
A Simple Entry to the 5,8-Disubstituted Indolizidine Skeleton via Hetero Diels-Alder Reaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
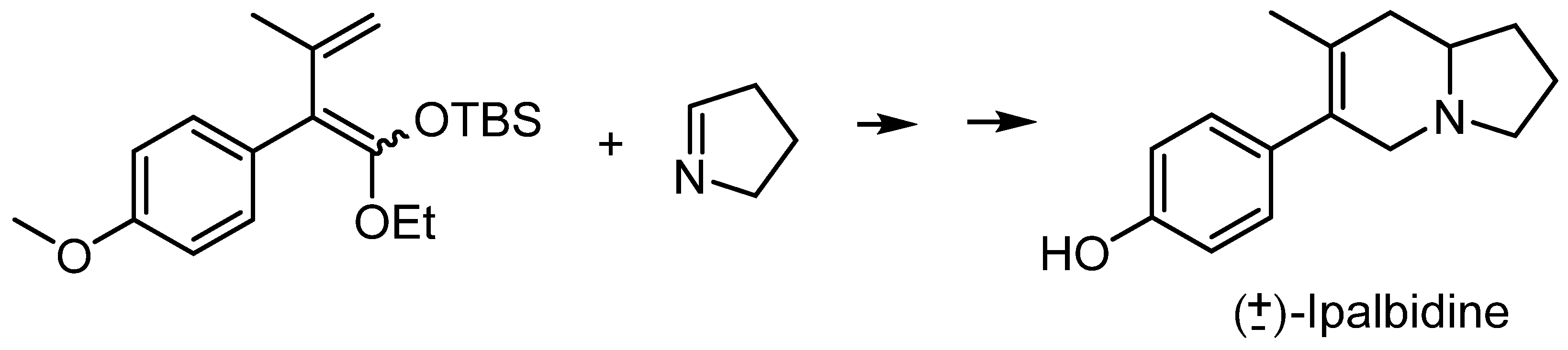
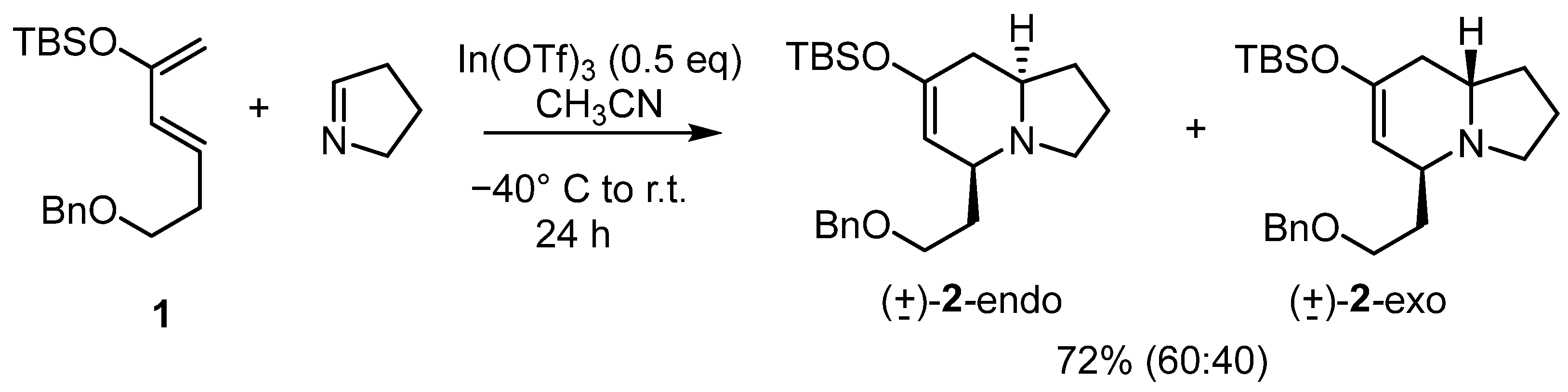
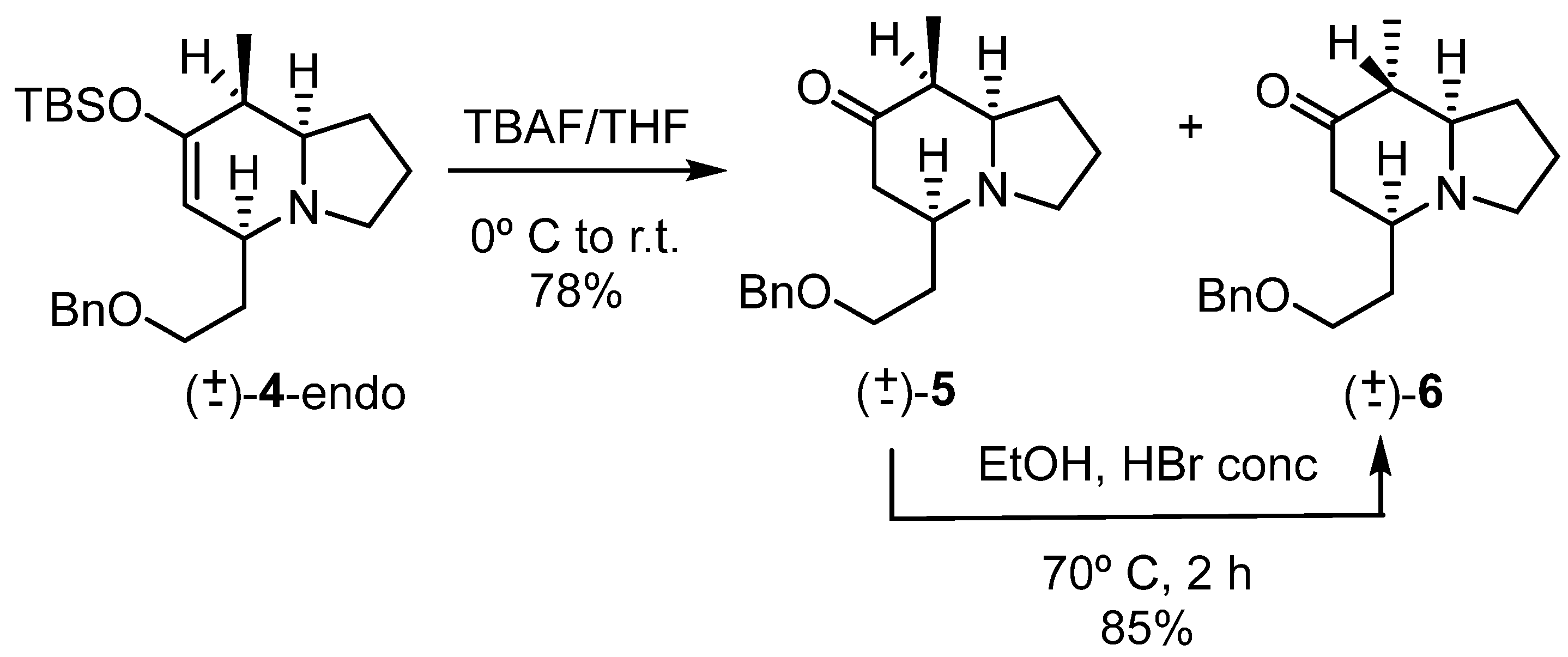
2.1. Synthesis of the First Model: Mono-Substituted Indolizidine Skeleton
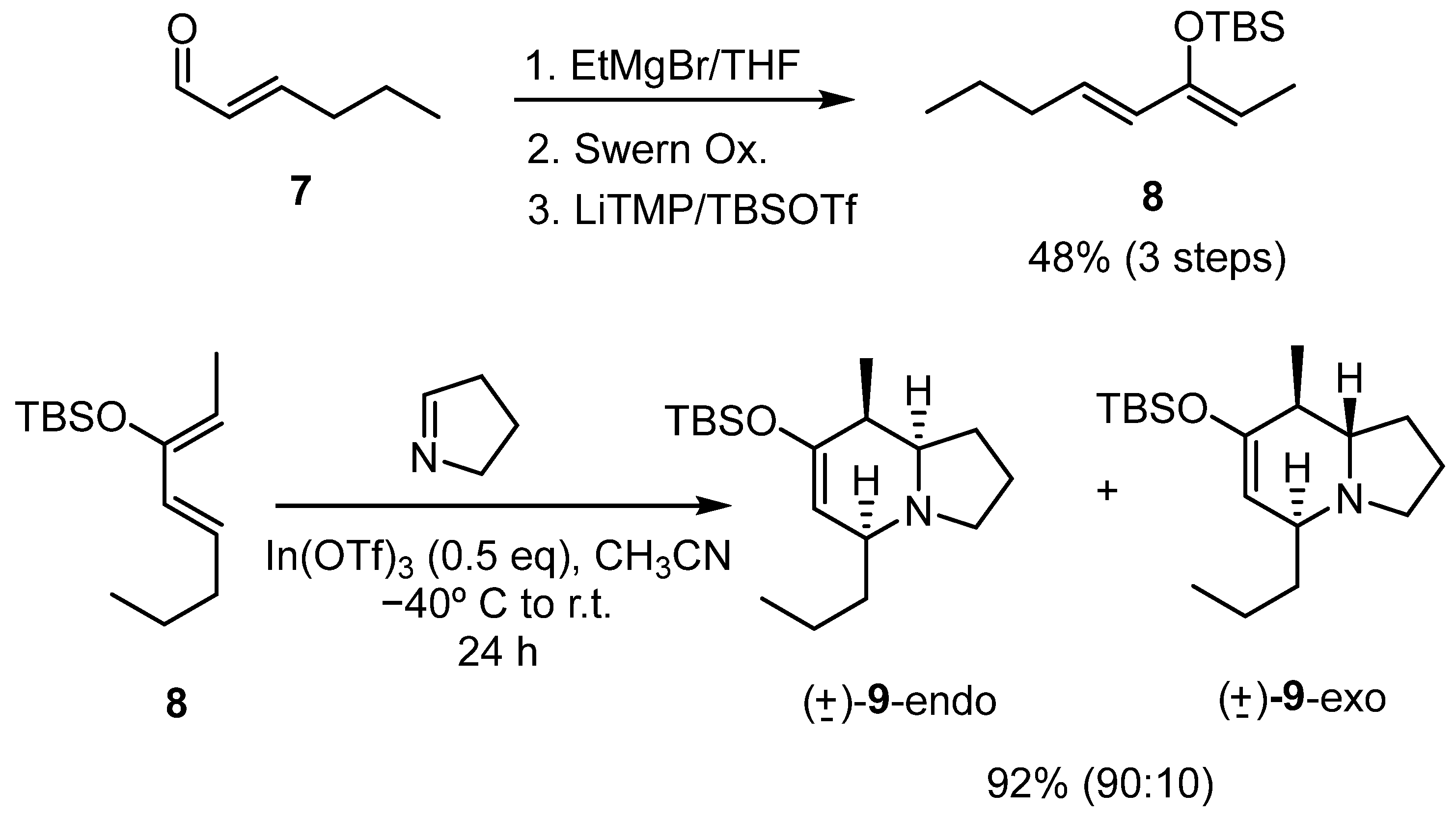
2.2. Synthesis of 8-Methyl-5-Alkyl Indolizidine Skeleton
2.3. Synthesis of Indolizidine Rac-181B
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Compound Synthesis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Michael, J.P. Indolizidine and quinolizidine alkaloids. Nat. Prod. Rep. 2008, 25, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.P. Chapter One-Simple Indolizidine and Quinolizidine Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 75, pp. 1–498. [Google Scholar] [CrossRef]
- De Rop, A.-S.; Rombaut, J.; Willems, T.; De Graeve, M.; Vanhaecke, L.; Hulpiau, P.; De Maeseneire, S.L.; De Mol, M.L.; Soetaert, W.K. Novel Alkaloids from Marine Actinobacteria: Discovery and Characterization. Mar. Drugs 2022, 20, 6. [Google Scholar] [CrossRef]
- Zhang, J.; Morris-Natschke, S.L.; Ma, D.; Shang, X.-F.; Yang, C.-J.; Liu, Y.-Q.; Lee, K.-H. Biologically active indolizidine alkaloids. Med. Res. Rev. 2021, 41, 928–960. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Kamal, R.; Kumar, D.; Kumar, V. Indolizidine Alkaloids: Prospective Lead Molecules in Medicinal Chemistry. Curr. Tradit. Med. 2021, 7, 45–56. [Google Scholar] [CrossRef]
- Daly, J.W.; Spande, T.F.; Garraffo, H.M. Alkaloids from Amphibian Skin: A Tabulation of Over Eight-Hundred Compounds. J. Nat. Prod. 2005, 68, 1556–1575. [Google Scholar] [CrossRef] [PubMed]
