
Evaluation of the Anticancer and Biological Activities of Istaroxime via Ex Vivo Analyses, Molecular Docking and Conceptual Density Functional Theory Computations

 , , , ,
, , , ,  , and
, and 
Abstract
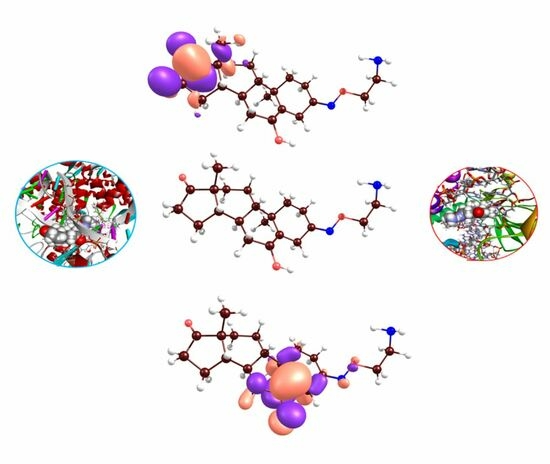
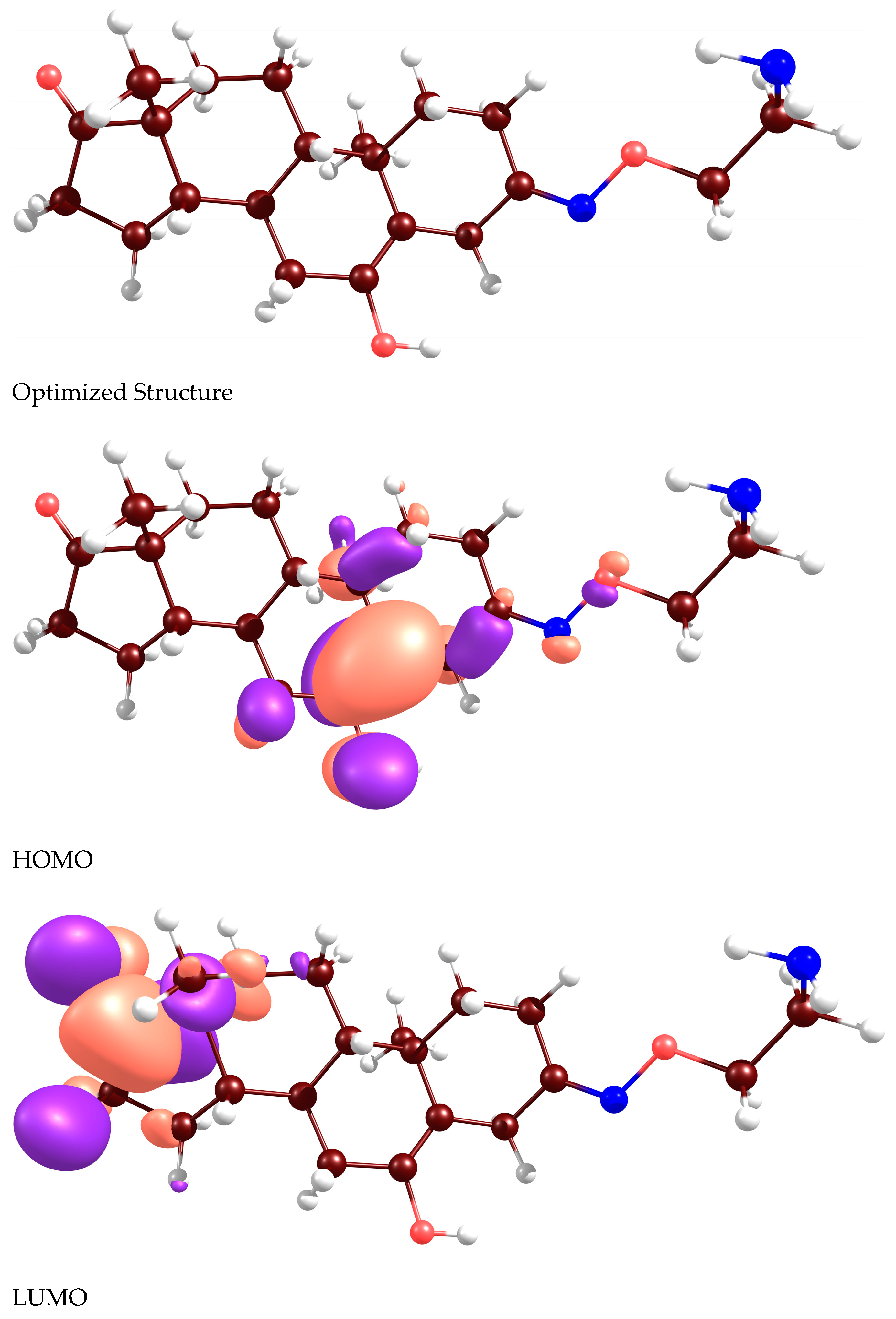
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Details of the Conceptual Density Functional Theory Based Computations
A = E(neutral) − E(anionic form)
2. Results
2.1. The Cell Proliferation Assay
2.2. Docking Analyses and CDFT Computations
2.3. Enyzme Activity Assays
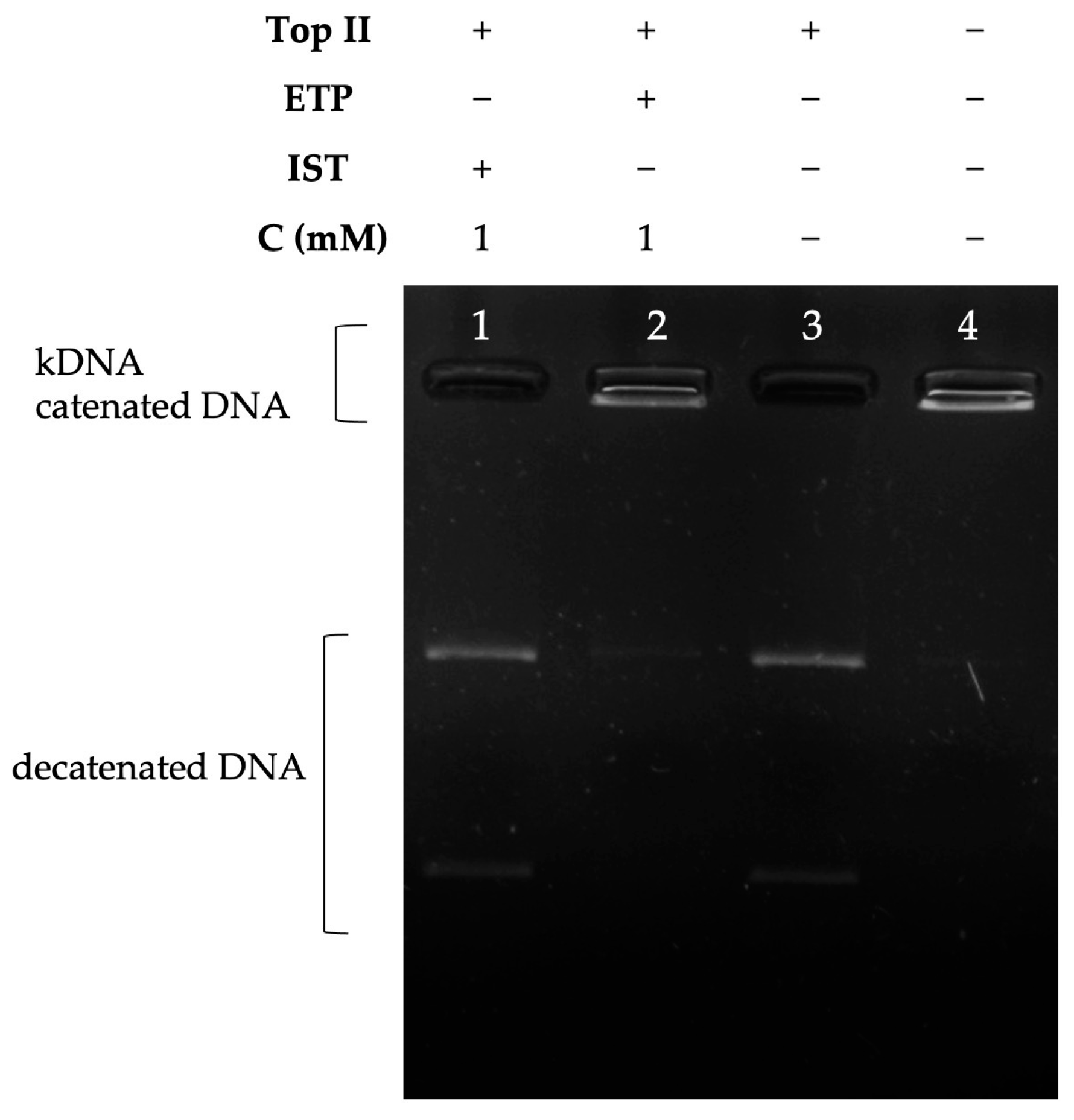
2.3.1. Supercoiled DNA Relaxation Analyses
2.3.2. Decatenation Analyses
3. Discussion
4. Materials and Methods
4.1. Cell Proliferation Assay
4.2. Molecular Docking
4.3. Enzyme Activity Tests
4.3.1. Supercoiled DNA Relaxation Assays
4.3.2. Decatenation Assays
4.4. Details of Density Functional Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, W.L. Targeted therapy in T-cell malignancies: Dysregulation of the cellular signaling pathways. Leukemia 2010, 24, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shindel, A.W.; Lin, G.; Lue, T.F. Cellular signaling pathways modulated by low-intensity extracorporeal shock wave therapy. Int. J. Impot. Res. 2019, 31, 170–176. [Google Scholar] [CrossRef]
- Nitiss, J.L. Investigating the Biological Functions of DNA Topoisomerases in Eukaryotic Cells. Biochim. Biophys. Acta 1998, 1400, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Interaction between DNA and an Escherichia Coli Protein Omega. J. Mol. Biol. 1971, 55, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as Anticancer Targets. Biochem. J. 2018, 475, 373. [Google Scholar] [CrossRef]
- Baglini, E.; Salerno, S.; Barresi, E.; Robello, M.; Da Settimo, F.; Taliani, S.; Marini, A.M. Multiple Topoisomerase I (TopoI), Topoisomerase II (TopoII) and Tyrosyl-DNA Phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur. J. Pharm. Sci. 2021, 156, 105594. [Google Scholar] [CrossRef]
- Madkour, M.M.; Ramadan, W.S.; Saleh, E.; El-Awady, R. Epigenetic modulations in cancer: Predictive biomarkers and potential targets for overcoming the resistance to topoisomerase I inhibitors. Ann. Med. 2023, 55, 2203946. [Google Scholar] [CrossRef]
