

Enhanced Activity and Stability of an Acetyl Xylan Esterase in Hydrophilic Alcohols through Site-Directed Mutagenesis
Abstract
:1. Introduction
2. Results and Discussion
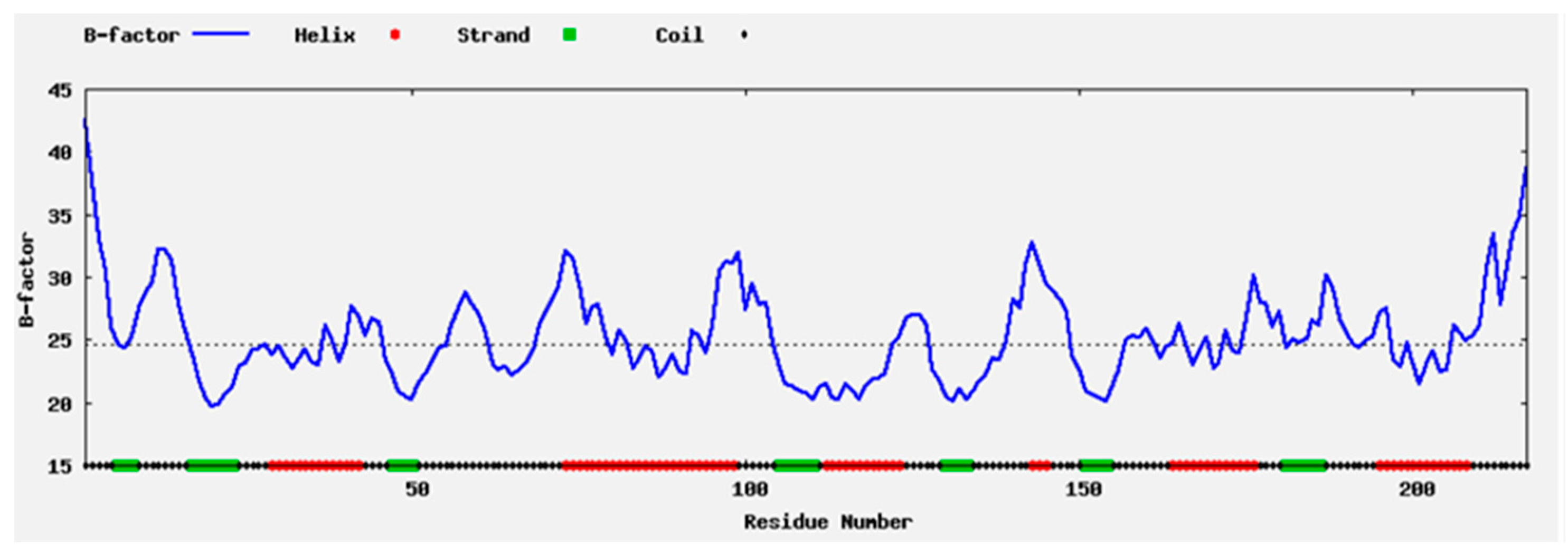
2.1. Enzyme Flexibility and Mutant Sites
2.2. Mutant Residues
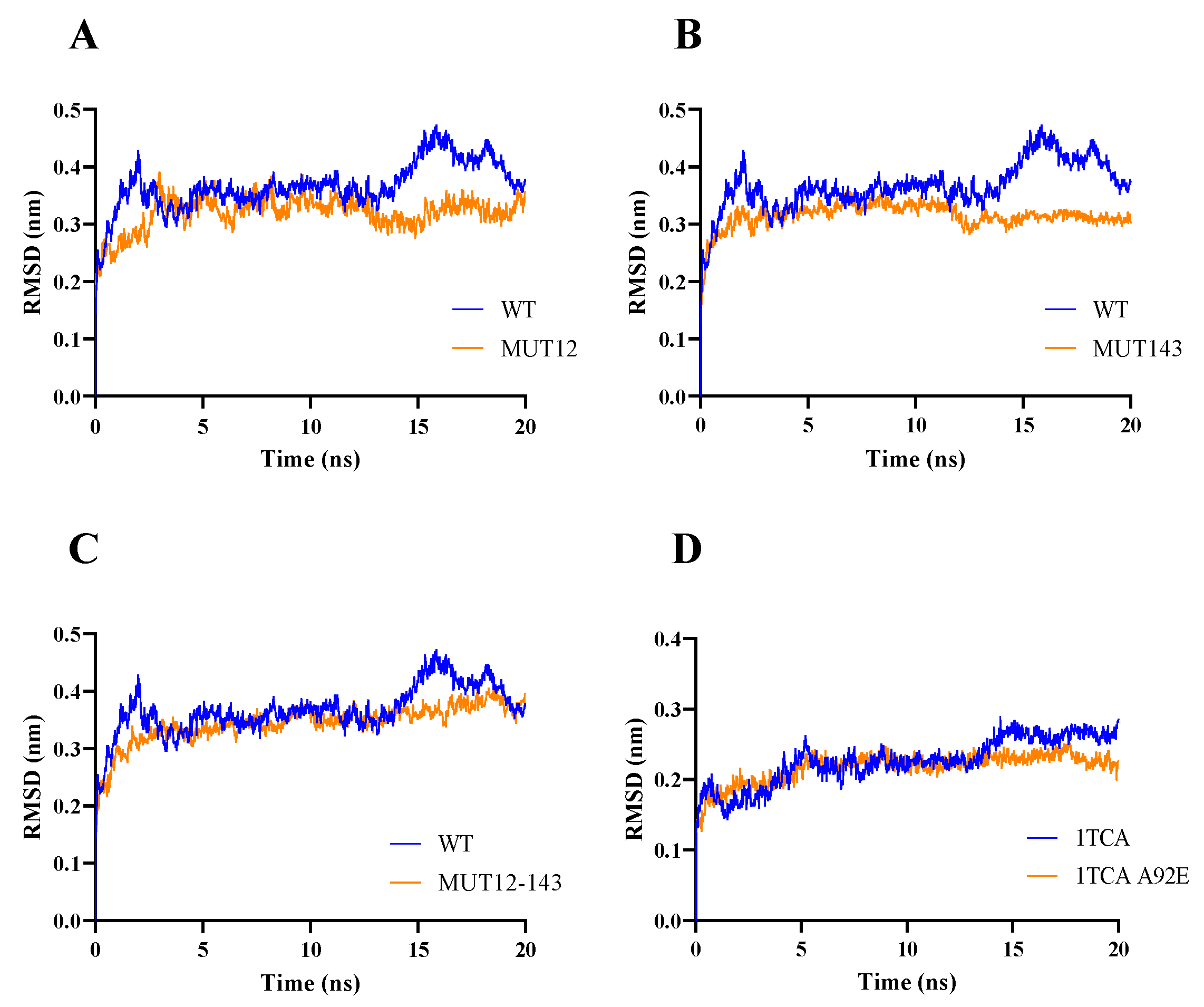
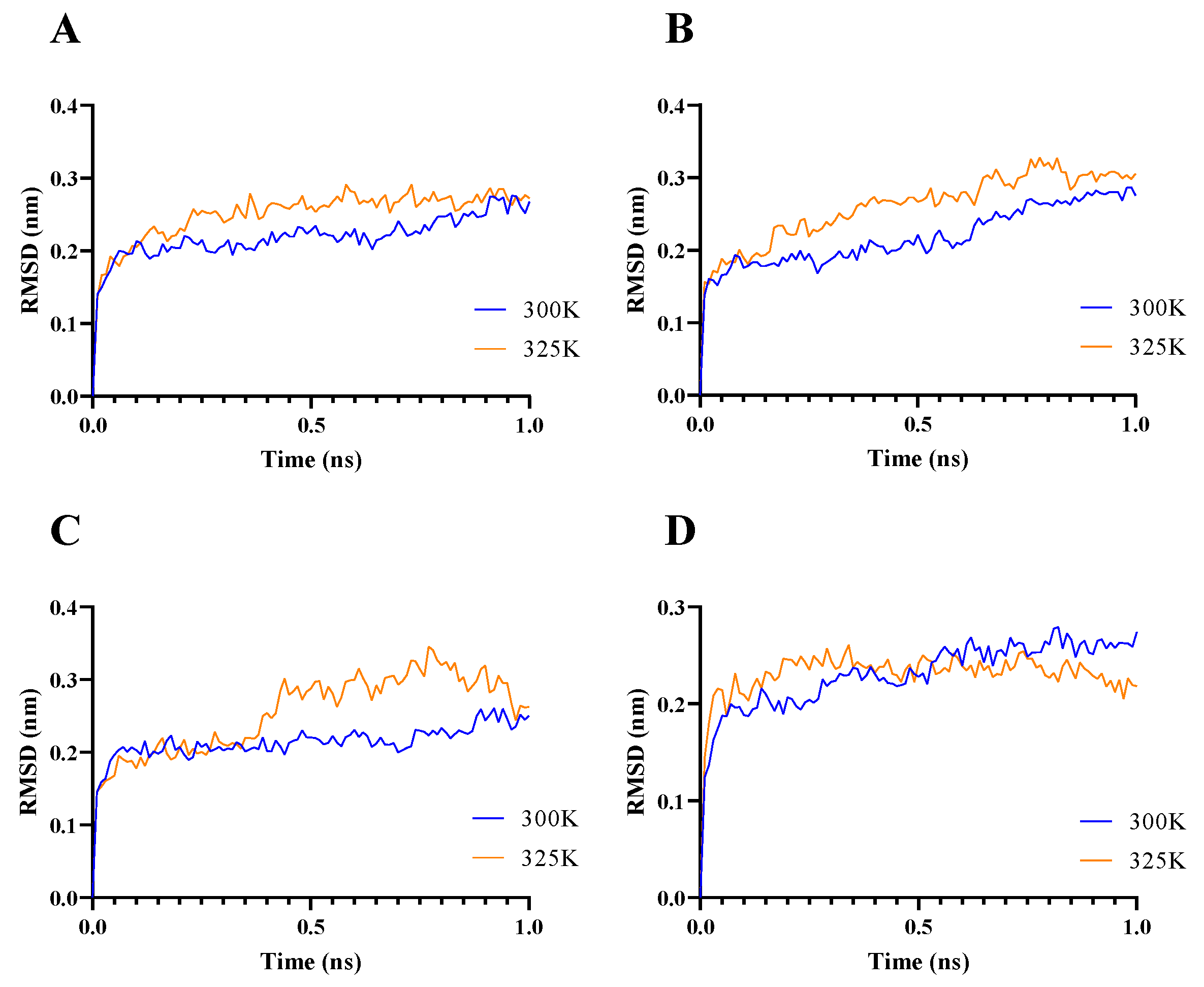
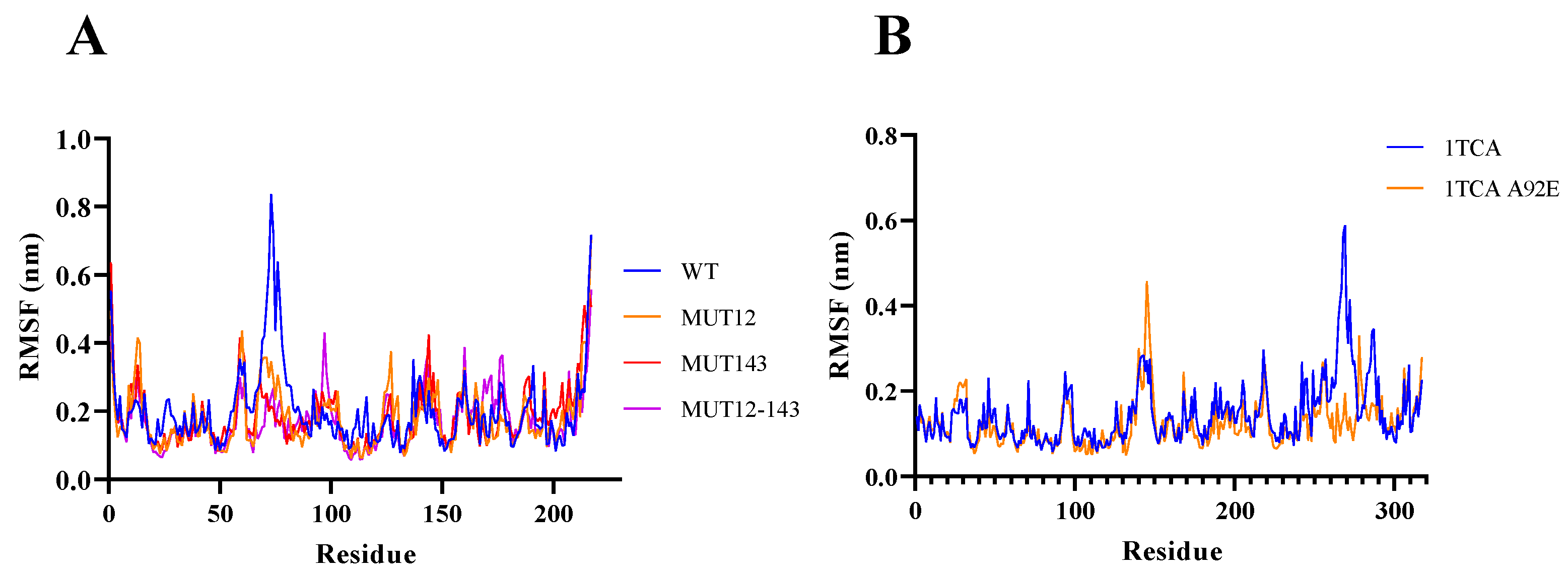
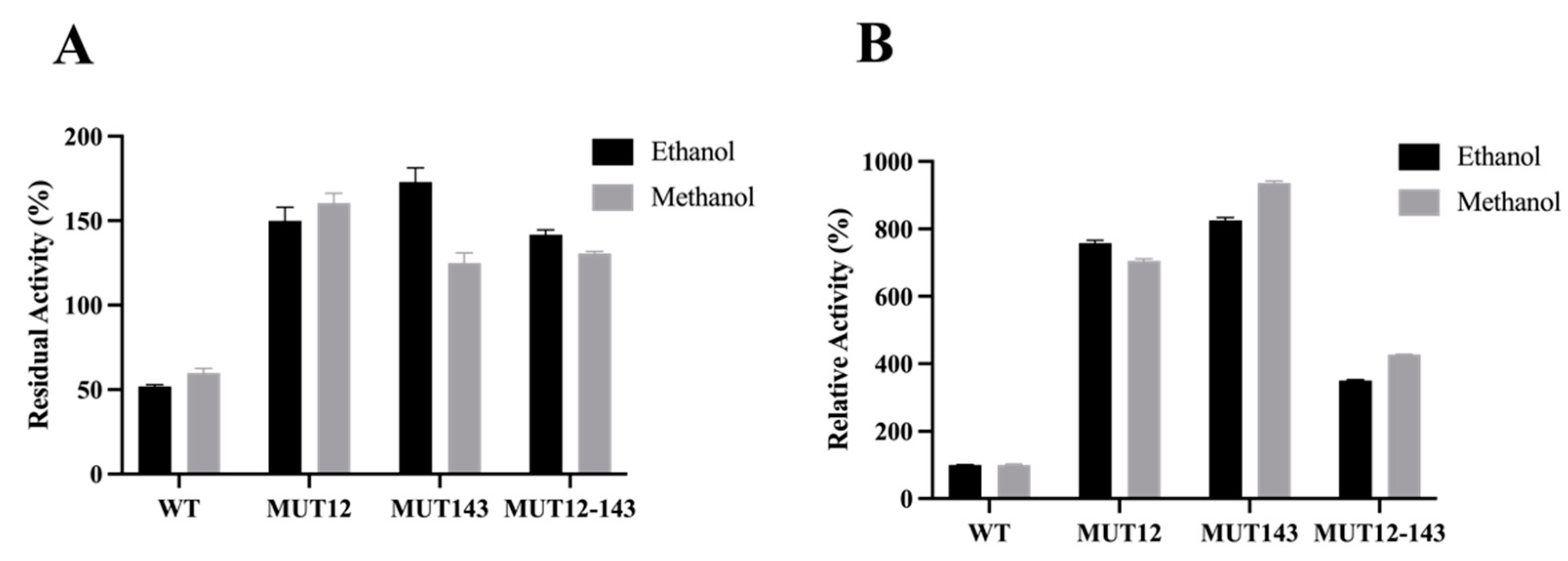
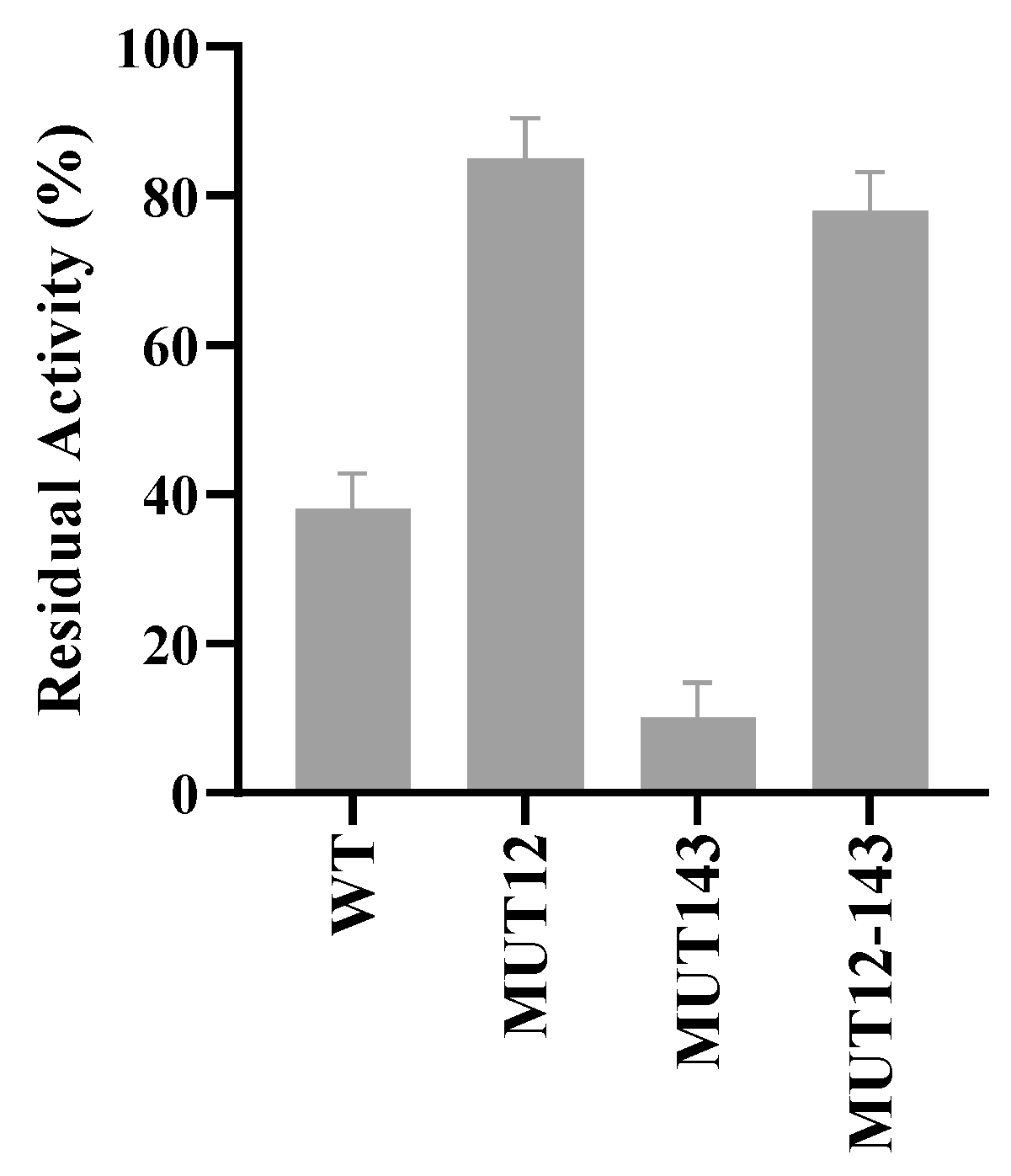
2.3. Enzyme Characterisics and Activity
3. Materials and Methods
3.1. Flexibility and Mutagenesis Analysis
3.2. Interatomic Contacts
3.3. Molecular Dynamic Simulation
3.4. Generating Mutants
3.5. Transformation, Expression, and Purification of Recombinant Proteins
3.6. Activity Assay
3.7. Stability Assay in Organic Solvents
3.8. Stability in Acidic pH
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sarno, M.; Iuliano, M. Active Biocatalyst for Biodiesel Production from Spent Coffee Ground. Bioresour. Technol. 2018, 266, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Yagonia, C.F.J.; Park, H.J.; Hong, S.Y.; Yoo, Y.J. Simultaneous Improvements in the Activity and Stability of Candida Antarctica Lipase B through Multiple-Site Mutagenesis. Biotechnol. Bioprocess Eng. 2015, 20, 218–224. [Google Scholar] [CrossRef]
- Ribitsch, D.; Acero, E.H.; Greimel, K.; Dellacher, A.; Zitzenbacher, S.; Marold, A.; Rodriguez, R.D.; Steinkellner, G.; Gruber, K.; Schwab, H.; et al. A New Esterase from Thermobifida Halotolerans Hydrolyses Polyethylene Terephthalate (PET) and Polylactic Acid (PLA). Polymers 2012, 4, 617–629. [Google Scholar] [CrossRef]
- Bollinger, A.; Molitor, R.; Thies, S.; Koch, R.; Coscolín, C.; Ferrer, M.; Jaeger, K.-E. Organic-Solvent-Tolerant Carboxylic Ester Hydrolases for Organic Synthesis. Appl. Environ. Microbiol. 2020, 86, e00106-20. [Google Scholar] [CrossRef]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, F.; de Castro, M.S.; Sanchez-Montero, J.M.; Sinisterra, J.V.; Valmaseda, M.; Elson, S.W.; Alvarez, E. Novel Microbial Lipases: Catalytic Activity in Reactions in Organic Media. Enzym. Microb. Technol. 