

Interaction of Chondroitin and Hyaluronan Glycosaminoglycans with Surfaces of Carboxylated Carbon Nanotubes Studied Using Molecular Dynamics Simulations
 , and
, and
Abstract
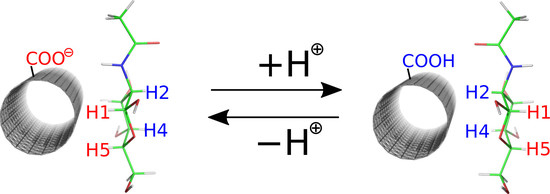
:
1. Introduction
2. Results and Discussion
2.1. Simulation Setup
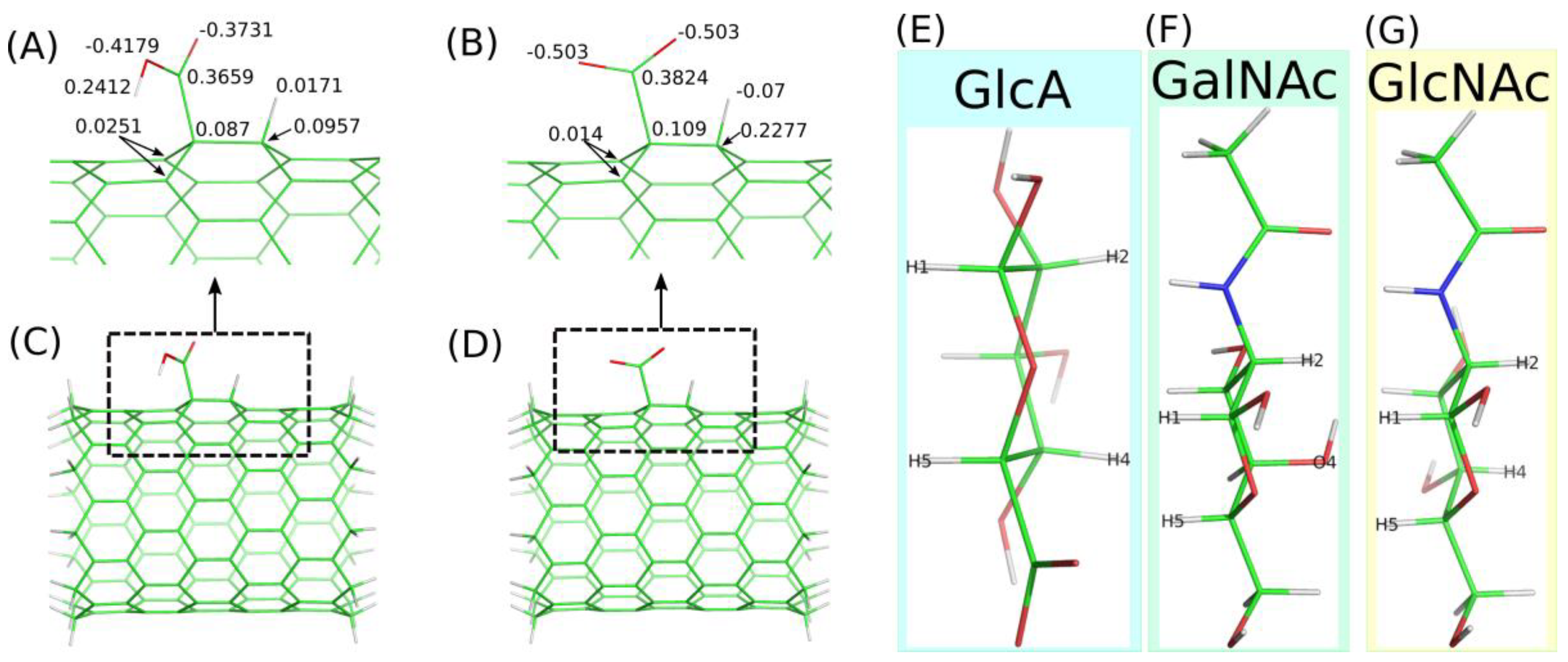
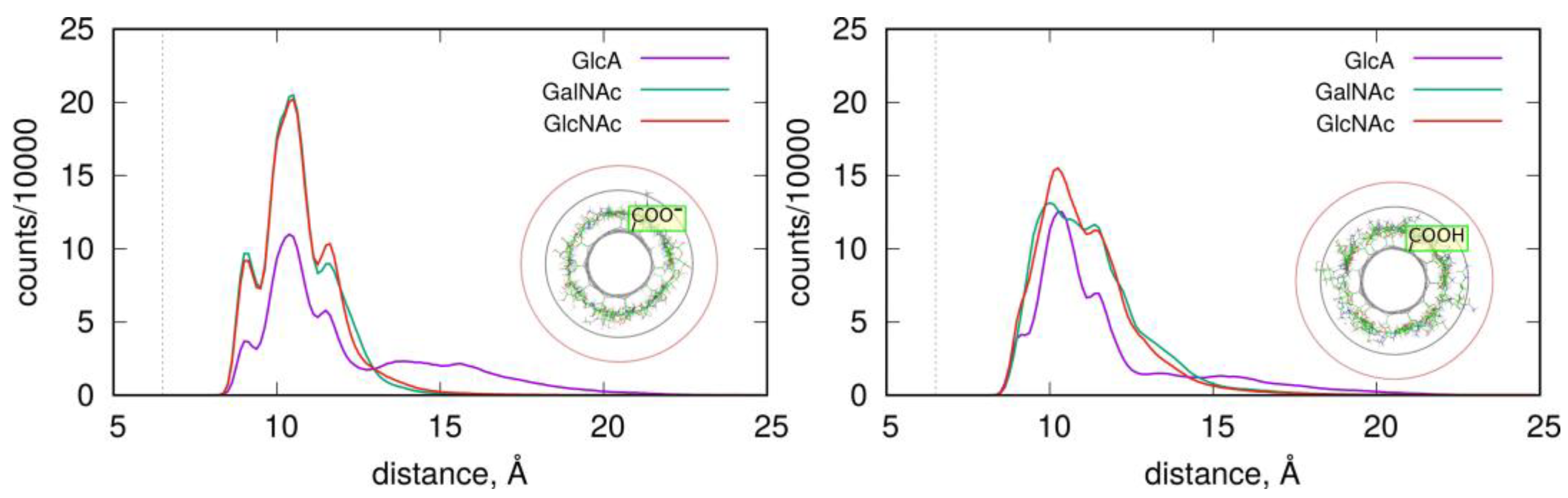
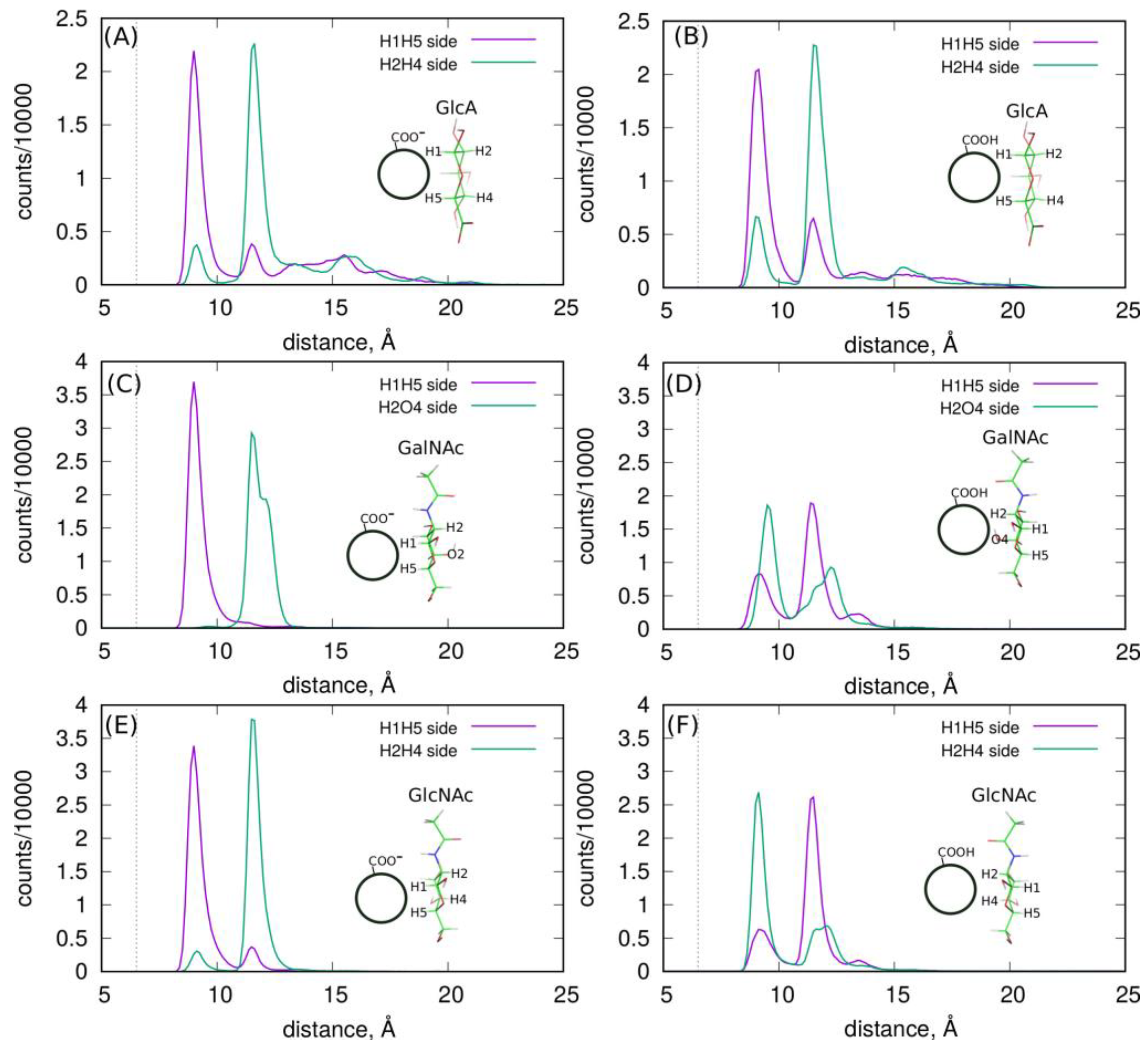
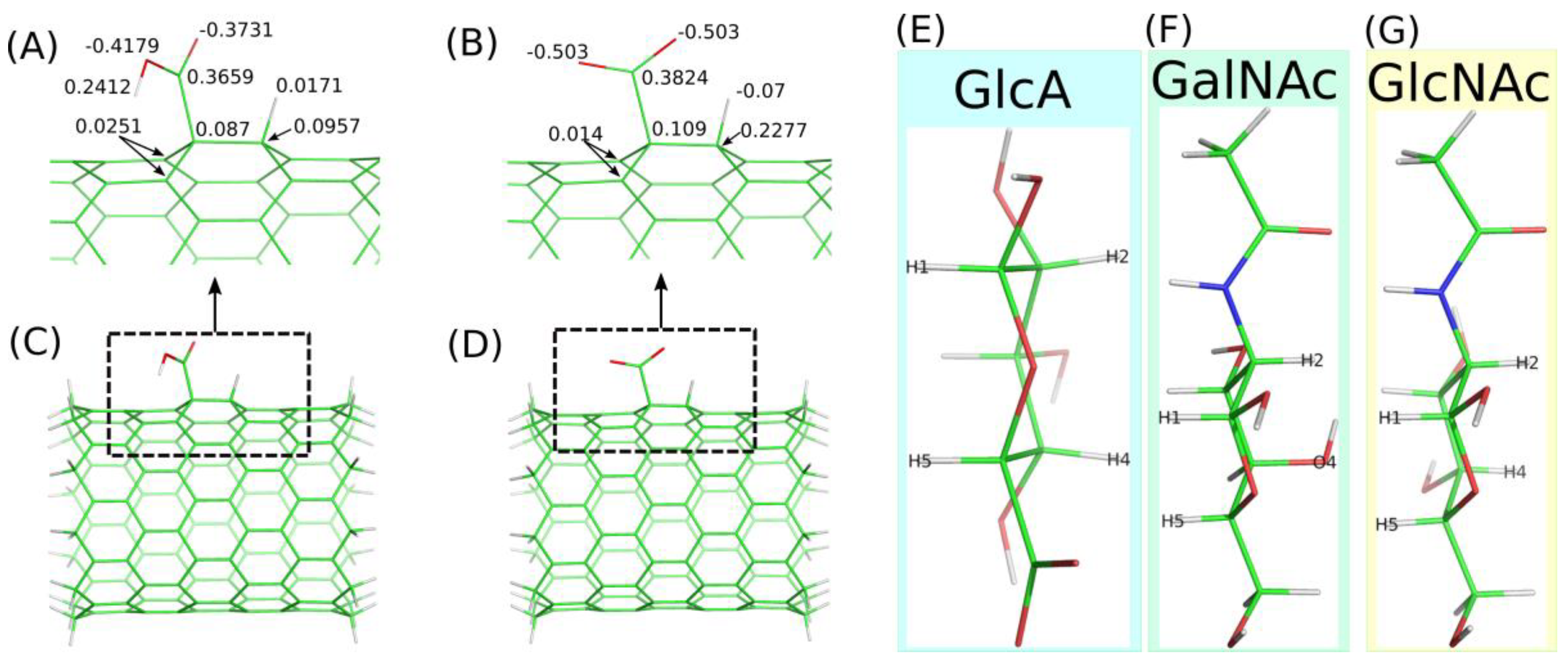
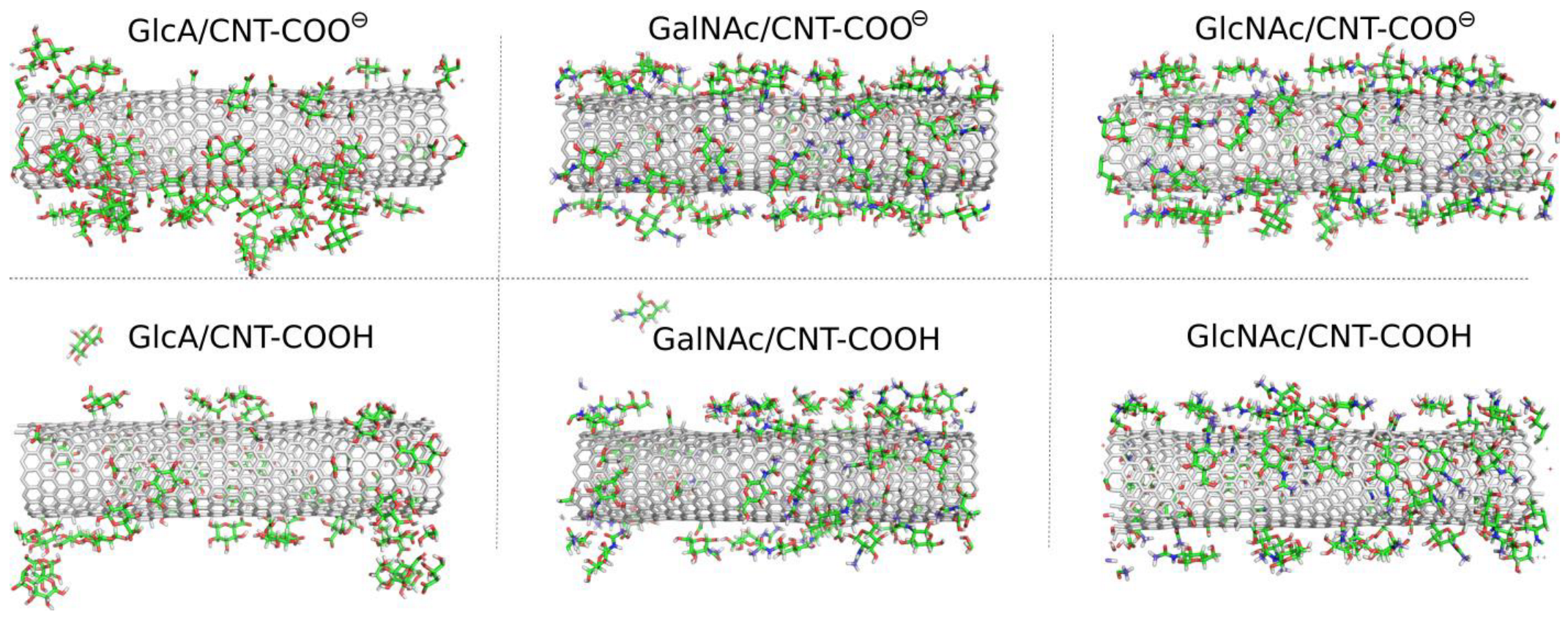
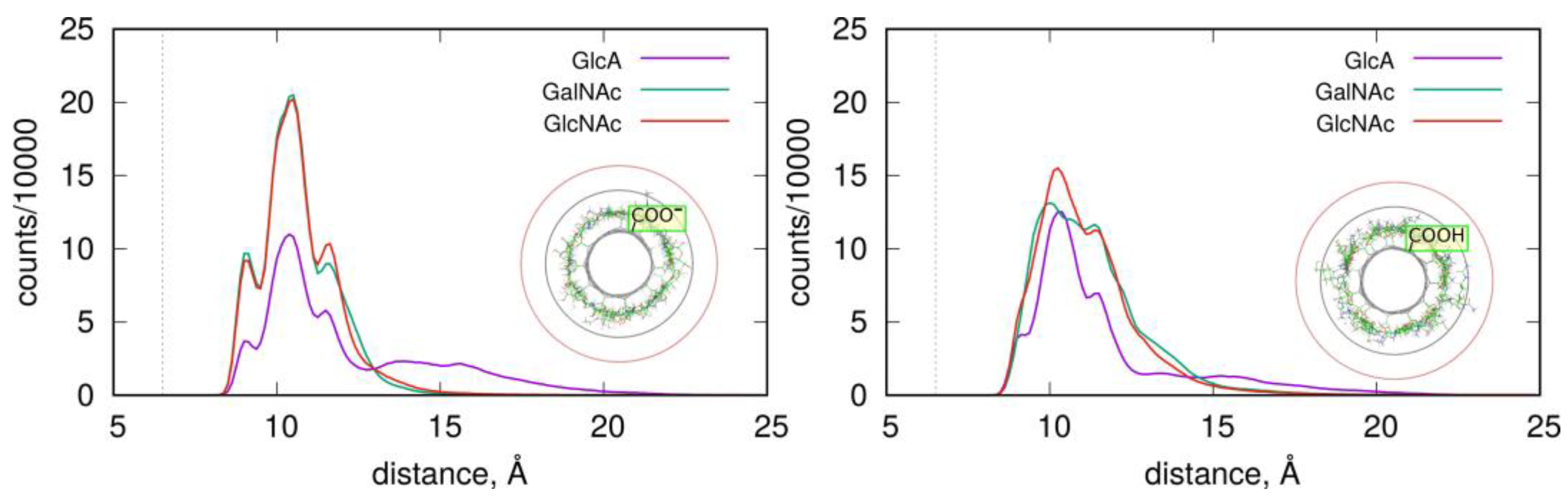
2.2. Adsorption of Monosaccharides on fCNT
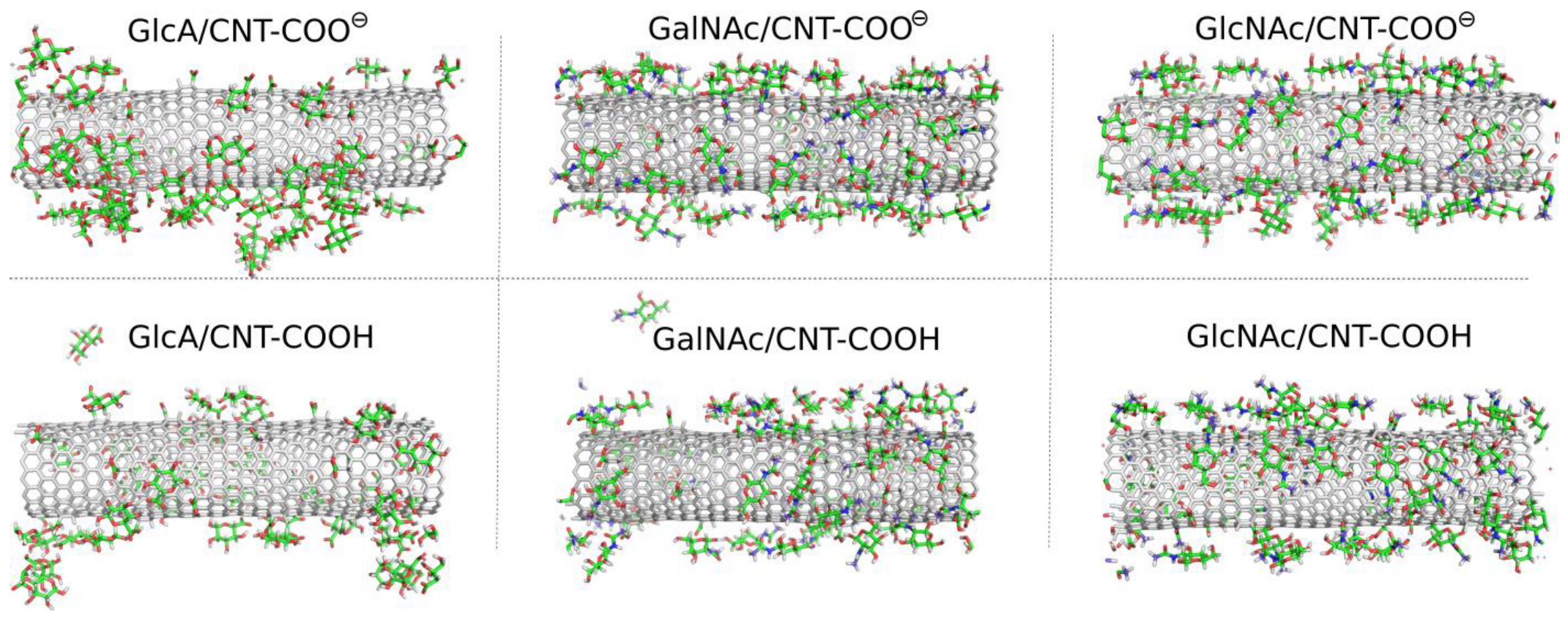
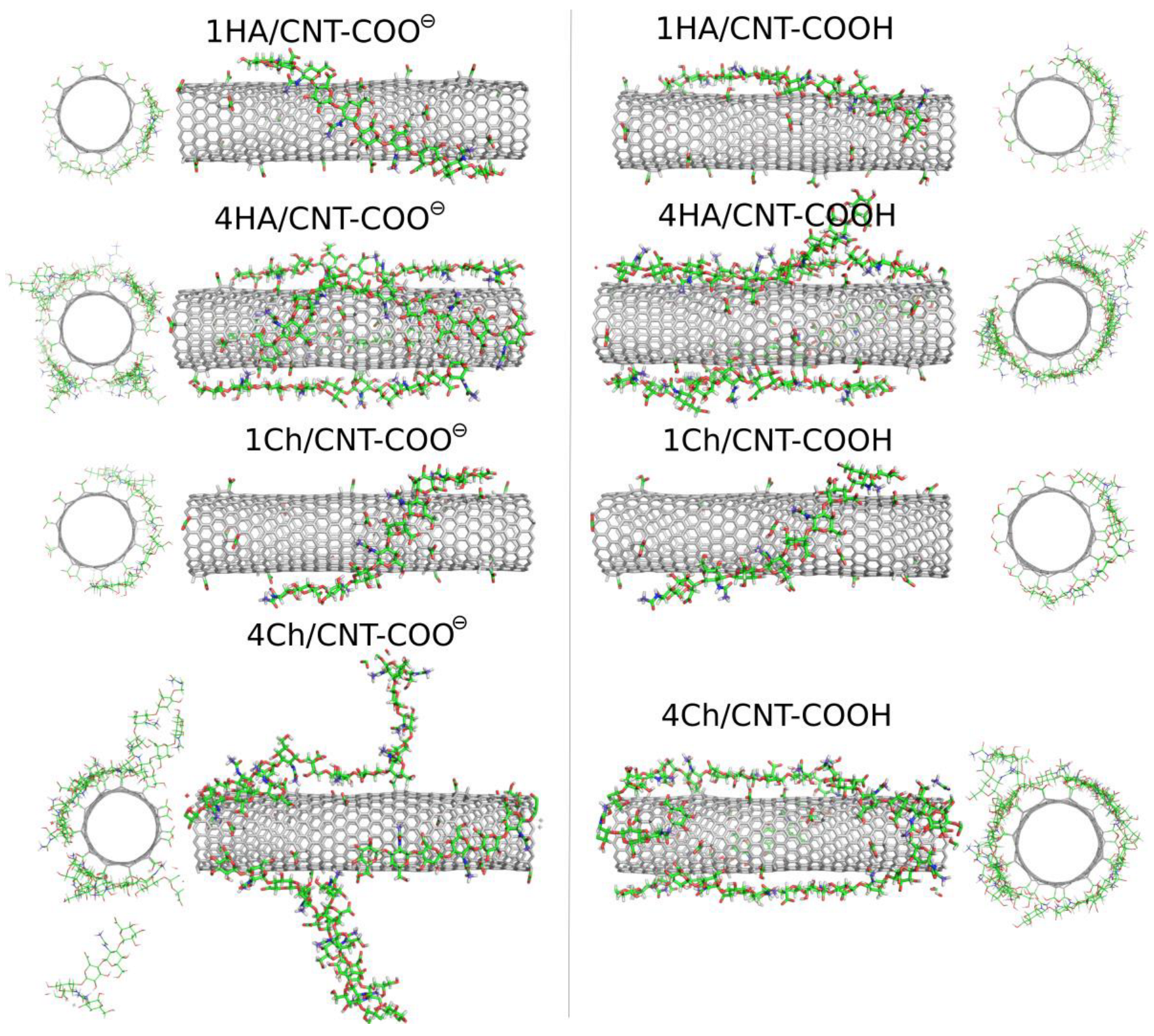
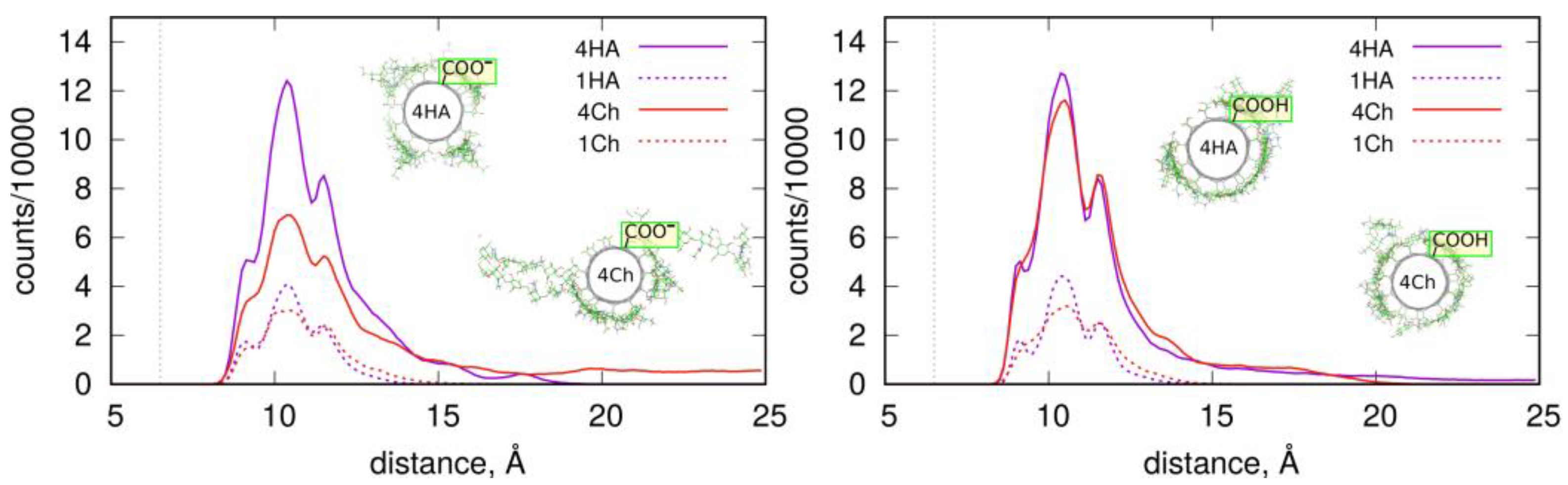
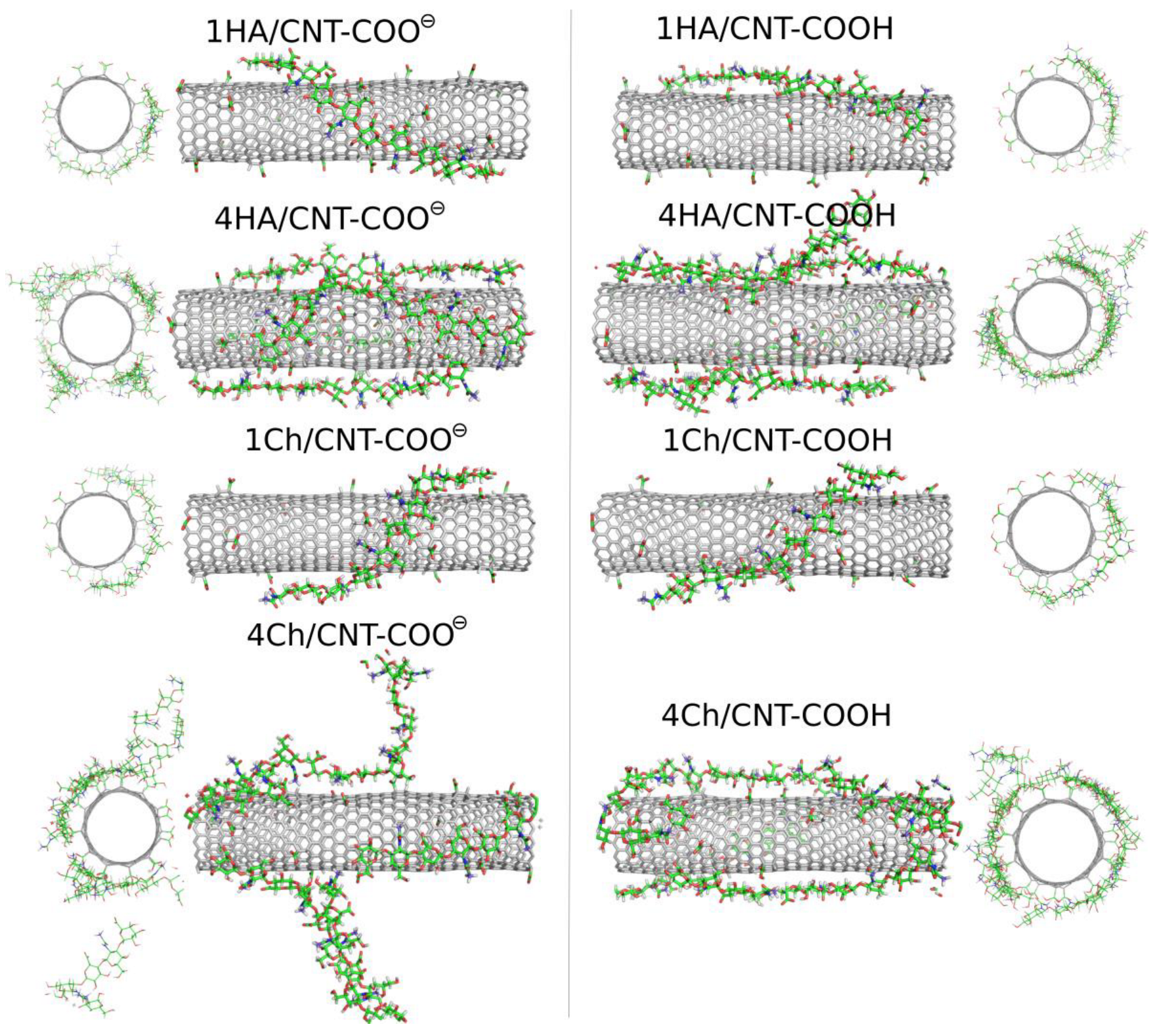
2.3. Adsorption of Oligosaccharides on fCNTs
2.4. Hydrogen Bonds (HB)
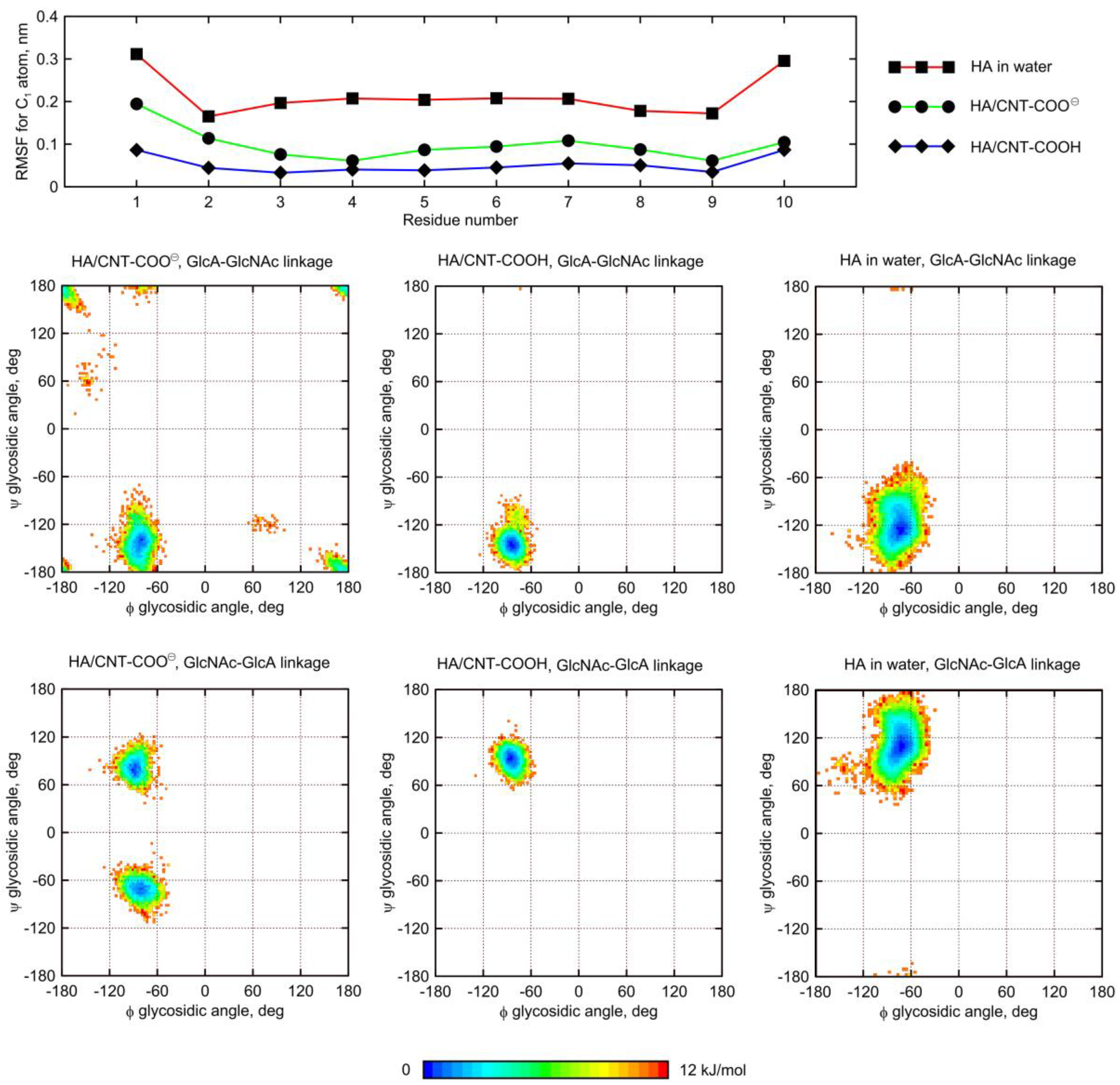
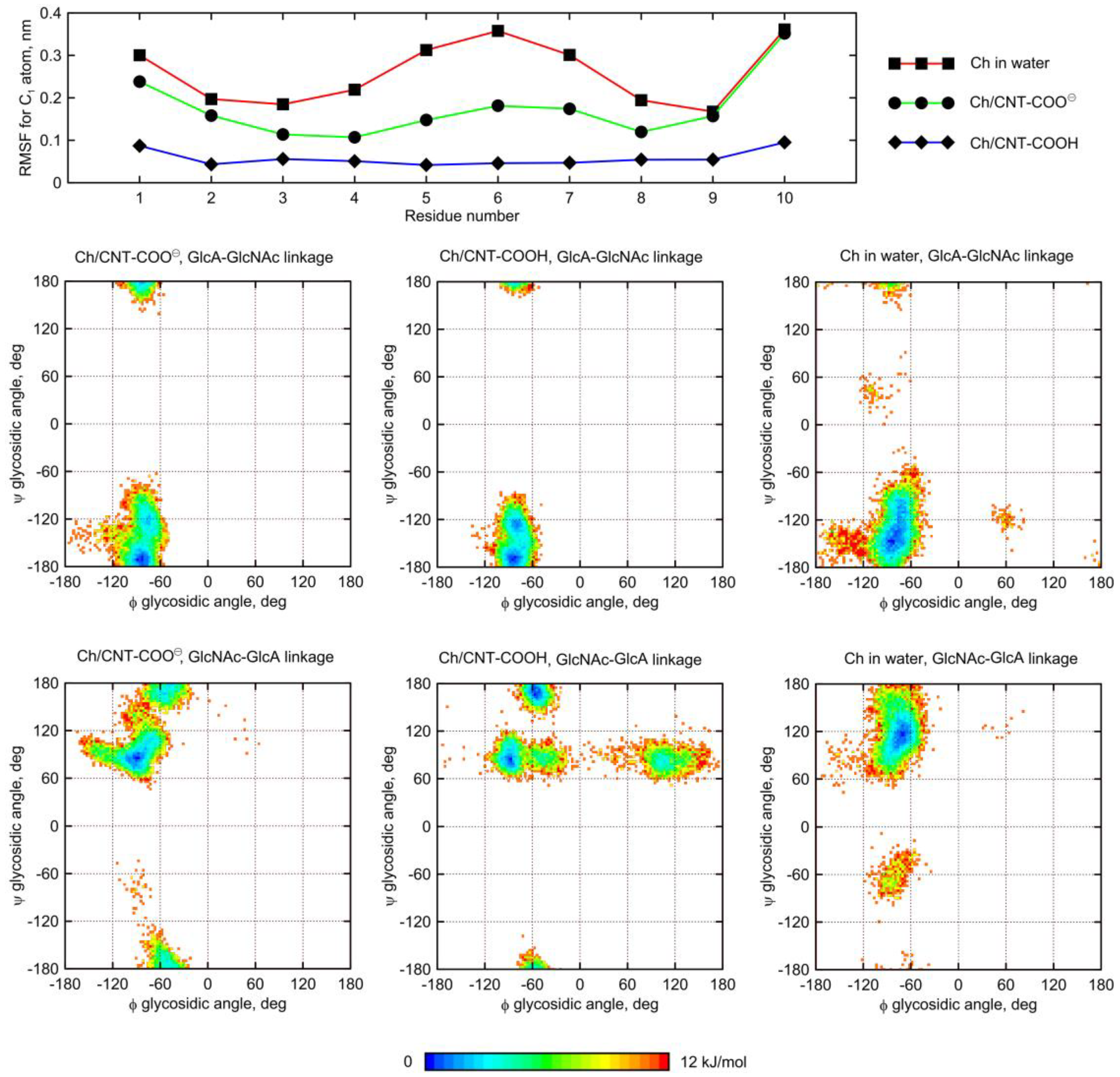
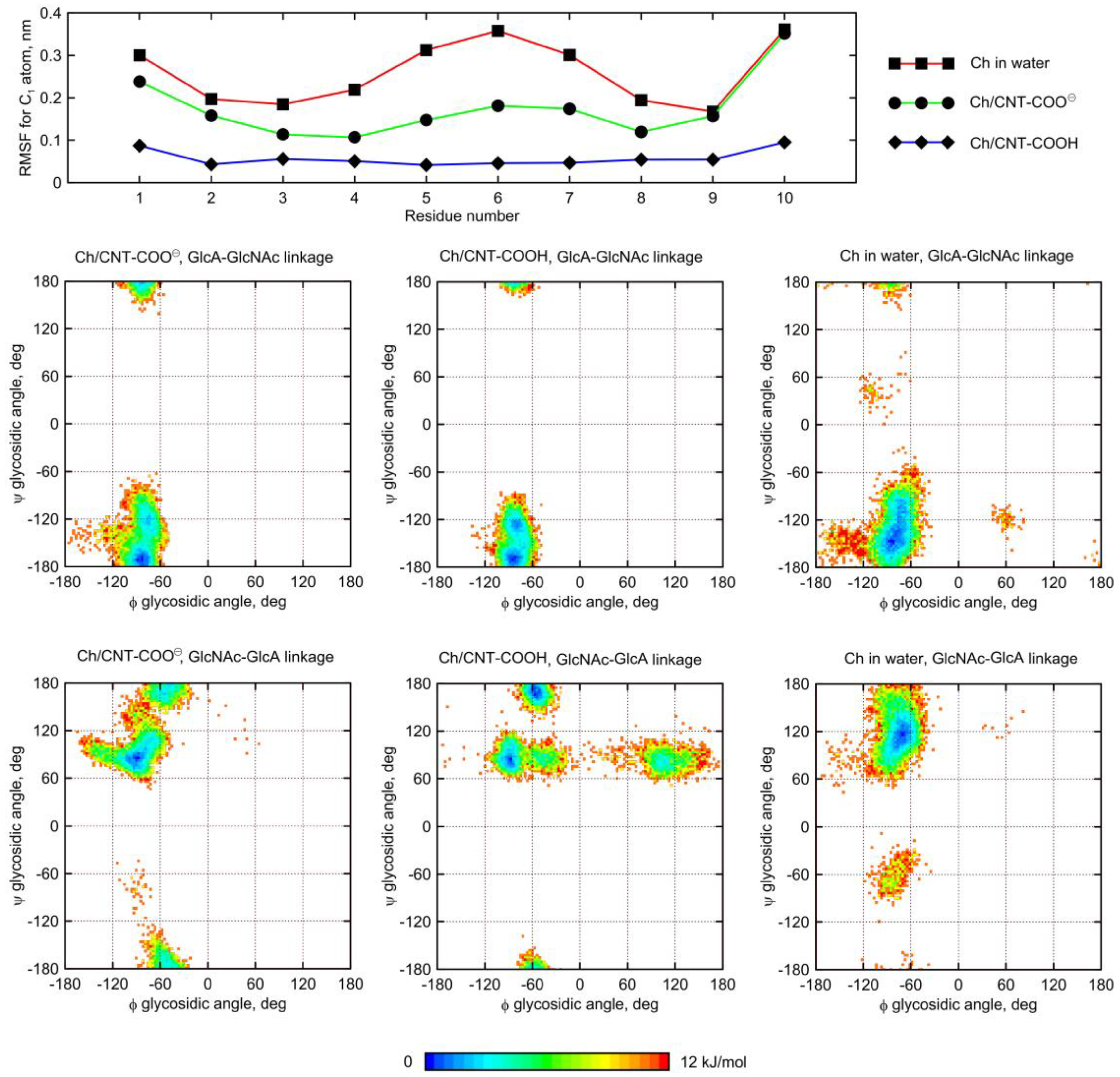
2.5. Influence of Glycan Adsorption on Its Conformation
3. Methods
3.1. Carbon Nanotubes
3.2. Carbohydrates
3.3. Computational Methodology
4. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Battigelli, A.; Ménard-Moyon, C.; Da Ros, T.; Prato, M.; Bianco, A. Endowing Carbon Nanotubes with Biological and Biomedical Properties by Chemical Modifications. Adv. Drug Deliv. Rev. 2013, 65, 1899–1920. [Google Scholar] [CrossRef] [PubMed]
- Morales-Torres, S.; Silva, T.L.S.; Pastrana-Martínez, L.M.; Brandão, A.T.S.C.; Figueiredo, J.L.; Silva, A.M.T. Modification of the Surface Chemistry of Single- and Multi-Walled Carbon Nanotubes by HNO3 and H2SO4 Hydrothermal Oxidation for Application in Direct Contact Membrane Distillation. Phys. Chem. Chem. Phys. 2014, 16, 12237–12250. [Google Scholar] [CrossRef] [PubMed]
- Kan, K.; Xia, T.; Li, L.; Bi, H.; Fu, H.; Shi, K. Amidation of Single-Walled Carbon Nanotubes by a Hydrothermal Process for the Electrooxidation of Nitric Oxide. Nanotechnology 2009, 20, 185502. [Google Scholar] [CrossRef] [PubMed]
- Kierkowicz, M.; Pach, E.; Santidrián, A.; Sandoval, S.; Gonçalves, G.; Tobías-Rossell, E.; Kalbáč, M.; Ballesteros, B.; Tobias, G. Comparative Study of Shortening and