
In Silico Identification of 1-DTP Inhibitors of Corynebacterium diphtheriae Using Phytochemicals from Andrographis paniculata

 ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussions
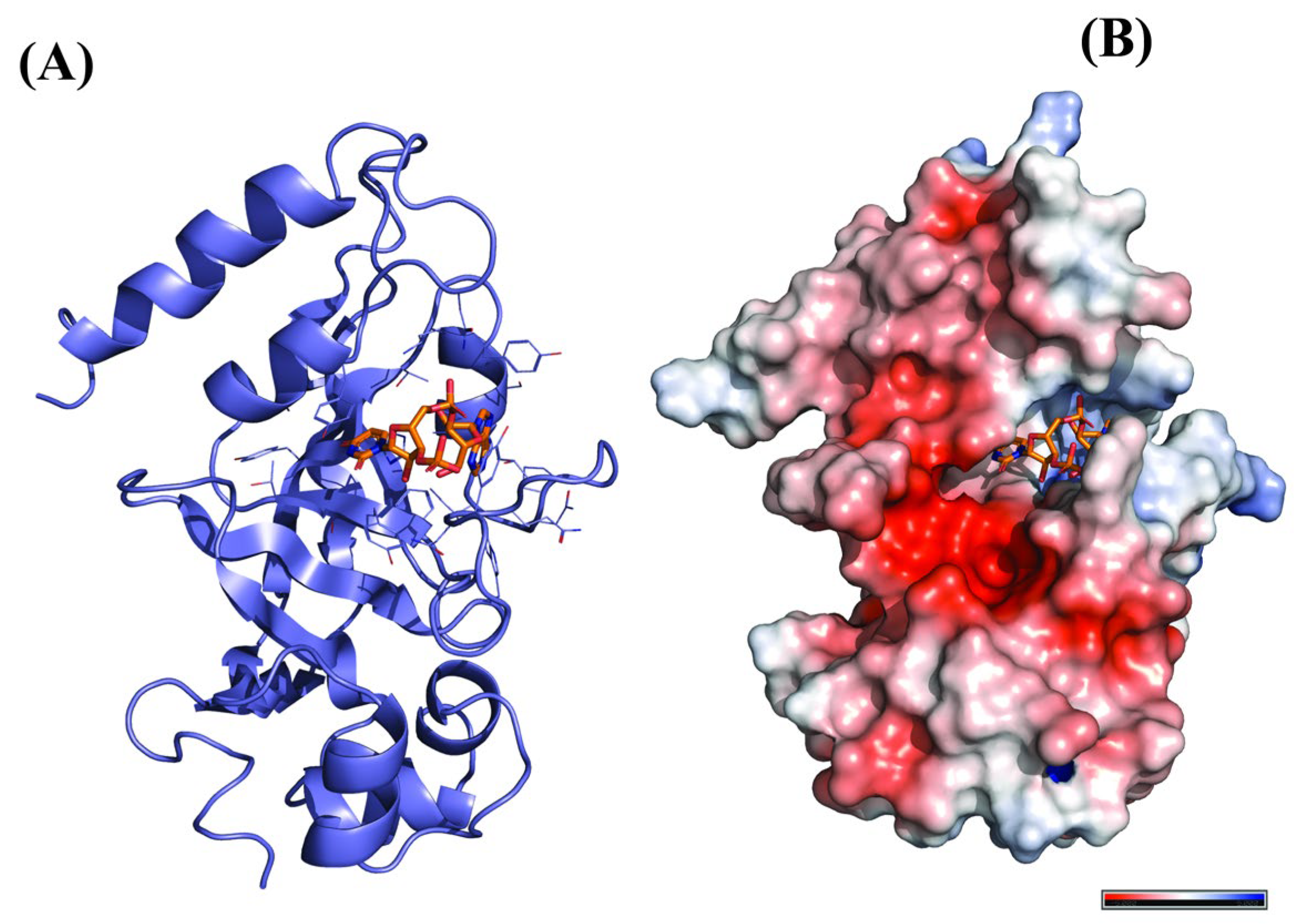
2.1. Molecular Docking Analysis
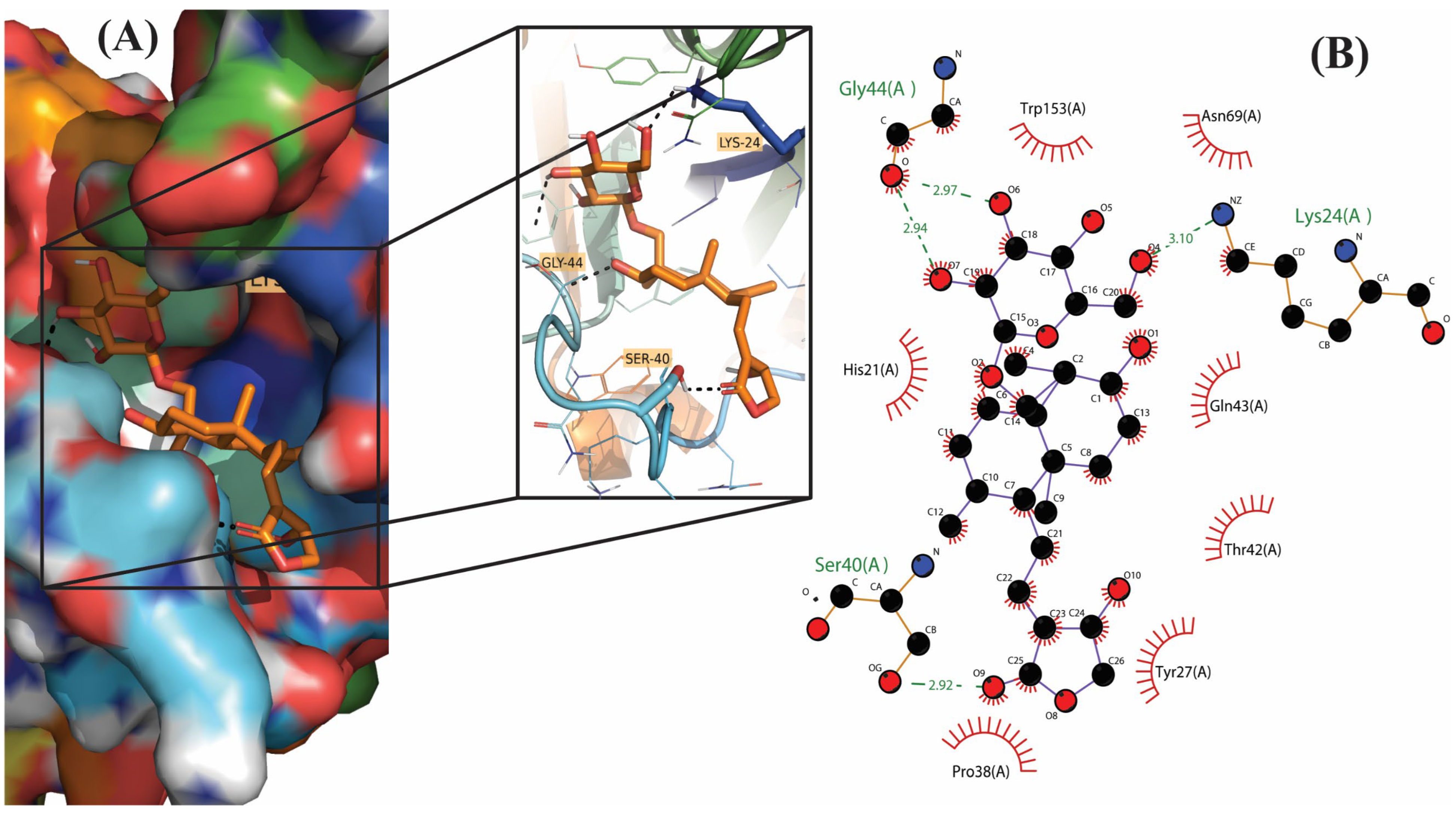
2.2. Molecular Interactions
2.3. Pharmacodynamic Analysis and Toxicity Studies
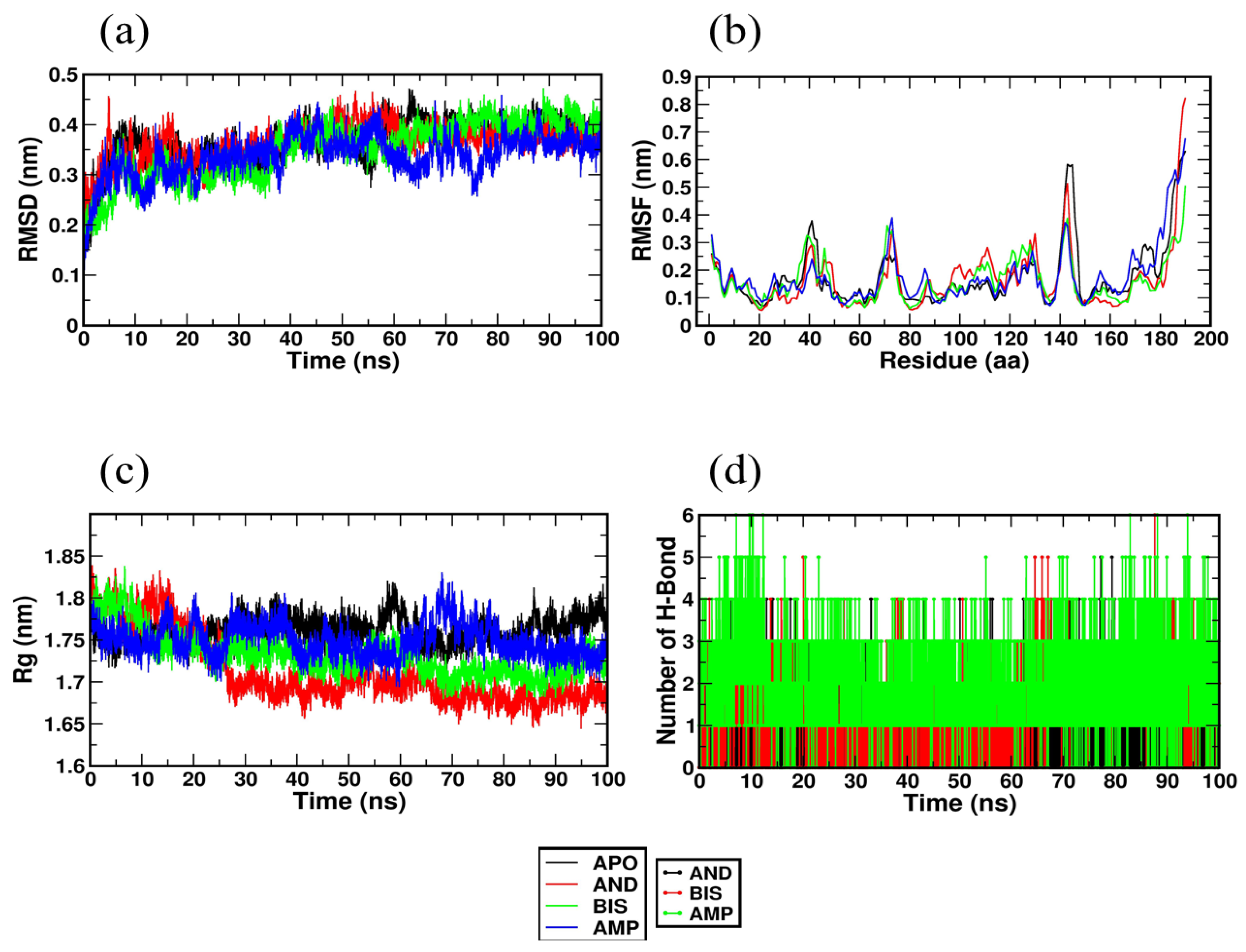
2.4. Molecular Dynamics Simulation Studies
3. Materials and Methods
3.1. Selection and Preparation of Ligands
3.2. Selection and Preparation of Receptor Protein
3.3. Analysis of Target Active Binding Sites
3.4. Molecular Docking
3.5. Pharmacodynamic Analysis and Toxicity Studies
3.6. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chugh, T.D. Emerging and Re-Emerging Bacterial Diseases in India. J. Biosci. 2008, 33, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotriffer, C.; Klebe, G. Identification and Mapping of Small-Molecule Binding Sites in Proteins: Computational Tools for Structure-Based Drug Design. Farmaco 2002, 57, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Sharff, A.; Jhoti, H. High-Throughput Crystallography to Enhance Drug Discovery. Curr. Opin. Chem. Biol. 2003, 7, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-Based Drug Design Strategies and Challenges. Curr. Top. Med. Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Salum, L.B.; Polikarpov, I.; Andricopulo, A.D. Structure-Based Approach for the Study of Estrogen Receptor Binding Affinity and Subtype Selectivity. J. Chem. Inf. Model. 2008, 48, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M. Computer-Aided Drug Discovery and Development (CADDD): In Silico-Chemico-Biological Approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [Green Version]
- Paramashivam, S.K.; Elayaperumal, K.; Natarajan, B.B.; Ramamoorthy, M.D.; Balasubramanian, S.; Dhiraviam, K.N. In Silico Pharmacokinetic and Molecular Docking Studies of Small Molecules Derived from Indigofera Aspalathoides Vahl Targeting Receptor Tyrosine Kinases. Bioinformation 2015, 11, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Enmozhi, S.K.; Raja, K.; Sebastine, I.; Joseph, J. Andrographolide as a Potential Inhibitor of SARS-CoV-2 Main Protease: An in Silico Approach. J. Biomol. Struct. Dyn. 2021, 39, 3092–3098. [Google Scholar] [CrossRef]
- Herowati, R.; Widodo, G.P. Molecular Docking Studies of Chemical Constituents of Tinospora cordifolia on Glycogen Phosphorylase. Procedia Chem. 2014, 13, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.F. Robbins and Cotran’s Pathologic Basis of Disease, 8th ed. Am. J. Surg. Pathol. 2010, 34, 132. [Google Scholar] [CrossRef] [Green Version]
