanoate 5: Compound 15 (0.57 g, 1.60 mmol) was heated at 75 °C in 6 M aqueous HCl (6 mL) for 3 h, after the reaction mixture was cooled to 0 °C, it was adjusted to pH 10–12 by the dropwise addition of 6 M aqueous solution of NaOH. Without further purification, the reaction mixture was diluted with THF (6 mL) and treated with Boc2O (0.45 mL, 2.10 mmol) at room temperature for 16 h. The reaction mixture was first extracted with diethyl ether (50 mL × 2). The organic extracts were discarded, while the aqueous layer was acidified to pH 1–2 at 0 °C with an 1 N aqueous solution of HCl and extracted with ethyl acetate (50 mL × 3). The combined organic layer was washed with brine (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was dissolved in CH2Cl2 (20 mL) at 0 °C, after K2CO3 (0.22 g, 1.59 mmol), triethylamine (0.44 mL, 3.2 mmol) and BnBr (0.27 mL, 2.3 mmol) were sequentially added, the resulting mixture was gradually warmed to room temperature and stirred for 4 h. The reaction mixture was then quenched with water (2 mL) and extracted with ethyl acetate (50 mL × 3). The combined organic layer was washed with saturated NH4Cl (10 mL × 2) and brine (10 mL), dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by flash chromatography to give compound 5 (0.47 g, 66% over 3 steps) as a colorless oil. [α]D23 = −69.5 (c 0.86, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.36–7.33 (m, 5H), 5.19 (q, J = 12.2 Hz, 2H), 4.39 (d, J = 8.9 Hz, 1H), 4.25 (t, J = 5.7 Hz, 1H), 3.83–3.81 (m, 2H), 3.67 (dt, J = 11.1, 5.8 Hz, 1H), 3.65–3.55 (m, 1H), 3.28 (t, J = 6.2 Hz, 1H), 1.73–1.54 (m, 5H), 1.42–1.39 (m, 12H), 1.27–1.21 (m, 1H), 0.93–0.88 (m, 9H) ppm. 13C NMR (125 MHz, CDCl3) δ 174.1 155.6, 135.5, 128.6, 128.4 (2C), 79.4, 79.0, 66.9, 59.8, 49.7, 39.3, 35.5, 32.6, 28.4, 25.1, 23.5, 21.9, 18.7, 13.8 ppm. HRMS (ESI, m/z) calculated for C25H42NO6+ [M+H]+: 452.3007, found: 452.2994.
Benzyl (S)-2-(((4S,8S,9S)-4-(4-(benzyloxy)benzyl)-9-isobutyl-13,13-dimethyl-5-((4-nitrophenyl)sulfonyl)-3,11-dioxo-2,12-dioxa-5,10-diazatetradecan-8-yl)oxy)pentanoate 16: Compounds 5 (0.85 g, 1.88 mmol), 4 (1.32 g, 2.82 mmol) and PPh3 (0.76 g, 2.90 mmol) were dissolved in THF (25 mL) at 0 °C, after DEAD (0.50 g, 2.90 mmol) was added, the reaction mixture was stirred for 6 h at room temperature. The reaction was quenched with water (20 mL). Volatiles were removed in vacuo, the aqueous residue was extracted with ethyl acetate (50 mL × 3). The combined organic layer was washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2SO4 and evaporated in vacuo. The residue was purified by flash chromatography to give compound 16 (1.33 g, 78%) as a colorless oil. [α]D23 = −32.9 (c 0.80, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 7.7 Hz, 1H), 7.60 (d, J = 7.0 Hz, 1H), 7.53 (d, J = 8.1 Hz, 2H), 7.47–7.38 (m, 5H), 7.38–7.32 (m, 5H), 7.21 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 5.15 (s, 2H), 5.04 (s, 2H), 4.90 (t, J = 7.7 Hz, 1H), 4.48 (d, J = 9.3 Hz, 1H), 4.23–4.15 (m, 1H), 4.12 (t, J = 6.0 Hz, 1H), 3.82–3.72 (m, 2H), 3.58 (s, 3H), 3.39–3.29 (m, 2H), 2.97 (dd, J = 14.4, 8.5 Hz, 1H), 1.93–1.67 (m, 5H), 1.46 (s, 9H), 1.31–1.27 (m, 4H), 0.95–0.91 (m, 9H) ppm. 13C NMR (100 MHz, CDCl3) δ 172.9, 170.8, 157.7, 155.6, 148.3, 137.1, 135.8, 133.1, 133.0, 131.4, 130.9, 130.3, 128.6, 128.5, 128.4, 128.2, 128.1, 127.9, 127.5, 123.7, 114.9, 79.3, 79.3, 70.0, 66.6, 66.4, 61.7, 52.2, 49.6, 43.8, 39.7, 35.5, 35.4, 31.1, 28.4, 25.0, 23.5, 22.0, 18.5, 13.9 ppm. HRMS (ESI, m/z) calculated for C48H62N3O12S+ [M+H]+: 904.4049, found: 904.4076.
