
The Optimization of the Synthesis Process and the Identification of Levobupivacaine Hydrochloride
Abstract
:
1. Introduction
2. Results
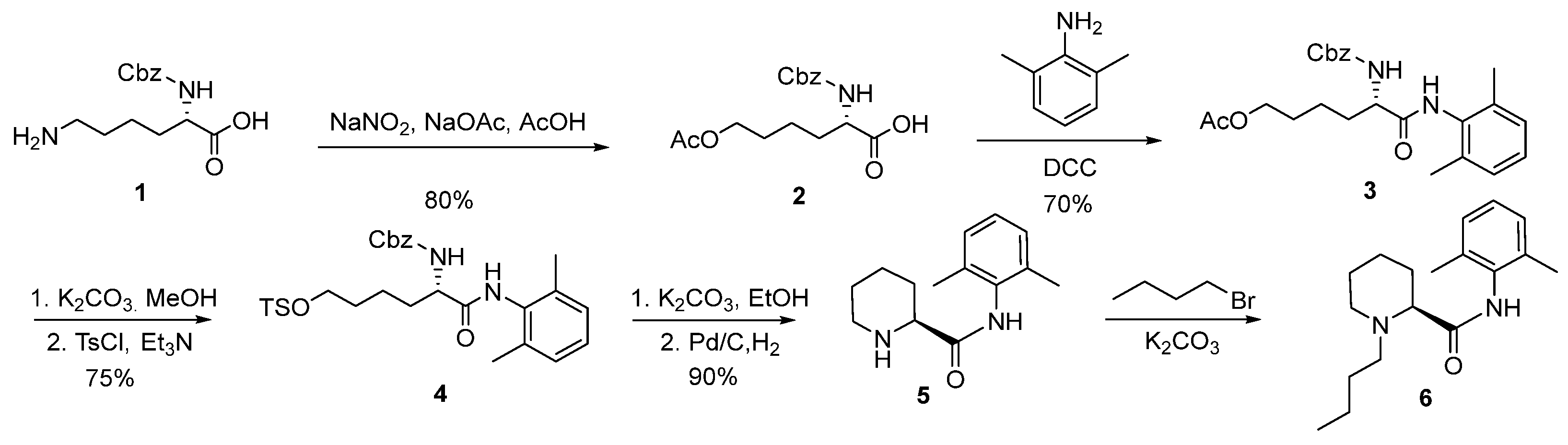
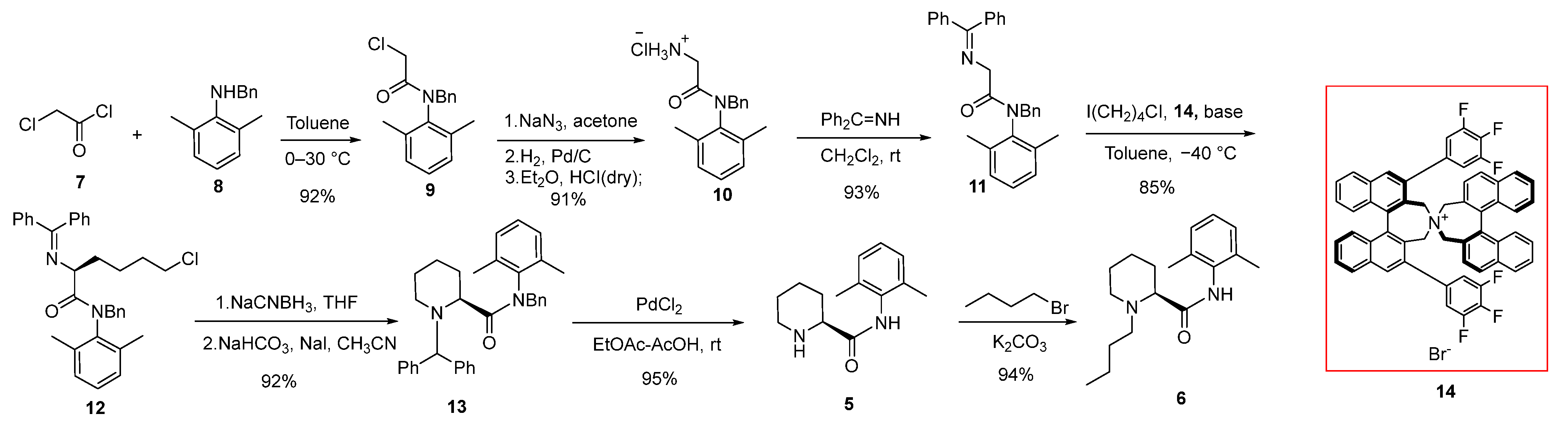
2.1. Synthesis of (2S)-N-(2,6-Dimethylphenyl)Piperidine-2-Carboxamide (5)
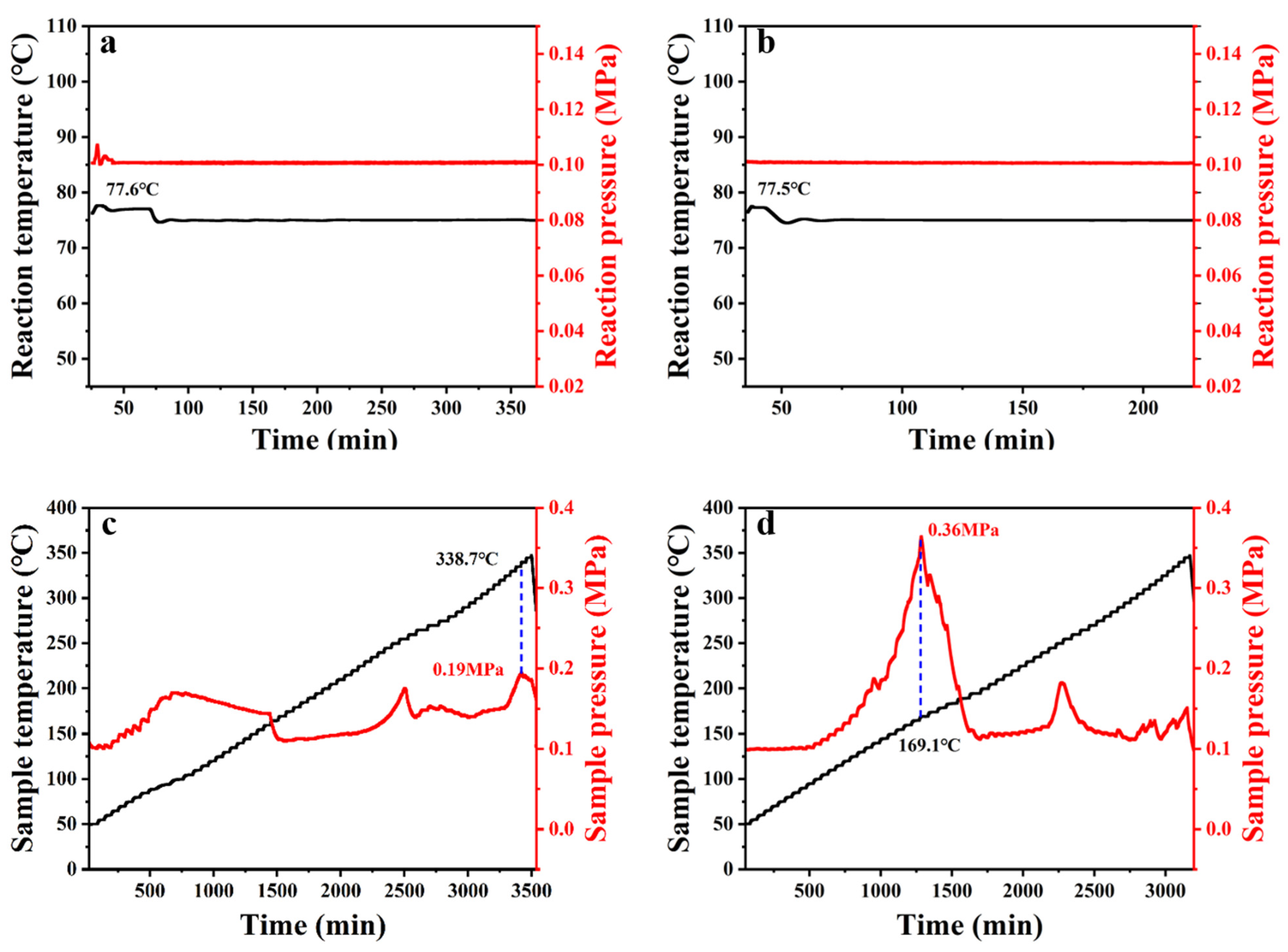
2.2. Synthesis and Safety Evaluation of Levobupivacaine (6)
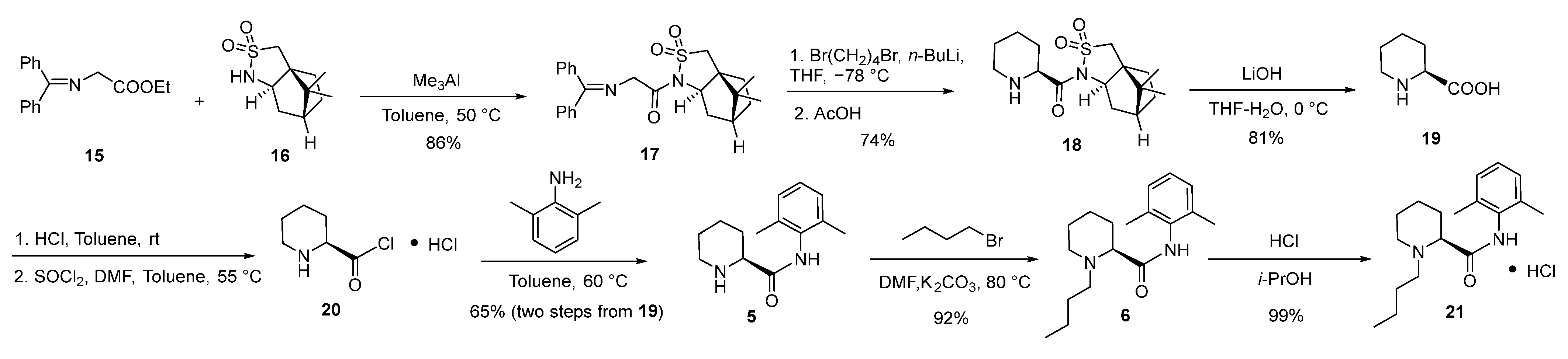
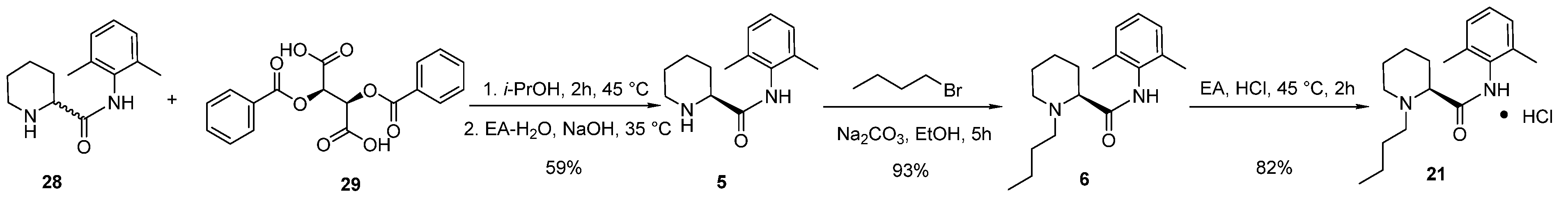
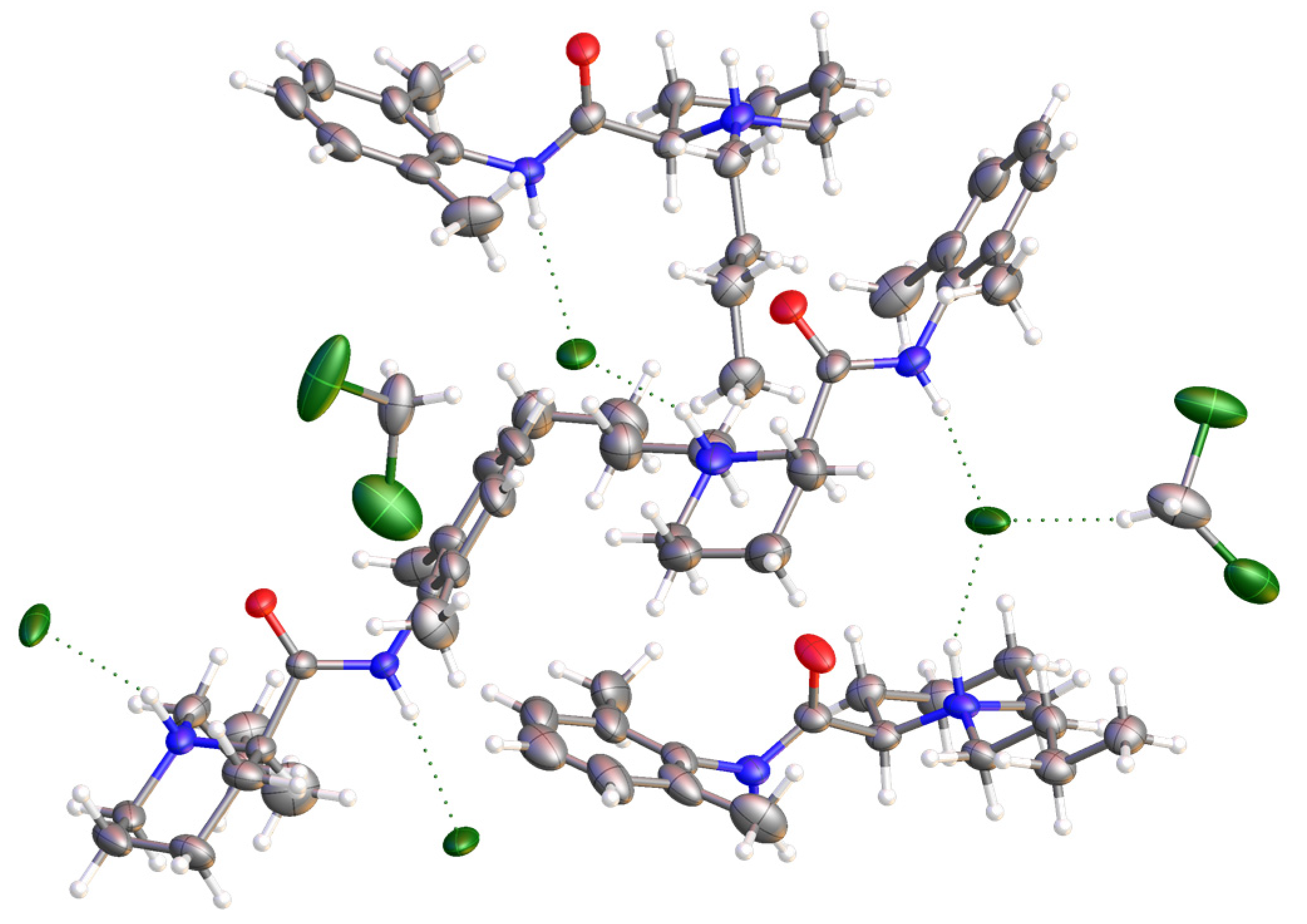
2.3. Synthesis and Crystal Structure Determination of Levobupivacaine Hydrochloride (21)
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. (2S)-N-(2,6-crystal)Piperidine-2-Carboxamide (5)
3.2.2. (2S)-1-Butyl-N-(2,6-Dimethylphenyl)-2-Piperidinecarboxamide (Levobupivacaine, 6)