- Cerdeño-Tárraga, A.M.; Efstratiou, A.; Dover, L.G.; Holden, M.T.G.; Pallen, M.; Bentley, S.D.; Besra, G.S.; Churcher, C.; James, K.D.; De Zoysa, A.; et al. The Complete Genome Sequence and Analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. 2003, 31, 6516–6523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadfield, T.L.; McEvoy, P.; Polotsky, Y.; Tzinserling, V.A.; Yakovlev, A.A. The Pathology of Diphtheria. J. Infect. Dis. 2000, 181 (Suppl. S1), S116–S120. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M.; Uchida, T.; Singer, R.A. Expression of Diphtheria Toxin Genes Carried by Integrated and Nonintegrated Phage Beta. Virology 1972, 50, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Kaczorek, M.; Delpeyroux, F.; Chenciner, N.; Streeck, R.E.; Murphy, J.R.; Boquet, P.; Tiollais, P. Nucleotide Sequence and Expression of the Diphtheria tox228 Gene in Escherichia coli. Science 1983, 221, 855–858. [Google Scholar] [CrossRef]
- Ratti, G.; Rappuoli, R.; Giannini, G. The Complete Nucleotide Sequence of the Gene Coding for Diphtheria Toxin in the Corynephage Omega (tox+) Genome. Nucleic Acids Res. 1983, 11, 6589–6595. [Google Scholar] [CrossRef] [Green Version]
- Draper, R.K.; Simon, M.I. The Entry of Diphtheria Toxin into the Mammalian Cell Cytoplasm: Evidence for Lysosomal Involvement. J. Cell Biol. 1980, 87, 849–854. [Google Scholar] [CrossRef]
- Drazin, R.; Kandel, J.; Collier, R.J. Structure and Activity of Diphtheria Toxin. J. Biol. Chem. 1971, 246, 1504–1510. [Google Scholar] [CrossRef]
- Clarke, K.E.N.; MacNeil, A.; Hadler, S.; Scott, C.; Tiwari, T.S.P.; Cherian, T. Global Epidemiology of Diphtheria, 2000–2017. Emerg. Infect. Dis. 2019, 25, 1834–1842. [Google Scholar] [CrossRef]
- Murhekar, M. Epidemiology of Diphtheria in India, 1996-2016: Implications for Prevention and Control. Am. J. Trop. Med. Hyg. 2017, 97, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Diphtheria—Chapter 4—2020 Yellow Book Travelers’ Health|CDC. Available online: https://wwwnc.cdc.gov/travel/yellowbook/2020/travel-related-infectious-diseases/diphtheria (accessed on 12 May 2021).