- Saporito, R.A.; Donnelly, M.A.; Roy, A.; Norton, R.A.; Garraffo, H.M.; Spande, T.F.; Daly, J.W. Oribatid mites as a major dietary source for alkaloids in poison frogs. Proc. Natl. Acad. Sci. USA 2007, 104, 8885–8890. [Google Scholar] [CrossRef]
- Hantak, M.M.; Grant, T.; Reinsch, S.; Mcginnity, D.; Loring, M.; Toyooka, N.; Saporito, R.A. Dietary Alkaloid Sequestration in a Poison Frog: An Experimental Test of Alkaloid Uptake in Melanophryniscus stelzneri (Bufonidae). J. Chem. Ecol. 2013, 39, 1400–1406. [Google Scholar] [CrossRef]
- Toyooka, N.; Tsuneki, H.; Kobayashi, S.; Dejun, Z.; Kawasaki, M.; Kimura, I.; Sasaoka, T.; Nemoto, H. Synthesis of Poison-Frog Alkaloids and Their Pharmacological Effects at Neuronal Nicotinic Acetylcholine Receptors. Curr. Chem. Biol. 2007, 1, 97–114. [Google Scholar] [CrossRef]
- Daly, J.W.; Nishizawa, Y.; Padgett, W.L.; Tokuyama, T.; Smith, A.L.; Holmes, A.B.; Kibayashi, C.; Aronstam, R.S. 5,8-Disubstituted indolizidines: A new class of noncompetitive blockers for nicotinic receptor-channels. Neurochem. Res. 1991, 16, 1213–1218. [Google Scholar] [CrossRef]
- Ratmanovaa, N.K.; Andreev, I.A.; Leontiev, A.V.; Momotova, D.; Novoselov, A.M.; Ivanova, O.A.; Trushkov, I.V. Strategic approaches to the synthesis of pyrrolizidine and indolizidine alkaloids. Tetrahedron 2020, 76, 131031. [Google Scholar] [CrossRef]
- Danishefsky, S.J.; Vogel, C. Concise Total Synthesis of (±)-Ipalbidine by Application of the Aldimine-Diene Cyclocondensation Reaction. J. Org. Chem. 1986, 51, 3915–3916. [Google Scholar] [CrossRef]