2001, 28, 145–154. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, S.J.; Park, S.; Kim, H.K. Characterization of Organic Solvent-Tolerant Lipolytic Enzyme from Marinobacter Lipolyticus Isolated from the Antarctic Ocean. Appl. Biochem. Biotechnol. 2019, 187, 1046–1060. [Google Scholar] [CrossRef]
- Bernal, C.; Rodríguez, K.; Martínez, R. Integrating Enzyme Immobilization and Protein Engineering: An Alternative Path for the Development of Novel and Improved Industrial Biocatalysts. Biotechnol. Adv. 2018, 36, 1470–1480. [Google Scholar] [CrossRef]
- Cui, H.; Stadtmüller, T.H.J.; Jiang, Q.; Jaeger, K.E.; Schwaneberg, U.; Davari, M.D. How to Engineer Organic Solvent Resistant Enzymes: Insights from Combined Molecular Dynamics and Directed Evolution Study. ChemCatChem 2020, 12, 4073–4083. [Google Scholar] [CrossRef]
- Dinmukhamed, T.; Huang, Z.; Liu, Y.; Lv, X.; Li, J.; Du, G.; Liu, L. Current Advances in Design and Engineering Strategies of Industrial Enzymes. Syst. Microbiol. Biomanuf. 2021, 1, 15–23. [Google Scholar] [CrossRef]
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223. [Google Scholar] [CrossRef]
- Cruz, G.; Acosta, J.; Mancheño, J.M.; del Arco, J.; Fernández-Lucas, J. Rational Design of a Thermostable 2′-Deoxyribosyltransferase for Nelarabine Production by Prediction of Disulfide Bond Engineering Sites. Int. J. Mol. Sci. 2022, 23, 11806. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S. Beyond Directed Evolution-Semi-Rational Protein Engineering and Design. Curr. Opin. Biotechnol. 2010, 21, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhang, Q.; Wu, W.; Pu, Z.; Yu, H. Rational Design of Enzyme Activity and Enantioselectivity. Front. Bioeng. Biotechnol. 2023, 11, 1129149. [Google Scholar] [CrossRef] [PubMed]