Benzyl (S)-2-(((4S,8S,9S)-4-(4-(benzyloxy)benzyl)-9-isobutyl-13,13-dimethyl-3,11-dioxo-2,12-dioxa-5,10-diazatetradecan-8-yl)oxy)pentanoate 3: At 0 °C, K2CO3 (0.80 g 5.88 mmol) and PhSH (0.45 mL, 4.38 mmol) were added to a solution of 16 (1.32 g, 1.46 mmol) in CH2Cl2 (20 mL). The reaction mixture was then stirred at room temperature for 3 h, and then filtered through a pad of silica gel. The filtrate was concentrated in vacuo, and the residue was purified by flash chromatography to give compound 3 (0.89 g, 85%) as a colorless oil. [α]D23 = −26.8 (c 1.3, CHCl3). 1H NMR (500 MHz, CDCl3) δ 7.43–7.27 (m, 10H), 7.06 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.5 Hz, 2H), 5.15 (s, 2H), 5.01 (s, 2H), 4.67 (d, J = 9.4 Hz, 1H), 4.03 (t, J = 6.2 Hz, 1H), 3.71 (dd, J = 14.5, 7.1 Hz, 1H), 3.62 (s, 3H), 3.46–3.37 (m, 2H), 2.88–2.77 (m, 2H), 2.69–2.61 (m, 1H), 2.55–2.47 (m, 1H), 1.70–1.51 (m, 5H), 1.42 (s, 9H), 1.40–1.23 (m, 4H), 0.93–0.88 (m, 9H) ppm. 13C NMR (125 MHz, CDCl3) δ 174.9, 172.9, 157.5, 155.6, 137.1, 135.6, 130.2, 129.8, 128.5, 128.4, 128.3, 128.3, 127.8, 127.3, 114.7, 79.7, 78.8, 78.2, 69.9, 66.3, 63.0, 51.4, 49.8, 44.5, 40.6, 38.7, 35.4, 31.0, 28.3, 28.3, 24.9, 23.1, 22.1, 18.5, 13.8 ppm. HRMS (ESI, m/z) calculated for C42H59N2O8+ [M+H]+: 719.4266, found: 719.4278.