Cutting Strategies of Single-Walled and Multi-Walled Carbon Nanotubes Assessed by Scanning Electron Microscopy. Carbon 2018, 139, 922–932. [Google Scholar] [CrossRef]
- Kostarelos, K. The Long and Short of Carbon Nanotube Toxicity. Nat. Biotechnol. 2008, 26, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Plaza, E.; Shaukat, A.; Lehtonen, I.; Kostiainen, M.A. Biomolecule-Directed Carbon Nanotube Self-Assembly. Adv. Healthc. Mater. 2021, 10, 2001162. [Google Scholar] [CrossRef]
- Mu, B.; Zhang, J.; McNicholas, T.P.; Reuel, N.F.; Kruss, S.; Strano, M.S. Recent Advances in Molecular Recognition Based on Nanoengineered Platforms. Acc. Chem. Res. 2014, 47, 979–988. [Google Scholar] [CrossRef]
- Di Crescenzo, A.; Ettorre, V.; Fontana, A. Non-Covalent and Reversible Functionalization of Carbon Nanotubes. Beilstein J. Nanotechnol. 2014, 5, 1675–1690. [Google Scholar] [CrossRef]
- Jagusiak, A.; Goclon, J.; Panczyk, T. Adsorption of Evans Blue and Congo Red on Carbon Nanotubes and Its Influence on the Fracture Parameters of Defective and Functionalized Carbon Nanotubes Studied Using Computational Methods. Appl. Surf. Sci. 2021, 539, 148236. [Google Scholar] [CrossRef]