- Gihaz, S.; Kanteev, M.; Pazy, Y.; Fishman, A. Filling the Void: Introducing Aromatic Interactions into Solvent Tunnels To Enhance Lipase Stability in Methanol. Appl. Environ. Microbiol. 2018, 84, e02143-18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, Z.; Xie, W.; Yu, Y.; Ning, C.; Yuan, M.; Mou, H. Improving Catalytic Efficiency and Maximum Activity at Low PH of Aspergillus Neoniger Phytase Using Rational Design. Int. J. Biol. Macromol. 2019, 131, 1117–1124. [Google Scholar] [CrossRef]
- Kato, T.; Shiono, Y.; Koseki, T. Identification and Characterization of an Acetyl Xylan Esterase from Aspergillus Oryzae. J. Biosci. Bioeng. 2021, 132, 337–342. [Google Scholar] [CrossRef]
- Carr, P.D.; Ollis, D.L. α/ΒHydrolase Fold: An Update. Protein Pept. Lett. 2009, 16, 1137–1148. [Google Scholar]
- Javed, S.; Azeem, F.; Hussain, S.; Rasul, I.; Siddique, M.H.; Riaz, M.; Afzal, M.; Kouser, A.; Nadeem, H. Bacterial Lipases: A Review on Purification and Characterization. Prog. Biophys. Mol. Biol. 2018, 132, 23–34. [Google Scholar] [CrossRef]
- Debeche, T.; Bliard, C.; Debeire, P.; O’donohue, M.J. Probing the Catalytically Essential Residues of the α-l-Arabinofuranosidase from Thermobacillus xylanilyticus. Protein Eng. 2002, 15, 21–28. [Google Scholar] [CrossRef]
- Madhavan, A.; Arun, K.B.; Binod, P.; Sirohi, R.; Tarafdar, A.; Reshmy, R.; Kumar Awasthi, M.; Sindhu, R. Design of Novel Enzyme Biocatalysts for Industrial Bioprocess: Harnessing the Power of Protein Engineering, High Throughput Screening and Synthetic Biology. Bioresour. Technol. 2021, 325, 124617. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, Y.; Lv, X.; Sun, G.; Zhang, H.; Chen, T.; Du, G.; Li, J.; Liu, L. Design and Construction of Novel Biocatalyst for Bioprocessing: Recent Advances and Future Outlook. Bioresour. Technol. 2021, 322, 125701. [Google Scholar] [CrossRef]
- Silva, C.; Martins, M.; Jing, S.; Fu, J.; Cavaco-Paulo, A. Practical Insights on Enzyme Stabilization. Crit. Rev. Biotechnol. 2018, 38, 335–350. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]