Benzyl (5S,13S,15S)-10-((S)-3-(4-(benzyloxy)phenyl)-1-methoxy-1-oxopropan-2-yl)-13-((S)-1-((tert-butoxycarbonyl)amino)-3-methylbutyl)-5-isobutyl-3,6,9-trioxo-1-phenyl-15-propyl-2,14-dioxa-4,7,10-triazahexadecan-16-oate 2a: Amine 3 (0.10 g, 0.14 mmol) and the dipeptide ((benzyloxy)carbonyl)-L-leucylglycine 17a (0.18 g, 0.56 mmol) were dissolved in DMF (10 mL), after HATU (0.16 g, 0.42 mmol), HOAT (19 mg, 0.14 mmol) and NMM (88 μL, 0.80 mmol) were sequentially added at 0 °C, the reaction mixture was stirred at room temperature for 16 h. Volatiles were removed in vacuo, the residue was dissolved in ethyl acetate (150 mL) and successively washed with saturated aqueous NH4Cl (10 mL × 2), water (10 mL × 3) and brine (10 mL). The organic phase was then dried over Na2SO4 and evaporated in vacuo. The residue was purified by flash chromatography to give the desired product 2a (121 mg, 85%). [α]D23 = −90.0 (c 1.4, CHCl3). 1H NMR (400 MHz, MeOH-d4) δ 7.45–7.28 (m, 15H), 7.15 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.3 Hz, 2H), 5.23–5.07 (m, 4H), 5.03 (s, 2H), 4.30–4.13 (m, 2H), 3.96 (dd, J = 51.9, 16.9 Hz, 2H), 3.69 (s, 3H), 3.54–3.40 (m, 1H), 3.29–3.20 (m, 2H), 3.17–3.07 (m, 1H), 2.58 (d, J = 10.1 Hz, 1H), 1.79–1.58 (m, 7H), 1.44 (s, 9H), 1.38–1.21 (m, 5H), 0.99–0.88 (m, 15H) ppm. 13C NMR (100 MHz, MeOH-d4) δ 174.0, 173.1, 171.1, 168.8, 157.7, 157.1, 156.7, 137.3, 136.8, 135.9, 130.2, 130.2, 128.2, 128.1, 128.0, 127.6, 127.4, 127.4, 127.1, 114.7, 78.7, 78.4, 76.9, 69.7, 66.4, 66.2, 63.0, 53.6, 51.3, 46.4, 40.7, 37.7, 35.0, 33.3, 28.8, 27.4, 24.8, 24.5, 22.6, 22.1, 20.7, 20.4, 18.1, 12.8 ppm. HRMS (ESI, m/z) calculated for C58H79N4O12+ [M+H]+: 1023.5689, found: 1023.5667.
Methyl (S)-2-((2S,5S,13S)-13-((S)-1-((tert-butoxycarbonyl)amino)-3-methylbutyl)-5-isobutyl-3,6,9-trioxo-2-propyl-1-oxa-4,7,10-triazacyclotridecan-10-yl)-3-(4-hydroxyphen-yl)propanoate 1a: Compound 2a (55.0 mg, 54 μmol) was dissolved in MeOH (10 mL) at room temperature. After Pd(OH)2/C (6.0 mg, 10% wt) was added, the reaction vessel was sealed and changed into a hydrogen atmosphere for the deprotection of both Cbz and Bn groups. Ten hours later, the catalyst was filtered off and the filtrate was concentrated in vacuo. The residue was dissolved in DMF (25 mL) and cooled to 0 °C. After HOAT (7.0 mg, 49 μmol), HATU (93.0 mg, 0.25 mmol), and NMM (32 μL, 0.29 mmol) were sequentially added to the solution, the reaction mixture was stirred at room temperature for 16 h. All volatiles were removed in vacuo, the residue was dissolved in ethyl acetate (100 mL) and washed with saturated aqueous NH4Cl (10 mL × 2), water (10 mL × 3) and brine (10 mL). The organic phase was then dried over Na2SO4 and evaporated in vacuo. The residue was purified by flash chromatography to give the corresponding product 1a (23.0 mg, 62%). [α]D23 = −124.4 (c 1.0, CHCl3). 1H NMR (500 MHz, MeOH-d4): δ 7.05 (d, J = 8.3 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 4.67–4.60 (m, 1H), 4.22–4.09 (m, 1H), 4.02–3.96 (m, 2H), 3.85 (d, J = 15.4 Hz, 1H), 3.75–3.70 (m, 1H), 3.65 (s, 3H), 3.41–3.34 (m, 1H), 3.18–3.08 (m, 3H), 2.96–2.91 (m, 1H), 2.36–2.27 (m, 1H), 1.91–1.79 (m, 1H), 1.75–1.52 (m, 7H), 1.42 (s, 9H), 1.35–1.22 (m, 3H), 1.15–1.11 (m, 1H), 0.96–0.88 (m, 15H) ppm; 13C NMR (125 MHz, MeOH-d4) δ 174.6, 172.2, 171.3, 169.3, 157.3, 156.0, 130.4, 128.5, 115.1, 84.7, 80.0, 78.6, 62.9, 51.3, 50.7, 50.6, 45.0, 40.7, 38.2, 35.2, 33.3, 31.5, 27.6, 24.9, 24.7, 22.3, 22.0, 20.9, 20.8, 18.3, 12.9 ppm. HRMS (ESI, m/z) calculated for C36H59N4O9+ [M+H]+: 691.4277 found: 691.4259.