- Carey, J.F.; Schultz, S.J.; Sisson, L.; Fazzio, T.G.; Champoux, J.J. DNA relaxation by human topoisomerase I occurs in the closed clamp conformation of the protein. Proc. Natl. Acad. Sci. USA 2003, 100, 5640–5645. [Google Scholar] [CrossRef]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Capranico, G.; Binaschi, M. DNA sequence selectivity of topoisomerases and topoisomerase poisons. Biochim. Biophys. Acta 1998, 1400, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, A.; Iacovelli, F.; Fiorani, P.; Desideri, A. Natural Compounds as Therapeutic Agents: The Case of Human Topoisomerase IB. Int. J. Mol. Sci. 2021, 22, 4138. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.A.; Osheroff, N. DNA Topoisomerase Protocols, 1st ed.; Humana: Totowa, NJ, USA, 1999; pp. 1–8. [Google Scholar]
- Alevizopoulos, K.; Dimas, K.; Papadopoulou, N.; Schmidt, E.M.; Tsapara, A.; Alkahtani, S.; Honisch, S.; Prousis, K.C.; Alarifi, S.; Calogeropoulou, T.; et al. Functional Characterization and Anti-Cancer Action of the Clinical Phase II Cardiac Na+/K+ ATPase Inhibitor Istaroxime: In Vitro and in Vivo Properties and Cross Talk with the Membrane Androgen Receptor. Oncotarget 2016, 7, 24415–24428. [Google Scholar] [CrossRef]
- Suter, T.M.; Ewer, M.S. Cancer Drugs and the Heart: Importance and Management. Eur. Heart J. 2013, 34, 1102–1111. [Google Scholar] [CrossRef]
- Racioppi, M.F.; Burgos, J.I.; Morell, M.; Gonano, L.A.; Petroff, M.V. Cellular Mechanisms Underlying the Low Cardiotoxicity of Istaroxime. J. Am. Heart Assoc. 2021, 10, e018833. [Google Scholar] [CrossRef]
- Wallner, M.; Khafaga, M.; Kolesnik, E.; Vafiadis, A.; Schwantzer, G.; Eaton, D.M.; Curcic, P.; Köstenberger, M.; Knez, I.; Rainer, P.P.; et al. Istaroxime, a Potential Anticancer Drug in Prostate Cancer, Exerts Beneficial Functional Effects in Healthy and Diseased Human Myocardium. Oncotarget 2017, 8, 49264. [Google Scholar] [CrossRef]
- Kislitsina, O.N.; Rich, J.D.; Wilcox, J.E.; Pham, D.T.; Churyla, A.; Vorovich, E.B.; Ghafourian, K.; Yancy, C.W. Shock—Classification and Pathophysiological Principles of Therapeutics. Curr. Cardiol. Rev. 2019, 15, 102–113. [Google Scholar] [CrossRef]