- Polaczek, E.; Stobinski, L.; Mazurkiewicz, J.; Tomasik, P.; Koloczek, H.; Lin, H.-M. Interactions of Anionic Polysaccharides with Carbon Nanotubes. Polimery 2007, 52, 34–38. [Google Scholar] [CrossRef]
- Panczyk, T.; Plazinski, W.; Brzyska, A.; Wolski, P. Adsorption of Hyaluronan Saccharides on the Surface of a Single Walled Carbon Nanotube. A Computational Study. Appl. Surf. Sci. 2022, 584, 152599. [Google Scholar] [CrossRef]
- Plazinska, A.; Plazinski, W. Comparison of Carbohydrate Force Fields in Molecular Dynamics Simulations of Protein–Carbohydrate Complexes. J. Chem. Theory Comput. 2021, 17, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Hudson, K.L.; Bartlett, G.J.; Diehl, R.C.; Agirre, J.; Gallagher, T.; Kiessling, L.L.; Woolfson, D.N. Carbohydrate–Aromatic Interactions in Proteins. J. Am. Chem. Soc. 2015, 137, 15152–15160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsyk, V.; Plazinski, W. Conformational Properties of Glycosaminoglycan Disaccharides: A Molecular Dynamics Study. J. Phys. Chem. B 2021, 125, 10900–10916. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.C.; Harris, P.J.F.; Green, M.L.H. Thinning and Opening of Carbon Nanotubes by Oxidation Using Carbon Dioxide. Nature 1993, 362, 520–522. [Google Scholar] [CrossRef]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. Tools: Advances in RESP and ESP Charge Derivation and Force Field Library Building. Phys. Chem. Chem. Phys. 2010, 12, 7821. [Google Scholar] [CrossRef] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.-Y.R.E.D. Server: A Web Service for Deriving RESP and ESP Charges and Building Force Field Libraries for New Molecules and Molecular Fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef] [Green Version]
- Spiwok, V. CH/π Interactions in Carbohydrate Recognition. Molecules 2017, 22, 1038. [Google Scholar] [CrossRef] [Green Version]
- Kalinichev, A.G.; Bass, J.D. Hydrogen Bonding in Supercritical Water. 2. Computer Simulations. J. Phys. Chem. A 1997, 101, 9720–9727. [Google Scholar] [CrossRef]
- Yu, Y.; Delbianco, M. Conformational Studies of Oligosaccharides. Chem. Eur. J. 2020, 26, 9814–9825. [Google Scholar] [CrossRef] [PubMed]
- Tipson, R.S. Advances in Carbohydrate Chemistry and Biochemistry, 47; Elsevier: Burlington, UK, 1989; ISBN 978-0-12-007247-7. [Google Scholar]
- Plazinski, W.; Plazinska, A. Molecular Dynamics Simulations of Hexopyranose Ring Distortion in Different Force Fields. Pure Appl. Chem. 2017, 89, 1283–1294. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A Reactive Potential for Hydrocarbons with Intermolecular Interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates: GLYCAM06. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
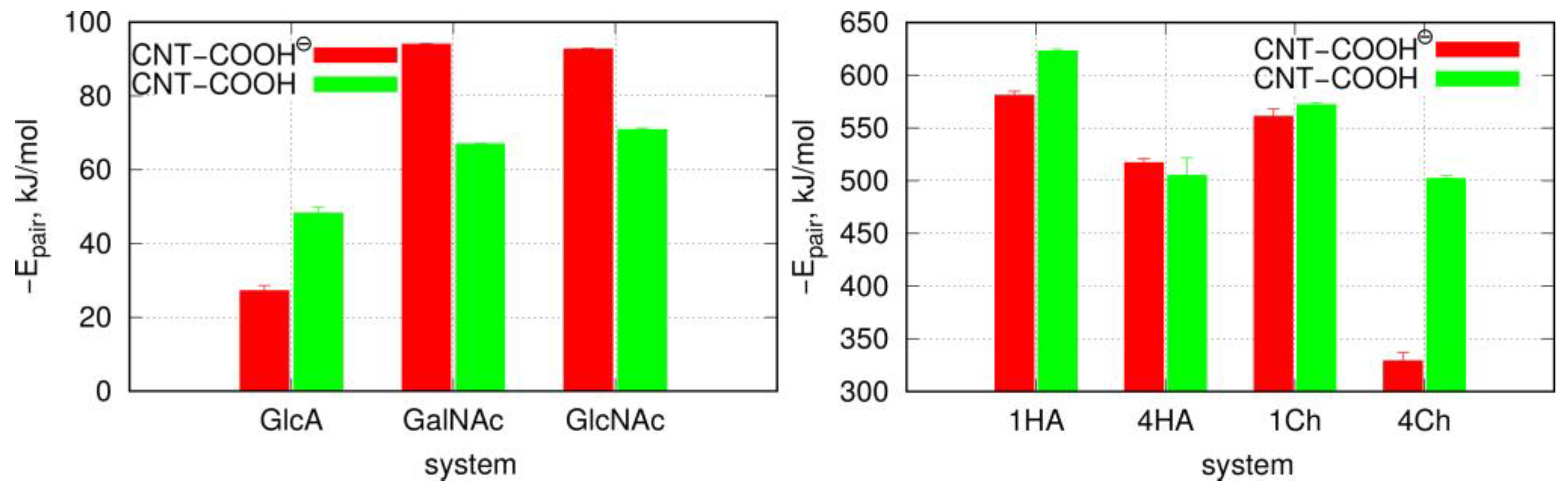
| Molecular Systems | Lennard–Jones (kJ mol−1) | Coulomb (kJ mol−1) | Total (kJ mol−1) |
|---|---|---|---|
| GlcA/CNT-COO⊖ | −40.1 ± 1.4 | 12.9 ± 1.4 | −27.2 ± 1.4 |
| GlcA/CNT-COOH | −49.6 ± 1.6 | 1.5 ± 0.4 | −48.2 ± 1.6 |
| GalNAc/CNT-COO⊖ | −67.1 ± 0.2 | −26.9 ± 0.2 | −94.0 ± 0.2 |
| GalNAc/CNT-COOH | −61.1 ± 0.1 | −6.0 ± 0.1 | −67.0 ± 0.1 |