- Shao, J.; Yang, J.-S. A Diastereoselective Cyclic Imine Cycloaddition Strategy To Access Polyhydroxylated Indolizidine Skeleton: Concise Syntheses of (+)-/(−)-Lentiginosines and (−)-2-epi-Steviamine. J. Org. Chem. 2012, 77, 7891–7900. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, E.; Leoni, S.; Abbiati, G.; Pirovano, V.; Rossi, E. Formal Aza-Diels-Alder Reactions of Spiroindolenines with Electronrich Dienes. Eur. J. Org. Chem. 2021, 2021, 2440–2447. [Google Scholar] [CrossRef]
- Del Fiandra, C.; Moccia, M.; Cerullia, V.; Adamo, M.F.A. Catalytic asymmetric conjugate addition of isocyanoacetate to (Z)-3-substituted-2-(4-pyridyl)-acrylonitrile, a reactive class of Michael acceptor. Chem. Commun. 2016, 52, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Szczesniak, P.; Stecko, S.; Maziarz, E.; Staszewska-Krajewska, O.; Furman, B. Synthesis of Polyhydroxylated Quinolizidine and Indolizidine Scaffolds from Sugar-Derived Lactams via a One-Pot Reduction/Mannich/Michael Sequence. J. Org. Chem. 2014, 79, 10487–10503. [Google Scholar] [CrossRef]
- Aoyagi, S.; Hakoishi, M.; Suzuki, M.; Nakanoya, Y.; Shimada, K.; Takikawa, Y. Synthesis of δ-thiolactams by the aza-Diels–Alder reaction of in situ generated allenyltrimethylsilylthioketenes with imines. Tetrahedron Lett. 2006, 47, 7763–7766. [Google Scholar] [CrossRef]
- Aoyagi, S.; Kikuchi, K.; Shimada, K.; Takikawa, Y. Convenient Synthesis of 4-Methylenecyclobutenones and Their Synthetic Utility as Allenylketene Precursors. Synlett 2007, 16, 2553–2556. [Google Scholar] [CrossRef]
- Martín, M.; Afonso, M.M.; Galindo, A.; Palenzuela, J.A. Enantioselective Synthesis of a Tetrasubstituted Oxocane via a Double Diastereoselective Hetero Diels-Alder Reaction. Synlett 2001, 2001, 117–119. [Google Scholar] [CrossRef]