- Forzano, I.; Mone, P.; Mottola, G.; Kansakar, U.; Salemme, L.; De Luca, A.; Tesorio, T.; Varzideh, F.; Santulli, G. Efficacy of the New Inotropic Agent Istaroxime in Acute Heart Failure. J. Clin. Med. 2022, 11, 7503. [Google Scholar] [CrossRef]
- Khalid Khan, S.; Rawat, A.; Khan, Z.; Reyaz, I.; Kumar, V.; Batool, S.; Yadav, R.; Hirani, S. Safety and Efficacy of Istaroxime in Patients with Acute Heart Failure: A Meta-Analysis of Randomized Controlled Trials. Cureus 2023, 15, e41084. [Google Scholar] [CrossRef]
- Metra, M.; Chioncel, O.; Cotter, G.; Davison, B.; Filippatos, G.; Mebazaa, A.; Novosadova, M.; Ponikowski, P.; Simmons, P.; Soffer, J.; et al. Safety and Efficacy of Istaroxime in Patients with Acute Heart Failure-Related Pre-Cardiogenic Shock—A Multicentre, Randomized, Double-Blind, Placebo-Controlled, Parallel Group Study (SEISMiC). Eur. J. Heart Fail. 2022, 24, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Stagno, M.J.; Zacharopoulou, N.; Bochem, J.; Tsapara, A.; Pelzl, L.; Al-Maghout, T.; Kallergi, G.; Alkahtani, S.; Alevizopoulos, K.; Dimas, K.; et al. Istaroxime Inhibits Motility and Down-Regulates Orai1 Expression, SOCE and FAK Phosphorylation in Prostate Cancer Cells. Cell. Physiol. Biochem. 2017, 42, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Chattaraj, P.K. Conceptual density functional theory based electronic structure principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Yildiz, S.; Canbaz, G.T.; Kaya, S.; Maslov, M.M. Experimental and Density Functional Theoretical Analyses on Degradation of Acid Orange 7 via UV Irradiation and Ultrasound Enhanced by Fenton Process. J. Mol. Struct. 2023, 1277, 134833. [Google Scholar] [CrossRef]
- Gázquez, J.L.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Pluhařová, E.; Slavíček, P.; Jungwirth, P. Modeling Photoionization of Aqueous DNA and Its Components. Acc. Chem. Res. 2015, 48, 1209–1217. [Google Scholar] [CrossRef]
- Liu, H.; Liu, D.; Jiang, M.Y.; Zhao, X.D.; Li, R.T.; Li, H.M. Iridoids from Valeriana Jatamansi with Anti-Inflammatory and Antiproliferative Properties. Phytochemistry 2021, 184, 112681. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455. [Google Scholar] [CrossRef]
- Podlivaev, A.I.; Openov, L.A. Specific features of the formation of defects in fullerene C46. Phys. Solid State 2012, 54, 1507–1513. [Google Scholar] [CrossRef]