- Adler, N.R.; Mahony, A.; Friedman, N.D. Diphtheria: Forgotten, but Not Gone. Intern. Med. J. 2013, 43, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Nishiuchi, Y.; Maekura, R.; Kitada, S.; Tamaru, A.; Taguri, T.; Kira, Y.; Hiraga, T.; Hirotani, A.; Yoshimura, K.; Miki, M.; et al. The Recovery of Mycobacterium Avium-Intracellulare Complex (MAC) from the Residential Bathrooms of Patients with Pulmonary MAC. Clin. Infect. Dis. 2007, 45, 347–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambray, G.; Guerout, A.-M.; Mazel, D. Integrons. Annu. Rev. Genet. 2010, 44, 141–166. [Google Scholar] [CrossRef]
- Karimi, A.; Majlesi, M.; Rafieian-Kopaei, M. Herbal versus Synthetic Drugs; Beliefs and Facts. J. Nephropharmacol. 2015, 4, 27–30. [Google Scholar]
- D’Onofrio, G.; Sancarlo, D.; Ruan, Q.; Yu, Z.; Panza, F.; Daniele, A.; Greco, A.; Seripa, D. Phytochemicals in the Treatment of Alzheimer’s Disease: A Systematic Review. Curr. Drug Targets 2017, 18, 1487–1498. [Google Scholar] [CrossRef]
- Ranjan, A.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Wright, S.; Srivastava, S.; Das, H.; Srivastava, S.; Prasad, S.; Srivastava, S.K. Role of Phytochemicals in Cancer Prevention. Int. J. Mol. Sci. 2019, 20, 4981. [Google Scholar] [CrossRef] [Green Version]
- Howes, M.-J.R.; Perry, E. The Role of Phytochemicals in the Treatment and Prevention of Dementia. Drugs Aging 2011, 28, 439–468. [Google Scholar] [CrossRef]
- Gaikwad, S.B.; Krishna Mohan, G.; Rani, M.S. Phytochemicals for Diabetes Management. Pharm. Crop. 2014, 5, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Pop, R.M.; Popolo, A.; Trifa, A.P.; Stanciu, L.A. Phytochemicals in Cardiovascular and Respiratory Diseases: Evidence in Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2018, 2018, 1603872. [Google Scholar] [CrossRef]
- Dai, Y.; Chen, S.-R.; Chai, L.; Zhao, J.; Wang, Y.; Wang, Y. Overview of Pharmacological Activities of and Its Major Compound Andrographolide. Crit. Rev. Food Sci. Nutr. 2019, 59, S17–S29. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Araújo, T.A.; de Melo, J.G.; Júnior, W.S.F.; Albuquerque, U.P. Medicinal plants. In Introduction to Ethnobiology; Springer International Publishing: Cham, Switzerland, 2016; pp. 143–149. ISBN 9783319281537. [Google Scholar]
- Cheung, H.-Y.; Cheung, S.-H.; Li, J.; Cheung, C.-S.; Lai, W.-P.; Fong, W.-F.; Leung, F.-M. Andrographolide Isolated from Andrographis paniculata Induces Cell Cycle Arrest and Mitochondrial-Mediated Apoptosis in Human Leukemic HL-60 Cells. Planta Med. 2005, 71, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Sheeja, K.; Shihab, P.K.; Kuttan, G. Antioxidant and Anti-Inflammatory Activities of the Plant Andrographis paniculata Nees. Immunopharmacol. Immunotoxicol. 2006, 28, 129–140. [Google Scholar] [CrossRef]