- Sun, Z.; Yu, S.; Ding, Z.; Ma, D. Enantioselective Addition of Activated Terminal Alkynes to 1-Acylpyridinium Salts Catalyzed by Cu-Bis(oxazoline) Complexes. J. Am. Chem. Soc. 2007, 129, 9300–9301. [Google Scholar] [CrossRef] [PubMed]
- Kapat, A.; Nyfeler, E.; Giuffredi, G.T.; Renaud, P. Intramolecular Schmidt Reaction Involving Primary Azidoalcohols under Nonacidic Conditions: Synthesis of Indolizidine (−)-167B. J. Am. Chem. Soc. 2009, 131, 17746–17747. [Google Scholar] [CrossRef]
- Yang, X.; Gu, X.; Bin, H.; Xie, J.; Zhou, Q. Asymmetric Synthesis of (−)-Indolizidine 167B and (+)-Coniine. Chin. J. Org. Chem. 2020, 40, 3963–3968. [Google Scholar] [CrossRef]
- Liu, C.; Wang, X.; Li, Z.; Cui, L.; Li, C. Silver-Catalyzed Decarboxylative Radical Azidation of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc. 2015, 137, 9820–9823. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.-H.; Chen, H.-Y. Synthesis of Dendrobatid Alkaloid (+)-167B and (+)-209D and the Investigation of Diastereoselectivity using DFT Calculations. RSC Adv. 2017, 7, 684–687. [Google Scholar] [CrossRef]
- Miao, P.; Li, R.; Lin, X.; Rao, L.; Sun, Z. Visible-light Induced Metal-free Cascade Wittig/Hydroalkylation Reactions. Green. Chem. 2021, 23, 1638–1641. [Google Scholar] [CrossRef]
- Lee, E.; Li, K.S.; Lim, J. Radical Cyclization of β-Aminoacrylates: Stereoselective Synthesis of Indolizidines 167B and 209D. Tetrahedron Lett. 1996, 37, 1448. [Google Scholar] [CrossRef]
- Ponpandian, T.; Muthusubramanian, S. Sequential deprotection–Cyclisation reaction: Stereoselective synthesis of azabicyclic β-enamino ester derivatives and (−) indolizidine 209D. Tetrahedron 2013, 69, 527–536. [Google Scholar] [CrossRef]