3.2.3. Levobupivacaine Hydrochloride (21)
3.3. Characterization Methods
3.3.1. Reaction Calorimeter
3.3.2. ARC
3.3.3. DSC
3.3.4. TGA
3.3.5. XRPD
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Af Ekenstam, B.; Egnér, B.; Pettersson, G. Local Anesthetics I: N-Alkyl Pyrrolidine and N-Alkyl Piperidine Carboxylic Acid Amides. Acta Chem. Scand. 1957, 11, 1183–1190. [Google Scholar] [CrossRef]
- Budharapu, A.; Sinha, R.; Uppada, U.K.; Subramanya Kumar, A.V.S.S. Ropivacaine: A New Local Anaesthetic Agent in Maxillofacial Surgery. Br. J. Oral Maxillofac. Surg. 2015, 53, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.H.; Markham, A. Levobupivacaine. Drugs 2000, 59, 551–579. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Saxena, R.; Dutta, A.; Ganguly, N.; Sood, J. Comparison of Ropivacaine with and without Fentanyl vs. Bupivacaine with Fentanyl for Postoperative Epidural Analgesia in Bilateral Total Knee Replacement Surgery. J. Clin. Anesth. 2017, 37, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Ruetsch, Y.; Boni, T.; Borgeat, A. From Cocaine to Ropivacaine: The History of Local Anesthetic Drugs. Curr. Top. Med. Chem. 2001, 1, 175–182. [Google Scholar] [CrossRef] [PubMed]
- McLure, H.A.; Rubin, A.P. Comparison of 0.75% Levobupivacaine with 0.75% Racemic Bupivacaine for Peribulbar Anaesthesia. Anaesthesia 1998, 53, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Adger, B.; Dyer, U.; Hutton, G.; Woods, M. Stereospecific Synthesis of the Anaesthetic Levobupivacaine. Tetrahedron Lett. 1996, 37, 6399–6402. [Google Scholar] [CrossRef]