- Yu, B.-C.; Hung, C.-R.; Chen, W.-C.; Cheng, J.-T. Antihyperglycemic Effect of Andrographolide in Streptozotocin-Induced Diabetic Rats. Planta Med. 2003, 69, 1075–1079. [Google Scholar] [PubMed]
- Misra, P.; Pal, N.L.; Guru, P.Y.; Katiyar, J.C.; Srivastava, V.; Tandon, J.S. Antimalarial Activity of Andrographis paniculata (Kalmegh) against Plasmodium berghei NK 65 in Mastomys Natalensis. Int. J. Pharmacogn. 1992, 30, 263–274. [Google Scholar] [CrossRef]
- Chao, W.-W.; Lin, B.-F. Isolation and Identification of Bioactive Compounds in Andrographis paniculata (Chuanxinlian). Chin. Med. 2010, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Naglich, J.G.; Metherall, J.E.; Russell, D.W.; Eidels, L. Expression Cloning of a Diphtheria Toxin Receptor: Identity with a Heparin-Binding EGF-like Growth Factor Precursor. Cell 1992, 69, 1051–1061. [Google Scholar] [CrossRef]
- Yates, S.P.; Jørgensen, R.; Andersen, G.R.; Merrill, A.R. Stealth and Mimicry by Deadly Bacterial Toxins. Trends Biochem. Sci. 2006, 31, 123–133. [Google Scholar] [CrossRef]
- Subramaniyan, V.; Sekar, R.; Praveenkumar, A.; Selvam, R. Molecular Modeling Studies of Repandusinic Acid as Potent Small Molecule for Hepatitis B Virus through Molecular Docking and ADME Analysis. Quant. Biol. 2019, 7, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Chakotiya, A.S.; Chawla, R.; Tanwar, A.; Sharma, A.; Sharma, R.K. Molecular Docking Analysis—An aid for Selection of Promising Natural Plant Products against Diphtheria Toxin. JSM Chem. 2016, 4, 1017. [Google Scholar]
- Acharya, D.R.R. A Review of Phytoconstituents and Their Pharmacological Properties of Andrographis paniculata (nees). Int. J. Pharma Bio Sci. 2017, 8, 77–83. [Google Scholar] [CrossRef]
- Jadhav, A.K.; Karuppayil, S.M. Andrographis paniculata (Burm. F) Wall Ex Nees: Antiviral Properties. Phytother. Res. 2021, 35, 5365–5373. [Google Scholar] [CrossRef] [PubMed]
- Thirumoorthy, G.; Tarachand, S.P.; Nagella, P.; Veerappa Lakshmaiah, V. Identification of Potential ZIKV NS2B-NS3 Protease Inhibitors from Andrographis paniculata: An Insilico Approach. J. Biomol. Struct. Dyn. 2021, 40, 11203–11215. [Google Scholar] [CrossRef] [PubMed]
- Ferreira de Freitas, R.; Schapira, M. A Systematic Analysis of Atomic Protein-Ligand Interactions in the PDB. Medchemcomm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nittinger, E.; Inhester, T.; Bietz, S.; Meyder, A.; Schomburg, K.T.; Lange, G.; Klein, R.; Rarey, M. Large-Scale Analysis of Hydrogen Bond Interaction Patterns in Protein-Ligand Interfaces. J. Med. Chem. 2017, 60, 4245–4257. [Google Scholar] [CrossRef]