- Parra-Cruz, R.; Jäger, C.M.; Lau, P.L.; Gomes, R.L.; Pordea, A. Rational Design of Thermostable Carbonic Anhydrase Mutants Using Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 8526–8536. [Google Scholar] [CrossRef]
- Priyanka, P.; Kinsella, G.; Henehan, G.T.; Ryan, B.J. Isolation, Purification and Characterization of a Novel Solvent Stable Lipase from Pseudomonas Reinekei. Protein Expr. Purif. 2019, 153, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Burton, S.G.; Cowan, D.A.; Woodley, J.M. The Search for the Ideal Biocatalyst. Nat. Biotechnol. 2002, 20, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Otten, R.; Pádua, R.A.P.; Bunzel, H.A.; Nguyen, V.; Pitsawong, W.; Patterson, M.; Sui, S.; Perry, S.L.; Cohen, A.E.; Hilvert, D. How Directed Evolution Reshapes the Energy Landscape in an Enzyme to Boost Catalysis. Science 2020, 370, 1442–1446. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, L.; Eltoukhy, L.; Jiang, Q.; Korkunc, S.K.; Jaeger, K.E.; Schwaneberg, U.; Davari, M.D. Enzyme Hydration Determines Resistance in Organic Cosolvents. ACS Catal. 2020, 10, 14847–14856. [Google Scholar] [CrossRef]
- Doukyu, N.; Ogino, H. Organic Solvent-Tolerant Enzymes. Biochem. Eng. J. 2010, 48, 270–282. [Google Scholar] [CrossRef]
- Xie, Y.; An, J.; Yang, G.; Wu, G.; Zhang, Y.; Cui, L.; Feng, Y. Enhanced Enzyme Kinetic Stability by Increasing Rigidity within the Active Site. J. Biol. Chem. 2014, 289, 7994–8006. [Google Scholar] [CrossRef]
- Tang, H.; Shi, K.; Shi, C.; Aihara, H.; Zhang, J.; Du, G. Enhancing Subtilisin Thermostability through a Modified Normalized B-Factor Analysis and Loop-Grafting Strategy. J. Biol. Chem. 2019, 294, 18398–18407. [Google Scholar] [CrossRef] [PubMed]
- Jain, D.; Mishra, S. Multifunctional Solvent Stable Bacillus Lipase Mediated Biotransformations in the Context of Food and Fuel. J. Mol. Catal. B Enzym. 2015, 117, 21–30. [Google Scholar] [CrossRef]