- Makov, G. Chemical hardness in density functional theory. J. Phys. Chem. 1995, 99, 9337–9339. [Google Scholar] [CrossRef]
- Ayers, P.W.; Parr, R.G. Variational principles for describing chemical reactions: The Fukui function and chemical hardness revisited. J. Am. Chem. Soc. 2000, 122, 2010–2018. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. The principle of maximum hardness. Acc. Chem. Res. 1993, 26, 250–255. [Google Scholar] [CrossRef]
- Parr, R.G.; Chattaraj, P.K. Principle of maximum hardness. J. Am. Chem. Soc. 1991, 113, 1854–1855. [Google Scholar] [CrossRef]
- Putz, M.V.; Russo, N.; Sicilia, E. About the Mulliken electronegativity in DFT. Theor. Chem. Acc. 2005, 114, 38–45. [Google Scholar] [CrossRef]
- Noorizadeh, S. Is there a minimum electrophilicity principle in chemical reactions? Chin. J. Chem. 2007, 25, 1439–1444. [Google Scholar] [CrossRef]
- Byl, J.A.W.; Fortune, J.M.; Burden, D.A.; Nitiss, J.L.; Utsugi, T.; Yamada, Y.; Osheroff, N. DNA Topoisomerases as Targets for the Anticancer Drug TAS-103: Primary Cellular Target and DNA Cleavage Enhancement. Biochemistry 1999, 38, 15573–15579. [Google Scholar]
- Nitiss, J.L.; Kiianitsa, K.; Sun, Y.; Nitiss, K.C.; Maizels, N. Topoisomerase Assays. Curr. Protoc. 2021, 1, e250. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure, Molecular Mechanisms, and Evolutionary Relationships in DNA Topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Her, C. Inhibition of Topoisomerase (DNA) I (TOP1): DNA Damage Repair and Anticancer Therapy. Biomolecules 2015, 5, 1652. [Google Scholar] [CrossRef] [PubMed]
- Jo, U.; Murai, Y.; Agama, K.K.; Sun, Y.; Saha, L.K.; Yang, X.; Arakawa, Y.; Gayle, S.; Jones, K.; Paralkar, V.; et al. TOP1-DNA Trapping by Exatecan and Combination Therapy with ATR Inhibitor. Mol. Cancer Ther. 2022, 21, 1090–1102. [Google Scholar] [CrossRef]
- Cinelli, M.A. Topoisomerase 1B Poisons: Over a Half-Century of Drug Leads, Clinical Candidates, and Serendipitous Discoveries. Med. Res. Rev. 2019, 39, 1294–1337. [Google Scholar] [CrossRef]
- Zhou, X.; Yao, G.; Zhang, J.; Bian, J.; Li, G.; Xu, J. An Integrated Multi-Omics Analysis of Topoisomerase Family in Pan-Cancer: Friend or Foe? PLoS ONE 2022, 17, e0274546. [Google Scholar] [CrossRef]