- Hendsch, Z.S.; Tidor, B. Do Salt Bridges Stabilize Proteins? A Continuum Electrostatic Analysis. Protein Sci. 1994, 3, 211–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldburger, C.D.; Schildbach, J.F.; Sauer, R.T. Are Buried Salt Bridges Important for Protein Stability and Conformational Specificity? Nat. Struct. Biol. 1995, 2, 122–128. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Chang, L.C.W.; Spanjersberg, R.F.; von Frijtag Drabbe Künzel, J.K.; Mulder-Krieger, T.; van den Hout, G.; Beukers, M.W.; Brussee, J.; Ijzerman, A.P. 2,4,6-Trisubstituted Pyrimidines as a New Class of Selective Adenosine A1 Receptor Antagonists. J. Med. Chem. 2004, 47, 6529–6540. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Clark, D.E. Rapid Calculation of Polar Molecular Surface Area and Its Application to the Prediction of Transport Phenomena. 1. Prediction of Intestinal Absorption. J. Pharm. Sci. 1999, 88, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Variya, B.C.; Modi, S.J.; Savjani, J.K.; Patel, S.S. In Silico Molecular Docking and Pharmacokinetic Prediction of Gallic Acid Derivatives as Ppar-γ Agonists. Int. J. Pharm. Pharm. Sci. 2016, 9, 102. [Google Scholar] [CrossRef]
- Ya’u Ibrahim, Z.; Uzairu, A.; Shallangwa, G.; Abechi, S. Molecular Docking Studies, Drug-Likeness and in-Silico ADMET Prediction of Some Novel β-Amino Alcohol Grafted 1,4,5-Trisubstituted 1,2,3-Triazoles Derivatives as Elevators of p53 Protein Levels. Sci. Afr. 2020, 10, e00570. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.; Price, A. The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects. Am. Fam. Physician. 2007, 76, 391–396. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Ligands | Binding Affinity (Kcal/Mol) |
|---|---|
| 7-methylwogonin | −8.5 |
| Apigenin | −8.4 |
| Luteolin | −7.6 |
| Andrographidine C | −8.7 |
| Andrographolide | −8.5 |
| Neoandrographolide | −9.1 |
| 3-O-beta-D-Glucopyranosyl-14,19-dideoxyandrographolide | −8.7 |
| 14-deoxyandrographolide | −8.6 |
| Andrograpanin | −8.5 |
| Bisandrographolide | −10.4 |
| Onysilin | −8.2 |
| Andrographidin A | −8.4 |
| 14-Deoxy-12-Hydroxyandrographolide | −8.4 |
| Andrographolactone | −8.6 |
| 8,17-Epoxy-14-Deoxyandrographolide | −8.5 |
| Andrographiside | −9.5 |
| 14-Deoxy-11-Hydroxyandrographolide | −8.3 |
| Isoandrographolide | −9.4 |
| Ampicillin | −7.4 |
| Names of Ligands | Binding Affinity (Kcal/mol) | Amino Acids Involved in Interaction | ||