Compound 1b–d was obtained from compound 3 and the corresponding Cbz- protected dipeptide by the same procedures described above.
Compound 1b was prepared from compound 3 and Cbz-Gly-Gly-OH: [α]D23 = −10.1 (c 0.30, CHCl3). 1H NMR (400 MHz, MeOH-d4) δ 7.11 (d, J = 8.4 Hz, 2H), 6.72 (d, J = 6.3 Hz, 2H), 4.60 (t, J = 8.4 Hz, 1H), 4.46 (d, J = 15.0 Hz, 1H), 4.30–4.20 (m, 2H), 3.79–3.74 (m, 1H), 3.69–3.62 (m, 4H), 3.57 (dd, J = 14.5, 9.8 Hz, 2H), 3.22–3.16 (m, 2H), 2.98–2.94 (m, 1H), 2.24–2.08 (m, 1H), 1.90–1.53 (m, 8H), 1.45 (s, 9H), 1.15 (s, 1H), 0.96–0.89 (m, 9H) ppm. 13C NMR (125 MHz, MeOH-d4) δ 174.3, 171.2, 169.4, 169.0, 130.5, 128.2, 115.0, 82.5, 80.1, 78.5, 62.2, 51.3, 43.8, 42.6, 41.2, 35.7, 34.1, 33.1, 30.4, 30.3, 27.5, 24.9, 22.2, 21.1, 18.0, 12.9 ppm. HRMS (ESI, m/z) calculated for C32H51N4O9+ [M+H]+: 635.3651, found: 635.3639.
Compound 1c was prepared from compound 3 and Cbz-L-Ala-Gly-OH: [α]D23 = −144.3 (c 0.34, CHCl3). 1H NMR (400 MHz, MeOH-d4): δ 7.08 (d, J = 8.3 Hz, 2H), 6.72 (d, J = 8.3 Hz, 2H), 4.66 (q, J = 7.0 Hz, 1H), 4.07–4.00 (m, 1H), 3.94 (q, J = 15.5 Hz, 2H), 3.77–3.71 (m, 1H), 3.67 (s, 3H), 3.41–3.35 (m, 1H), 3.20–3.13 (m, 3H), 3.02–2.95 (m, 1H), 2.32 (td, J = 13.8, 4.3 Hz, 1H), 1.97–1.85 (m, 1H), 1.78–1.52 (m, 5H), 1.44 (s, 9H), 1.36–1.27 (m, 4H), 1.20–1.12 (m, 1H), 0.96–0.90 (m, 9H) ppm; 13C NMR (100 MHz, MeOH-d4): δ 174.3, 172.2, 171.1, 169.2, 157.1, 155.9, 130.3, 128.3, 114.8, 84.5, 79.9, 78.4, 62.6, 51.1, 50.5, 44.9, 40.5, 35.0, 33.2, 31.3, 27.4, 24.8, 22.2, 20.8, 18.2, 14.1, 12.8 ppm. HRMS (ESI, m/z) calculated for C33H53N4O9+ [M+H]+: 649.3807, found: 649.3794.