- Yang, L.; Dordick, J.S.; Garde, S. Hydration of Enzyme in Nonaqueous Media Is Consistent with Solvent Dependence of Its Activity. Biophys. J. 2004, 87, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Newberry, R.W.; Bartlett, G.J.; VanVeller, B.; Woolfson, D.N.; Raines, R.T. Signatures of N→π* Interactions in Proteins. Protein Sci. 2014, 23, 284–288. [Google Scholar] [CrossRef]
- Priyanka, P.; Tan, Y.; Kinsella, G.K.; Henehan, G.T.; Ryan, B.J. Solvent Stable Microbial Lipases: Current Understanding and Biotechnological Applications. Biotechnol. Lett. 2019, 41, 203–220. [Google Scholar] [CrossRef]
- Markel, U.; Zhu, L.; Frauenkron-Machedjou, V.J.; Zhao, J.; Bocola, M.; Davari, M.D.; Jaeger, K.E.; Schwaneberg, U. Are Directed Evolution Approaches Efficient in Exploring Nature’s Potential to Stabilize a Lipase in Organic Cosolvents? Catalysts 2017, 7, 142. [Google Scholar] [CrossRef]
- Dordick, J.S. Designing Enzymes for Use in Organic Solvents. Biotechnol. Prog. 1992, 8, 259–267. [Google Scholar] [CrossRef]
- Beaufils, C.; Man, H.M.; de Poulpiquet, A.; Mazurenko, I.; Lojou, E. From Enzyme Stability to Enzymatic Bioelectrode Stabilization Processes. Catalysts 2021, 11, 497. [Google Scholar] [CrossRef]
- Maester, T.C.; Pereira, M.R.; Gibertoni Malaman, A.M.; Borges, J.P.; Pereira, P.A.M.; Lemos, E.G.M. Exploring Metagenomic Enzymes: A Novel Esterase Useful for Short-Chain Ester Synthesis. Catalysts 2020, 10, 1100. [Google Scholar] [CrossRef]
- Alex, D.; Shainu, A.; Pandey, A.; Sukumaran, R.K. Esterase Active in Polar Organic Solvents from the Yeast Pseudozyma sp. NII 08165. Enzym. Res. 2014, 2014, 494682. [Google Scholar] [CrossRef]
- Park, J.M.; Kang, C.H.; Won, S.M.; Oh, K.H.; Yoon, J.H. Characterization of a Novel Moderately Thermophilic Solvent-Tolerant Esterase Isolated From a Compost Metagenome Library. Front. Microbiol. 2020, 10, 3069. [Google Scholar] [CrossRef] [PubMed]
- Madubuike, H.; Ferry, N. Characterisation of a Novel Acetyl Xylan Esterase (BaAXE) Screened from the Gut Microbiota of the Common Black Slug (Arion Ater). Molecules 2022, 27, 2999. [Google Scholar] [CrossRef]