|---|---|---|---|---|
| Hydrogen Bond | Hydrophobic Interactions | Salt-Bridge Interactions | ||
| Neoandrographolide | −9.1 | Tyr (2.68), His (2.74), Gly (1.88), Ser (1.98), Ser (2.41) | Tyr (3.1) | Absent |
| Bisandrographolide | −10.4 | Gln (2.29), Thr (2.09), Asn (2.66) | Tyr (3.68), Ile (3.57), Pro (3.69), Thr (3.98), Tyr (3.66) | His (4.12), His (5.12), Lys (5.12) |
| Andrographiside | −9.5 | Lys (2.38), Lys (2.89), Ser (2.93), Ser (2.15), Gly (2.75), Gly (2.44) | Tyr (3.75), Pro (3.33), Thr (3.89), Trp (3.72) | Absent |
| Isoandrographolide | −9.4 | Gln (2.09), Asn (3.49) | Tyr (3.58), Ile (3.31), Pro (3.64), Trp (3.66) | His (5.35), Lys (4.88), Lys (2.86) |
| Ampicillin | −7.4 | Lys (2.64), Lys (2.17), His (2.17), Asn (3.18) | Thr (3.59), Tyr (3.75), Tyr (3.71), Pro (3.88), Tyr (3.76) | Lys (4.19), Lys (4.12) |
| Name of Ligands | MW (g/mol) | #H-Bond Acceptors | #H-Bond Donors | TPSA (Ų) | iLOGP | ESOL Log S | Lipinski Violations | Lead Likeness Violations |
|---|---|---|---|---|---|---|---|---|
| 7-methylwogonin | 298.29 | 5 | 1 | 68.9 | 2.99 | −4.12 | 0 | 0 |
| Apigenin | 270.24 | 5 | 3 | 90.9 | 1.89 | −3.94 | 0 | 0 |
| Luteolin | 286.24 | 6 | 4 | 111.13 | 1.86 | −3.71 | 0 | 0 |
| Andrographidine C | 460.43 | 10 | 4 | 148.05 | 2 | −3.26 | 0 | 1 |
| Andrographolide | 350.45 | 5 | 3 | 86.99 | 2.45 | −3.18 | 0 | 1 |
| Neoandrographolide | 480.59 | 8 | 4 | 125.68 | 3.27 | −4.01 | 0 | 1 |
| 3-O-beta-D-Glucopyranosyl-14,19-dideoxyandrographolide | 480.59 | 8 | 4 | 125.68 | 3 | −4 | 0 | 1 |
| 14-deoxyandrographolide | 334.45 | 4 | 2 | 66.76 | 2.91 | −3.81 | 0 | 0 |
| Andrograpanin | 318.45 | 3 | 1 | 46.53 | 3.34 | −4.21 | 0 | 1 |
| Bisandrographolide | 664.87 | 8 | 4 | 133.52 | 4.5 | −7.26 | 1 | 3 |
| Onysilin | 300.31 | 5 | 1 | 64.99 | 2.88 | −3.82 | 0 | 0 |
| Andrographidin A | 462.45 | 10 | 4 | 144.14 | 2.13 | −3.01 | 0 | 1 |
| 14-Deoxy-12-Hydroxyandrographolide | 350.45 | 5 | 3 | 86.99 | 2.61 | −3.44 | 0 | 1 |
| Andrographolactone | 296.4 | 2 | 0 | 26.3 | 3.46 | −4.66 | 1 | 1 |
| 8,17-Epoxy-14-Deoxyandrographolide | 350.45 | 5 | 2 | 79.29 | 2.76 | −3.26 | 0 | 1 |
| Andrographiside | 512.59 | 10 | 6 | 166.14 | 2.68 | −2.63 | 2 | 1 |
| 14-Deoxy-11-Hydroxyandrographolide | 350.45 | 5 | 3 | 86.99 | 2.87 | −3.18 | 0 | 1 |
| Isoandrographolide | 350.45 | 5 | 2 | 75.99 | 2.67 | −3.35 | 0 | 1 |
| Ampicillin | 349.40 | 5 | 3 | 138.03 | 1.14 | −1.15 | 0 | 0 |
| Sr. No. | Name of Ligand | Absorption | Distribution | Metabolism | Excretion | Toxicity | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyp | ||||||||||||||||||