- Dimas, K.; Papadopoulou, N.; Baskakis, C.; Prousis, K.; Tsakos, M.; Alkahtani, S.; Honisch, S.; Lang, F.; Calogeropoulou, T.; Alevizopoulos, K.; et al. Steroidal Cardiac Na+/K+ ATPase Inhibitors Exhibit Strong Anti-Cancer Potential in Vitro and in Prostate and Lung Cancer Xenografts in Vivo. Anticancer Agents Med. Chem. 2014, 14, 762–770. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. The SERCA Pump as a Therapeutic Target: Making a “Smart Bomb” for Prostate Cancer. Cancer Biol. Ther. 2005, 4, 21–29. [Google Scholar] [CrossRef]
- Gélébart, P.; Kovács, T.; Brouland, J.P.; Van Gorp, R.; Grossmann, J.; Rivard, N.; Panis, Y.; Martin, V.; Bredoux, R.; Enouf, J.; et al. Expression of Endomembrane Calcium Pumps in Colon and Gastric Cancer Cells. Induction of SERCA3 Expression during Differentiation. J. Biol. Chem. 2002, 277, 26310–26320. [Google Scholar] [CrossRef]
- Christensen, S.B.; Skytte, D.M.; Denmeade, S.R.; Dionne, C.; Moller, J.V.; Nissen, P.; Isaacs, J.T. A Trojan Horse in Drug Development: Targeting of Thapsigargins towards Prostate Cancer Cells. Anticancer Agents Med. Chem. 2009, 9, 276–294. [Google Scholar] [CrossRef]
- Torre, E.; Arici, M.; Lodrini, A.M.; Ferrandi, M.; Barassi, P.; Hsu, S.C.; Chang, G.J.; Boz, E.; Sala, E.; Vagni, S.; et al. SERCA2a Stimulation by Istaroxime Improves Intracellular Ca2+ Handling and Diastolic Dysfunction in a Model of Diabetic Cardiomyopathy. Cardiovasc. Res. 2022, 118, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Lunelli, M.; Hurwitz, R.; Lambers, J.; Kolbe, M. Crystal Structure of PrgI-SipD: Insight into a Secretion Competent State of the Type Three Secretion System Needle Tip and Its Interaction with Host Ligands. PLoS Pathog. 2011, 7, e1002163. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Li, Y.C.; Wang, Y.R.; Li, T.K.; Chan, N.L. On the Structural Basis and Design Guidelines for Type II Topoisomerase-Targeting Anticancer Drugs. Nucleic Acids Res. 2013, 41, 10630–10640. [Google Scholar] [CrossRef] [PubMed]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. 2007, 623, 83–97. [Google Scholar] [CrossRef]
- Senarisoy, M.; Canturk, P.; Zencir, S.; Baran, Y.; Topcu, Z. Gossypol Interferes with Both Type I and Type II Topoisomerase Activities without Generating Strand Breaks. Cell Biochem. Biophys. 2013, 66, 199–204. [Google Scholar] [CrossRef]