Compound 1d was prepared from compound 3 and Cbz-Gly-L-Ala-OH: [α]D23 = −84.0 (c 0.25, MeOH). 1H NMR (500 MHz, MeOH-d4) δ 7.12 (d, J = 8.4 Hz, 2H), 6.69 (d, J = 8.4 Hz, 2H), 4.56 (q, J = 7.5 Hz, 1H), 4.13–4.07 (m, 1H), 3.90 (dd, J = 53.8, 15.3 Hz, 2H), 3.65– 3.56 (m, 4H), 3.42–3.36 (m, 1H), 3.20–3.13 (m, 3H), 3.02–2.97 (m, 1H), 2.26 (td, J = 14.0, 4.0 Hz, 1H), 1.88 (td, J = 14.4, 3.9 Hz, 1H), 1.73 (dt, J = 12.6, 6.5 Hz, 1H), 1.66–1.48 (m, 5H), 1.43 (s, 9H), 1.39 (d, J = 7.6 Hz, 3H), 1.35–1.28 (m, 1H), 1.17–1.11 (m, 1H), 0.95–0.90 (m, 9H) ppm. 13C NMR (125 MHz, MeOH-d4) δ 172.0, 171.2, 169.8, 157.2, 156.1, 130.8, 128.2, 114.8, 83.4, 81.1, 78.5, 62.6, 52.5, 51.2, 50.3, 43.2, 41.0, 34.8, 33.3, 30.0, 27.6, 24.9, 22.3, 21.0, 18.2, 16.8, 12.8 ppm. HRMS (ESI, m/z) calculated for C33H53N4O9+ [M+H]+: 649.3807, found: 649.3789.
Methyl (S)-2-((2S,5S,13S)-13-((S)-1-amino-3-methylbutyl)-5-isobutyl-3,6,9-trioxo-2-propyl-1-oxa-4,7,10-triazacyclotridecan-10-yl)-3-(4-hydroxyphenyl)propanoate 18: Compound 1a (9.0 mg, 0.13 mmol) was dissolved in DCM (2 mL) at 0 °C. After TFA (0.2 mL) was dropwise added, the reaction mixture was stirred for 2 h and then concentrated in vacuo. The residue was purified by preparative HPLC to afford the desired cyclic peptide 18 (6.0 mg, 78%). The preparative HPLC was performed on an Agilent 1200 system equipped with a reverse-phase Agilent SB-C18 column (21.2 × 250 mm), and it was eluted with H2O-MeOH (contains 0.1% of TFA) (42:58) at a flow rate of 10 mL/min. [α]D23 = −123 (c 0.10, MeOH). 1H NMR (400 MHz, MeOH-d4) δ 7.01 (d, J = 8.4 Hz, 2H), 6.74 (d, J = 8.5 Hz, 2H), 4.67–4.57 (m, 1H), 3.99 (q, J = 15.5 Hz, 2H), 3.87–3.75 (m, 2H), 3.71 (s, 3H), 3.25–3.17 (m, 3H), 2.93–2.84 (m, 1H), 2.27–2.15 (m, 1H), 1.88–1.59 (m, 7H), 1.50–1.30 (m, 4H), 1.00–0.95 (m, 9H), 0.94–0.88 (m, 6H) ppm. 13C NMR (100 MHz, MeOH-d4) δ 173.2, 172.4, 170.9, 169.5, 156.1, 130.1, 128.6, 115.1, 84.9, 75.7, 63.6, 51.9, 51.3, 50.5, 46.3, 44.8, 38.9, 37.7, 34.6, 33.1, 30.8, 24.5, 23.9, 21.9, 21.2, 21.1, 20.5, 17.9, 12.7 ppm. HRMS (ESI, m/z) calculated for C31H51N4O7+ [M+H]+: 591.3752, found: 591.3757.
3.3. Two-Dimensional COSY and NOESY Analysis of Compound 15
The COSY and NOESY experiments were recorded in acetone-d6/methanol-d4 (2:1). The co-solvent system was used here to distinguish the signals of Ha from H2″, Hc from H1″ (which were found severely overlapped in either CDCl3 or Acetone-d6). From the 1H NMR and COSY spectra, Ha was assigned to 3.80 ppm (1H, dt, J1 = 10 Hz, J2 = 5.2 Hz), Hc were assigned to 1.69–1.71 ppm (2H, m) and Hb was assigned to 4.03 ppm (1H, dd, J1 = 8.0 Hz, J2 = 4.4 Hz). The NOESY spectrum showed clear correlations of Ha-Hc and Hc-Hb, no direct correlation between Ha and Hb, and these correlation signals unambiguously proved that Hb is at the equatorial position (six-membered ring’s chair-confirmation), and thus the stereochemistry at C-2 was determined as 2S.