| Substrate | Inhibitor | |||||||||||||||||
| Water Solubility | Intestinal Absorption | Skin Permeability | Blood Brain Permeability | Cyp2d6 | Cyp3a4 | Cyp1a2 | Cyp2c19 | Cyp2c9 | Cyp2d6 | Cyp3a4 | Total Clearance | Ames Toxicity | Herg | Max Tolerated Dose | Hepatotoxicity | Skin Sensitization | ||
| 1 | 7-Methylwogonin | −4.12 | High | −5.76 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | 0.46 | No | No/Yes | 0.24 | No | No |
| 2 | Apigenin | −3.94 | High | −5.8 | No | Yes | Yes | Yes | No | No | Yes | Yes | 0.693 | No | No/Yes | 0.713 | No | No |
| 3 | Luteolin | −3.71 | High | −6.25 | No | Yes | Yes | Yes | No | No | Yes | Yes | 0.407 | No | No/Yes | 0.49 | No | No |
| 4 | Andrographidine C | −3.26 | Low | −8.43 | No | No | Yes | No | No | No | No | Yes | 0.55 | No | No/Yes | 0.801 | No | No |
| 5 | Andrographolide | −3.18 | High | −6.9 | No | No | No | No | No | No | No | No | 1.18 | No | No | −0.10 | No | No |
| 6 | Neoandrographolide | −4.01 | High | −7.36 | No | No | Yes | No | No | No | No | Yes | 0.952 | Yes | No | −0.436 | No | No |
| 7 | 3-O-Beta-D-Glucopyranosyl-14,19-Dideoxyandrographolide | −4 | High | −7.46 | No | No | Yes | No | No | No | No | Yes | 0.91 | No | No/Yes | −0.15 | No | No |
| 8 | 14-Deoxyandrographolide | −3.81 | High | −5.9 | Yes | No | No | No | No | No | No | No | −0.84 | Yes | No | 1.37 | No | No |
| 9 | Andrograpanin | −4.21 | High | −5.25 | Yes | No | No | No | Yes | Yes | No | No | 1.11 | No | No | −0.61 | No | No |
| 10 | Bisandrographolide | −7.26 | Low | −6.04 | No | No | No | No | No | No | No | No | 0.225 | No | No | −0.191 | No | No |
| 11 | Onysilin | −3.82 | High | −5.97 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | −30.9 | Yes | No | 0.438 | No | No |
| 12 | Andrographidin A | −3.01 | Low | −8.63 | No | No | No | No | No | No | No | No | −32.9 | Yes | No | 0.438 | No | No |
| 13 | 14-Deoxy-12-Hydroxyandrographolide | −3.44 | High | −6.53 | No | No | No | No | No | No | No | No | 1.2 | No | No | 0.02 | No | No |
| 14 | Andrographolactone | −4.66 | High | −4.76 | Yes | No | No | No | No | Yes | No | No | 1.2 | No | No | 0.2 | No | Yes |
| 15 | 8,17-Epoxy-14-Deoxyandrographolide | −3.26 | High | −6.73 | No | No | No | No | No | No | No | No | −0.43 | Yes | No | 1.39 | No | No |
| 16 | Andrographiside | −2.63 | Low | −9.41 | No | No | No | No | No | No | No | No | 1.033 | Yes | No/Yes | −0.728 | No | No |
| 17 | 14-Deoxy-11-Hydroxyandrographolide | −3.18 | High | −6.83 | No | No | No | No | No | No | No | No | 1.18 | No | No | −0.4 | No | No |