- Mujica, M.T.; Afonso, M.M.; Galindo, A.; Palenzuela, J.A. Hetero diels-alder vs Mukaiyama aldol pathways in the reaction of monoactivated dienes and aldehydes. A Lewis acid study. Tetrahedron 1996, 52, 2167–2176. [Google Scholar] [CrossRef]
- Struve, C.; Christophersen, C. Structural Equilibrium and Ring-Chain Tautomerism of Aqueous Solutions of 4-Aminobutyraldehyde. Heterocycles 2003, 60, 1907–1914. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Michel, P.; Rassat, A.; Daly, J.W.; Spande, T.F. A Stereospecific Synthesis of (±)-5,8-Disubstituted Indolizidines and (±)-1,4-Disubstituted Quinolizidines Found in Poison Frog Skins. J. Org. Chem. 2000, 65, 8908–8918. [Google Scholar] [CrossRef]
- Reddy, C.R.; Ramesh, P.; Latha, B. Formal Syntheses of 5,8-Disubstituted Indolizidine Alkaloids (–)-205A, (–)-207A, and (–)-235B. Synlett 2017, 28, 481–484. [Google Scholar] [CrossRef]
- Zhou, D.-J.; Wang, Z.-H.; Zhang, Y.-R.; Cui, Z.-G. Flexible Syntheses of 5,8-Disubstituted Indolizidine Poisonous-Frog Alkaloids via a Michael-Type Conjugate Addition. J. Chem. Res. 2017, 41, 98–105. [Google Scholar] [CrossRef]
- Liou, B.-S.; Jhang, R.-F.; Chou, S.-S.P. Formal synthesis of (±)-indolizidine 209B. Tetrahedron 2014, 70, 7458–7463. [Google Scholar] [CrossRef]
- Michael, J.P.; Gravestock, D. An Expeditious Synthesis of the Dendrobatid Indolizidine Alkaloid 167B. Eur. J. Org. Chem. 1998, 1998, 865–870. [Google Scholar] [CrossRef]
- Takashima, K.; Okada, T.; Kato, A.; Yamasaki, Y.; Sugouchi, T.; Akanuma, S.-I.; Kubo, Y.; Hosoya, K.-I.; Morita, H.; Ito, T.; et al. Divergent Synthesis of Decahydroquinoline-Type Poison-Frog Alkaloids. ChemistrySelect 2022, 7, e202104533. [Google Scholar] [CrossRef]