4. Conclusions
The development of new methods for the helical stabilization of a linear peptide could provide additional opportunities for the discovery of peptide-based therapeutics targeting PPI. Design and synthesis of hydrogen-bond surrogates (HBSs) are one of the most successful approaches. In this paper, we developed a new helix template based on the replacement of the weak (i, i + 4) H-bond with a covalent carbon–carbon linkage; and also employed a covalent ether bond to mimic one amide-bond. In comparing the precedent hydrogen-bond surrogates in which the hydrogen bond was replaced with hydra-zine, carbon–carbon links, and thioether linkage, our new helix template could be placed in a position other than the N-terminus of a short peptide. The CD studies show that the helix template adopts stable conformations in aqueous buffer at exceptionally high temperatures. We also found that the stereogenic centers presented in the macrocycle are vital for the stability of helical conformation. The scaffold obtained has been proven as a single-turn helical mimetic. The further coupling of additional short peptide fragments and beta-strand mimetic aiming to develop inhibitors of p53 degradation based on the PIP motif of PCNA interaction proteins will be reported in due course.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28020780/s1, Copies of NMR spectra (1H and 13C) of 8–10, 6, 14, 15, 5, 16, 3, 2a, 1a, 1b, 1c, 1d, 18.
Author Contributions
J.L., S.T., J.-L.Y. and T.Y. conceived and designed this research; J.L., S.T. and J.-L.Y. prepared the compounds and collected their spectral data and analyzed the experimental data; J.L., S.T., J.-L.Y. and T.Y. prepared the manuscript. Conceptualization, T.Y.; Data curation, J.L., S.T. and J.-L.Y.; Funding acquisition, J.L., J.-L.Y. and T.Y.; Supervision, T.Y.; Writing—original draft, J.L., S.T. and J.-L.Y.; Writing—review and editing, T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Guangdong Department of Education (2021ZDJS097), Guangdong Basic and Applied Basic Research Foundation (2021A1515010188, 2022A1515110592), Science, Technology & Innovation Bureau of Longgang District (RCTDPT-2019-008).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Watkins, A.M.; Wuo, M.G.; Arora, P.S. Protein–Protein Interactions Mediated by Helical Tertiary Structure Motifs. J. Am. Chem. Soc. 2015, 137, 11622–11630. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Sable, R.; Jois, S. Surfing the Protein-Protein Interaction Surface Using Docking Methods: Application to the Design of PPI Inhibitors. Molecules 2015, 20, 11569–11603. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef]
- Milroy, L.-G.; Grossmann, T.N.; Hennig, S.; Brunsveld, L.; Ottmann, C. Modulators of Protein–Protein Interactions. Chem. Rev. 2014, 114, 4695–4748. [Google Scholar] [CrossRef]
- Wilson, A.J. Inhibition of protein–protein interactions using designed molecules. Chem. Soc. Rev. 2009, 38, 3289–3300. [Google Scholar] [CrossRef]