| 18 | Isoandrographolide | −3.18 | High | −6.78 | Yes | No | No | No | No | No | No | No | 0.784 | Yes | No | 0.076 | No | No |
| Control | Ampicillin | −2.577 | High | −2.735 | No | No | No | No | No | No | No | No | 0.455 | No | No | 1.606 | Yes | No |
| Name of Ligand | Pubchem ID | Structure |
|---|---|---|
| 7-methylwogonin | 188316 | 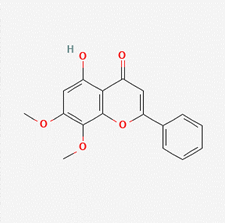 |
| Apigenin | 5280443 | 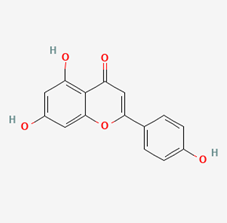 |
| Luteolin | 5280445 |  |
| Andrographidine C | 5318484 |  |
| Andrographolide C | 5318517 |  |
| Neoandrographolide | 9848024 |  |
| 3-O-beta-D-Glucopyranosyl-14,19-dideoxyandrographolide | 11576609 |  |
| 14-deoxyandrographolide | 11624161 |  |
| Andrograpanin | 11666871 | 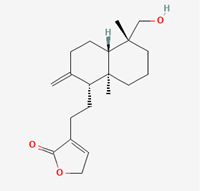 |
| Bisandrographolide | 12000062 |  |
| Onysilin | 12041831 | 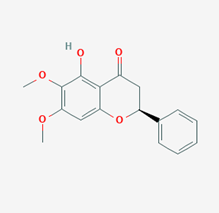 |
| Andrographidin A | 13963762 |  |
| 14-Deoxy-12-Hydroxyandrographolide | 38350563 |  |
| Andrographolactone | 44206466 | 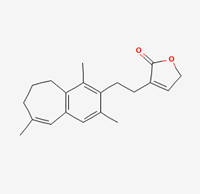 |
| 8,17-Epoxy-14-Deoxyandrographolide | 44575263 |  |
| Andrographiside | 44593583 |  |
| 14-Deoxy-11-Hydroxyandrographolide | 91884987 |  |
| Isoandrographolide | 101563019 |  |
| Ampicillin | 6249 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Khayri, J.M.; Dubey, S.; Thirumoorthy, G.; Nagella, P.; Rezk, A.A.-S.; Shehata, W.F. In Silico Identification of 1-DTP Inhibitors of Corynebacterium diphtheriae Using Phytochemicals from Andrographis paniculata. Molecules 2023, 28, 909. https://doi.org/10.3390/molecules28020909
Al-Khayri JM, Dubey S, Thirumoorthy G, Nagella P, Rezk AA-S, Shehata WF. In Silico Identification of 1-DTP Inhibitors of Corynebacterium diphtheriae Using Phytochemicals from Andrographis paniculata. Molecules. 2023; 28(2):909. https://doi.org/10.3390/molecules28020909
Chicago/Turabian StyleAl-Khayri, Jameel M., Sakshi Dubey, Gopishankar Thirumoorthy, Praveen Nagella, Adel Abdel-Sabour Rezk, and Wael Fathi Shehata. 2023. "In Silico Identification of 1-DTP Inhibitors of Corynebacterium diphtheriae Using Phytochemicals from Andrographis paniculata" Molecules 28, no. 2: 909. https://doi.org/10.3390/molecules28020909
APA StyleAl-Khayri, J. M., Dubey, S., Thirumoorthy, G., Nagella, P., Rezk, A. A.-S., & Shehata, W. F. (2023). In Silico Identification of 1-DTP Inhibitors of Corynebacterium diphtheriae Using Phytochemicals from Andrographis paniculata. Molecules, 28(2), 909. https://doi.org/10.3390/molecules28020909