- Joynson, R.; Pritchard, L.; Osemwekha, E.; Ferry, N. Metagenomic Analysis of the Gut Microbiome of the Common Black Slug Arion Ater in Search of Novel Lignocellulose Degrading Enzymes. Front. Microbiol. 2017, 8, 2181. [Google Scholar] [CrossRef]
- Joynson, R.; Swamy, A.; Bou, P.A.; Chapuis, A.; Ferry, N. Characterization of Cellulolytic Activity in the Gut of the Terrestrial Land Slug Arion Ater: Biochemical Identification of Targets for Intensive Study. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2014, 177, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Vedder, M.; Zhang, L.; Jaeger, K.E.; Schwaneberg, U.; Davari, M.D. Polar Substitutions on the Surface of a Lipase Substantially Improve Tolerance in Organic Solvents. ChemSusChem 2022, 15, e202102551. [Google Scholar] [CrossRef]
- Kawata, T.; Ogino, H. Enhancement of the Organic Solvent-Stability of the LST-03 Lipase by Directed Evolution. Biotechnol. Prog. 2009, 25, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.S. Defying the Activity–Stability Trade-off in Enzymes: Taking Advantage of Entropy to Enhance Activity and Thermostability. Crit. Rev. Biotechnol. 2017, 37, 309–322. [Google Scholar] [CrossRef]
- Xie, N.Z.; Du, Q.S.; Li, J.X.; Huang, R.B. Exploring Strong Interactions in Proteins with Quantum Chemistry and Examples of Their Applications in Drug Design. PLoS ONE 2015, 10, e0137113. [Google Scholar] [CrossRef]
- Rahim, A.; Saha, P.; Jha, K.K.; Sukumar, N.; Sarma, B.K. Reciprocal Carbonyl-Carbonyl Interactions in Small Molecules and Proteins. Nat. Commun. 2017, 8, 78. [Google Scholar] [CrossRef]
- Jain, A.; Purohit, C.S.; Verma, S.; Sankararamakrishnan, R. Close Contacts between Carbonyl Oxygen Atoms and Aromatic Centers in Protein Structures: Π⋯π or Lone-Pair⋯π Interactions? J. Phys. Chem. B 2007, 111, 8680–8683. [Google Scholar] [CrossRef]