- Jacquemin, D.; Wathelet, V.; Perpète, E.A.; Adamo, C. Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. [Google Scholar] [CrossRef]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y. Benchmark Calculations of the Adsorption of Aromatic Molecules on Graphene. J. Comput. Chem. 2015, 36, 1763–1771. [Google Scholar] [CrossRef]
- Celik, S.; Akyuz, S.; Ozel, A.E. Molecular Modeling, DFT Quantum Chemical Analysis, and Molecular Docking on Edotecarin, an Indolocarbazole Anticancer Agent. Mol. Cryst. Liq. Cryst. 2022, 753, 27–49. [Google Scholar] [CrossRef]
- Chudinov, G.E.; Napolov, D.V.; Basilevsky, M.V. Quantum-Chemical Calculations of the Hydration Energies of Organic Cations and Anions in the Framework of a Continuum Solvent Approximation. Chem. Phys. 1992, 160, 41–54. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gok, E.; Unal, N.; Gungor, B.; Karakus, G.; Kaya, S.; Canturk, P.; Katin, K.P. Evaluation of the Anticancer and Biological Activities of Istaroxime via Ex Vivo Analyses, Molecular Docking and Conceptual Density Functional Theory Computations. Molecules 2023, 28, 7458. https://doi.org/10.3390/molecules28227458
Gok E, Unal N, Gungor B, Karakus G, Kaya S, Canturk P, Katin KP. Evaluation of the Anticancer and Biological Activities of Istaroxime via Ex Vivo Analyses, Molecular Docking and Conceptual Density Functional Theory Computations. Molecules. 2023; 28(22):7458. https://doi.org/10.3390/molecules28227458
Chicago/Turabian StyleGok, Ege, Naz Unal, Burcin Gungor, Gulderen Karakus, Savas Kaya, Pakize Canturk, and Konstantin P. Katin. 2023. "Evaluation of the Anticancer and Biological Activities of Istaroxime via Ex Vivo Analyses, Molecular Docking and Conceptual Density Functional Theory Computations" Molecules 28, no. 22: 7458. https://doi.org/10.3390/molecules28227458
APA StyleGok, E., Unal, N., Gungor, B., Karakus, G., Kaya, S., Canturk, P., & Katin, K. P. (2023). Evaluation of the Anticancer and Biological Activities of Istaroxime via Ex Vivo Analyses, Molecular Docking and Conceptual Density Functional Theory Computations. Molecules, 28(22), 7458. https://doi.org/10.3390/molecules28227458