- Robertson, N.S.; Spring, D.R. Using Peptidomimetics and Constrained Peptides as Valuable Tools for Inhibiting Protein–Protein Interactions. Molecules 2018, 23, 959. [Google Scholar] [CrossRef]
- Sawyer, N.; Watkins, A.M.; Arora, P.S. Protein Domain Mimics as Modulators of Protein–Protein Interactions. Acc. Chem. Res. 2017, 50, 1313–1322. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of α-helix-mediated protein–protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, Q.-S.; Geng, H.; Tian, Y.; Cheng, M.; Jiang, Y.-H.; Xie, M.-S.; Niu, X.-G.; Jiang, F.; Zhang, Y.-O.; et al. Crosslinked Aspartic Acids as Helix-Nucleating Templates. Angew. Chem. Int. Ed. 2016, 55, 12088–12093. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Wang, D.; Li, Z. Design and Synthetic Strategies for Helical Peptides. In Cyclic Peptide Design; Goetz, G., Ed.; Springer: New York, NY, USA, 2019; pp. 107–131. [Google Scholar]
- Andrews, M.J.I.; Tabor, A.B. Forming stable helical peptides using natural and artificial amino acids. Tetrahedron 1999, 55, 11711–11743. [Google Scholar] [CrossRef]
- Garner, J.; Harding, M.M. Design and synthesis of α-helical peptides and mimetics. Org. Biomol. Chem. 2007, 5, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, H.E.; Grubbs, R.H. Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angew. Chem. Int. Ed. 1998, 37, 3281–3284. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Cabezas, E.; Satterthwait, A.C. The Hydrogen Bond Mimic Approach: Solid-Phase Synthesis of a Peptide Stabilized as an α-Helix with a Hydrazone Link. J. Am. Chem. Soc. 1999, 121, 3862–3875. [Google Scholar] [CrossRef]
- Patgiri, A.; Jochim, A.L.; Arora, P.S. A Hydrogen Bond Surrogate Approach for Stabilization of Short Peptide Sequences in α-Helical Conformation. Acc. Chem. Res. 2008, 41, 1289–1300. [Google Scholar] [CrossRef]
- Chapman, R.N.; Dimartino, G.; Arora, P.S. A Highly Stable Short α-Helix Constrained by a Main-Chain Hydrogen-Bond Surrogate. J. Am. Chem. Soc. 2004, 126, 12252–12253. [Google Scholar] [CrossRef]
- Koo, N.; Sharma, A.K.; Narayan, S. Therapeutics Targeting p53-MDM2 Interaction to Induce Cancer Cell Death. Int. J. Mol. Sci. 2022, 23, 5005. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction to Reactivate p53 Function: A Novel Approach for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-Molecule Inhibitors of the MDM2–p53 Protein–Protein Interaction (MDM2 Inhibitors) in Clinical Trials for Cancer Treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Banks, D.; Wu, M.; Higa, L.A.; Gavrilova, N.; Quan, J.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. L2DTL/CDT2 and PCNA Interact with p53 and Regulate p53 Polyubiquitination and Protein Stability through MDM2 and CUL4A/DDB1 Complexes. Cell Cycle 2006, 5, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Wang, B.; Ye, T. Cyp11B, Cyp17, and/or Cyp21 Inhibitors. WO 2012083112 A2 2012-06-21, WO 2011088160 A2 2011-07-21, US 2010216699 A1 2010-08-26, WO 2010065174 A1 2010-06-10, GB2464617 A 2010-04-28. Available online: https://patents.google.com/patent/US20130252930 (accessed on 9 January 2023).