- Pal, T.K.; Sankararamakrishnan, R. Quantum Chemical Investigations on Intraresidue Carbonyl—Carbonyl Contacts in Aspartates of High-Resolution Protein Structures. J. Phys. Chem. B 2010, 114, 1038–1049. [Google Scholar] [CrossRef]
- Deane, C.M.; Allen, F.H.; Taylor, R.; Blundell, T.L. Carbonyl-Carbonyl Interactions Stabilize the Partially Allowed Ramachandran Conformations of Asparagine and Aspartic Acid. Protein Eng. 1999, 12, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Pal, T.K.; Sankararamakrishnan, R. Self-Contacts in Asx and Glx Residues of High-Resolution Protein Structures: Role of Local Environment and Tertiary Interactions. J. Mol. Graph. Model. 2008, 27, 20–33. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. The N→π∗ Interaction. Acc. Chem. Res. 2017, 50, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Dhar, J.; Chakrabarti, P. Defining the Loop Structures in Proteins Based on Composite β-Turn Mimics. Protein Eng. Des. Sel. 2014, 28, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Baalham, C.A.; Lommerse, J.P.M.; Raithby, P.R. Carbonyl-Carbonyl Interactions Can Be Competitive with Hydrogen Bonds. Acta Crystallogr. Sect. B Struct. Sci. 1998, 54, 320–329. [Google Scholar] [CrossRef]
- Cui, H.; Eltoukhy, L.; Zhang, L.; Markel, U.; Jaeger, K.E.; Davari, M.D.; Schwaneberg, U. Less Unfavorable Salt Bridges on the Enzyme Surface Result in More Organic Cosolvent Resistance. Angew. Chem.—Int. Ed. 2021, 60, 11448–11456. [Google Scholar] [CrossRef]
- Sohail Siddiqui, K.; Mushir Shemsi, A.; Ahmad Anwar, M.; Hamid Rashid, M.; Ibrahim Rajoka, M. Partial and Complete Alteration of Surface Charges of Carboxymethylcellulase by Chemical Modification: Thermostabilization in Water-Miscible Organic Solvent. Enzym. Microb. Technol. 1999, 24, 599–608. [Google Scholar] [CrossRef]