| GlcNAc/CNT-COO⊖ | −67.2 ± 0.2 | −25.6 ± 0.1 | −92.7 ± 0.2 |
| GlcNAc/CNT-COOH | −65.0 ± 0.4 | −5.9 ± 0.1 | −70.9 ± 0.4 |
| 4HA/CNT-COO⊖ | −486 ± 4.0 | −30.6 ± 0.1 | −517 ± 4.0 |
| 1HA/CNT-COO⊖ | −570 ± 1.5 | −11.0 ± 3.5 | −581 ± 3.8 |
| 4HA/CNT-COOH | −486 ± 16.5 | −18.3 ± 1.8 | −505 ± 16.6 |
| 1HA/CNT-COOH | −606 ± 1.0 | −17.0 ± 1.6 | −623 ± 1.9 |
| 4Ch/CNT-COO⊖ | −299 ± 3.3 | −30.0 ± 7.3 | −329 ± 8.0 |
| 1Ch/CNT-COO⊖ | −519 ± 5.2 | −42.0 ± 4.8 | −561 ± 7.1 |
| 4Ch/CNT-COOH | −479 ± 2.7 | −24.0 ± 1.0 | −502 ± 2.9 |
| 1Ch/CNT-COOH | −546 ± 1.6 | −26.0 ± 0.9 | −572 ± 1.8 |
| CNT-COO⊖ | CNT-COOH | ||
|---|---|---|---|
| HB Acceptor | HB Acceptor | HB Donor | |
| GlcA | 2.51 | 0.47 | 0.02 |
| GalNAc | 8.71 | 1.03 | 0.02 |
| GlcNAc | 9.10 | 1.01 | 0.03 |
| 4HA | 3.24 | 0.51 | 0.02 |
| 1HA | 0.62 | 0.05 | 0.002 |
| 4Ch | 2.61 | 0.56 | 0.03 |
| 1Ch | 0.89 | 0.19 | 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panczyk, T.; Plazinski, W.; Dupradeau, F.-Y.; Brzyska, A.; Wolski, P. Interaction of Chondroitin and Hyaluronan Glycosaminoglycans with Surfaces of Carboxylated Carbon Nanotubes Studied Using Molecular Dynamics Simulations. Molecules 2023, 28, 826. https://doi.org/10.3390/molecules28020826
Panczyk T, Plazinski W, Dupradeau F-Y, Brzyska A, Wolski P. Interaction of Chondroitin and Hyaluronan Glycosaminoglycans with Surfaces of Carboxylated Carbon Nanotubes Studied Using Molecular Dynamics Simulations. Molecules. 2023; 28(2):826. https://doi.org/10.3390/molecules28020826
Chicago/Turabian StylePanczyk, Tomasz, Wojciech Plazinski, François-Yves Dupradeau, Agnieszka Brzyska, and Pawel Wolski. 2023. "Interaction of Chondroitin and Hyaluronan Glycosaminoglycans with Surfaces of Carboxylated Carbon Nanotubes Studied Using Molecular Dynamics Simulations" Molecules 28, no. 2: 826. https://doi.org/10.3390/molecules28020826
APA StylePanczyk, T., Plazinski, W., Dupradeau, F. -Y., Brzyska, A., & Wolski, P. (2023). Interaction of Chondroitin and Hyaluronan Glycosaminoglycans with Surfaces of Carboxylated Carbon Nanotubes Studied Using Molecular Dynamics Simulations. Molecules, 28(2), 826. https://doi.org/10.3390/molecules28020826