- Liu, J.; Chen, Y.; Luesch, H.; Ye, T. Total Synthesis of des-Thiomethyllooekeyolide A. Org. Lett. 2022, 24, 7260–7264. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Yu, J.; Meng, J.; Guo, Y.; Ye, T. Total Synthesis of Pagoamide A. Molecules 2021, 26, 4224. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, J.; Gao, B.; Zhao, M.; Yan, J.-L.; Xu, Z.; Choi, S.; Ye, T. Total Synthesis of Hoiamide A Using an Evans–Tishchenko Reaction as a Key Step. Org. Lett. 2019, 21, 5471–5474. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, B.; Xu, Z.; Ye, T. Total Synthesis and Stereochemical Assignment of Callyspongiolide. J. Am. Chem. Soc. 2016, 138, 6948–6951. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhang, J.; Xu, Z.; Ye, T. The total synthesis and stereochemical assignment of scytonemin A. Chem. Commun. 2016, 52, 1002–1005. [Google Scholar] [CrossRef]
- Liao, L.; Zhou, J.; Xu, Z.; Ye, T. Concise Total Synthesis of Nannocystin A. Angew. Chem. Int. Ed. 2016, 55, 13263–13266. [Google Scholar] [CrossRef]
- Qu, S.; Chen, Y.; Wang, X.; Chen, S.; Xu, Z.; Ye, T. Total synthesis of largamide B. Chem. Commun. 2015, 51, 2510–2513. [Google Scholar] [CrossRef]
- Lei, H.; Yan, J.; Yu, J.; Liu, Y.; Wang, Z.; Xu, Z.; Ye, T. Total Synthesis and Stereochemical Reassignment of Mandelalide A. Angew. Chem. Int. Ed. 2014, 53, 6533–6537. [Google Scholar] [CrossRef]
- Wang, D.; Chen, K.; Kulp, J.L.; Arora, P.S. Evaluation of Biologically Relevant Short α-Helices Stabilized by a Main-Chain Hydrogen-Bond Surrogate. J. Am. Chem. Soc. 2006, 128, 9248–9256. [Google Scholar] [CrossRef] [PubMed]
- Mahon, A.B.; Arora, P.S. Design, synthesis and protein-targeting properties of thioether-linked hydrogen bond surrogate helices. Chem. Commun. 2012, 48, 1416–1418. [Google Scholar] [CrossRef] [PubMed]
- Rittle, K.E.; Homnick, C.F.; Ponticello, G.S.; Evans, B.E. A synthesis of statine utilizing an oxidative route to chiral .alpha.-amino aldehydes. J. Org. Chem. 1982, 47, 3016–3018. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bischoff, A.; Cappiello, J. Asymmetric Total Synthesis of the Gastroprotective Microbial Agent AI-77-B. Eur. J. Org. Chem. 2003, 2003, 821–832. [Google Scholar] [CrossRef]
- Vara Prasad, J.V.N.; Rich, D.H. Addition of Allylic Metals to α-Aminoaldehydes. Application to the Synthesis of Statine, Ketomethylene and Hydroxyethylene Dipeptide Isosteres. Tetrahedron Lett. 1990, 31, 1803–1806. [Google Scholar] [CrossRef]
- Qabar, M.N.; Meara, J.P.; Ferguson, M.D.; Lum, C.; Kim, H.-O.; Kahn, M. Synthesis of 3-alkoxyazetidin-2-ones: Dipeptide mimics. Tetrahedron Lett. 1998, 39, 5895–5898. [Google Scholar] [CrossRef]
- Philippe, C.; Milcent, T.; Crousse, B.; Bonnet-Delpon, D. Non Lewis acid catalysed epoxide ring opening with amino acid esters. Org. Biomol. Chem. 2009, 7, 2026–2028. [Google Scholar] [CrossRef]
- Snider, B.B.; Lin, H. Biomimetic Total Syntheses of (−)-TAN1251A, (+)-TAN1251B, (+)-TAN1251C, and (+)-TAN1251D. Org. Lett. 2000, 2, 643–646. [Google Scholar] [CrossRef]
- Wünsch, E.; Fries, G.; Zwick, A. Beiträge zur Peptidsynthese, V. Darstellung und peptidsynthetische Verwendung von O-Benzyl-L-tyrosin. Chem. Ber. 1958, 91, 542–547. [Google Scholar] [CrossRef]
- Fukuyama, T.; Jow, C.-K.; Cheung, M. 2- and 4-Nitrobenzenesulfonamides: Exceptionally versatile means for preparation of secondary amines and protection of amines. Tetrahedron Lett. 1995, 36, 6373–6374. [Google Scholar] [CrossRef]
- Holzwarth, G.; Doty, P. The Ultraviolet Circular Dichroism of Polypeptides. J. Am. Chem. Soc. 1965, 87, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Sears, D.W.B.S. Physical Principles and Techniques of Protein Chemistry; Academic Press: New York, NY, USA, 1973; pp. 460–466. [Google Scholar]
- Patgiri, A.; Joy, S.T.; Arora, P.S. Nucleation Effects in Peptide Foldamers. J. Am. Chem. Soc. 2012, 134, 11495–11502. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).