- Park, H.J.; Joo, J.C.; Park, K.; Kim, Y.H.; Yoo, Y.J. Prediction of the Solvent Affecting Site and the Computational Design of Stable Candida Antarctica Lipase B in a Hydrophilic Organic Solvent. J. Biotechnol. 2013, 163, 346–352. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Zhang, Y. ResQ: An Approach to Unified Estimation of B-Factor and Residue-Specific Error in Protein Structure Prediction. J. Mol. Biol. 2016, 428, 693–701. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue Number | Amino Acid | SASA (%) a | nBF b | SS c | |
|---|---|---|---|---|---|
| Original | Mutant | ||||
| 12 | A | D | 44.8 | 0.92 | Loop |
| 143 | Q | E | 71.6 | 0.99 | Loop |
| Residue | Atom (s) | Contact Residue | Contact Atom (s) | Ave. Distance |
|---|---|---|---|---|
| ASP12 | Various | ASP45 | Various | 3.307 ± 0.270 |
| ASP12 | Various | SER10 | Various | 3.021 ± 0.503 |
| ASP12 | Various | LYS16 | NZ | 5.44 ± 0.07 |
| ASP12 | Various | PRO14 | Various | 3.26 ± 0.26 |
| ASP12 | Various | PRO44 | Various | 3.10 ± 0.33 |
| ALA12 | C | PRO14 | CD | 3.31 |
| ALA12 | N & CB | PRO44 | O | 3.02 ± 0.07 |
| ALA12 | N | SER10 | C & OG | 3.192 ± 0.20 |
| GLU143 | CB | GLN140 | NE2 | 3.421 |
| GLU143 | C | ALA145 | N | 3.614 |
| GLN143 | Various | GLN140 | Various | 3.344 ± 0.43 |
| GLN143 | Various | ALA145 | Various | 3.614 |
| Mutant ID | Primer Sequence (5′–3′) |
|---|---|
| MUT12 | FP: CAGAGTCCTGatACACCCGCAA RP: GACAACAAAGTGGTCATGTTTC |
| MUT143 | FP: GCAGCTTCCAgAAAAAGCATC RP: GCGTAGCGACCATTAAAAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madubuike, H.; Ferry, N. Enhanced Activity and Stability of an Acetyl Xylan Esterase in Hydrophilic Alcohols through Site-Directed Mutagenesis. Molecules 2023, 28, 7393. https://doi.org/10.3390/molecules28217393
Madubuike H, Ferry N. Enhanced Activity and Stability of an Acetyl Xylan Esterase in Hydrophilic Alcohols through Site-Directed Mutagenesis. Molecules. 2023; 28(21):7393. https://doi.org/10.3390/molecules28217393
Chicago/Turabian StyleMadubuike, Henry, and Natalie Ferry. 2023. "Enhanced Activity and Stability of an Acetyl Xylan Esterase in Hydrophilic Alcohols through Site-Directed Mutagenesis" Molecules 28, no. 21: 7393. https://doi.org/10.3390/molecules28217393