- Stoye, A.; Quandt, G.; Brunnhöfer, B.; Kapatsina, E.; Baron, J.; Fischer, A.; Weymann, M.; Kunz, H. Stereoselective Synthesis of Enantiomerically Pure Nupharamine Alkaloids from Castoreum. Angew. Chem. Int. Ed. 2009, 48, 2228–2230. [Google Scholar] [CrossRef]
- Abels, F.; Lindemann, C.; Schneider, C. A General Strategy for the Catalytic, Highly Enantio- and Diastereoselective Synthesis of Indolizidine-Based Alkaloid. Chem. Eur. J. 2014, 20, 1964–1979. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLview20; Université de Sherbrooke: Sherbrooke, QC, USA, 2020; Available online: http://www.cylview.org (accessed on 6 October 2023).










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Caro, J.F.; Afonso, M.M.; Palenzuela, J.A. A Simple Entry to the 5,8-Disubstituted Indolizidine Skeleton via Hetero Diels-Alder Reaction. Molecules 2023, 28, 7316. https://doi.org/10.3390/molecules28217316
Rodríguez-Caro JF, Afonso MM, Palenzuela JA. A Simple Entry to the 5,8-Disubstituted Indolizidine Skeleton via Hetero Diels-Alder Reaction. Molecules. 2023; 28(21):7316. https://doi.org/10.3390/molecules28217316
Chicago/Turabian StyleRodríguez-Caro, Juan Francisco, María M. Afonso, and José Antonio Palenzuela. 2023. "A Simple Entry to the 5,8-Disubstituted Indolizidine Skeleton via Hetero Diels-Alder Reaction" Molecules 28, no. 21: 7316. https://doi.org/10.3390/molecules28217316
APA StyleRodríguez-Caro, J. F., Afonso, M. M., & Palenzuela, J. A. (2023). A Simple Entry to the 5,8-Disubstituted Indolizidine Skeleton via Hetero Diels-Alder Reaction. Molecules, 28(21), 7316. https://doi.org/10.3390/molecules28217316