- Kumar, S.; Ramachandran, U. Studies Directed towards Asymmetric Synthesis of Levobupivacaine. Tetrahedron Lett. 2005, 46, 19–21. [Google Scholar] [CrossRef]
- Yang, Y.; Li, H.; You, Z.; Zhang, X. A Convenient and Highly Enantioselective Synthesis of (S)-2-Pipecolic Acid: An Efficient Access to Caine Anesthetics. Synth. Commun. 2021, 51, 3084–3089. [Google Scholar] [CrossRef]
- Frampton, G.A.C.; Zavareh, H.S. Process for Preparation Levobupivacaine and Analogues Thereof. WO 9612700, 2 May 1996. [Google Scholar]
- Wang, S.C.; Feng, Q.G.; Huo, L.G. A Method for Preparing Levobupivacaine. CN 113105385A, 13 July 2021. [Google Scholar]
- Shankaraiah, N.; Pilli, R.A.; Santos, L.S. Enantioselective Total Syntheses of Ropivacaine and Its Analogues. Tetrahedron Lett. 2008, 49, 5098–5100. [Google Scholar] [CrossRef]
- Soni, R.R.; Koftis, T.; Georgopoulou, I.; Karagiannidou, E. Process for Producing Pipecolic-2-acid-2’,6’-xylidide Useful as an Intermediate for the Preparation of Local Anesthetics. WO 2009089842, 23 July 2009. [Google Scholar]
- Niederwanger, V.; Gozzo, F.; Griesser, U.J. Characterization of Four Crystal Polymorphs and a Monohydrate of S-Bupivacaine Hydrochloride (Levobupivacaine Hydrochloride). J. Pharm. Sci. 2009, 98, 1064–1074. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Yield (%) | Chemical Purity (%) |
|---|---|---|---|
| 1 | Acetone | 58 | 98.31 |
| 2 | EtOH | 30 | 99.99 |
| 3 | i-PrOH | 40 | 99.95 |
| 4 | 4-Methyl-2-pentanone | 60 | 97.53 |
| 5 | EA | 59 | 99.98 |
| Entry | Solvent | Base | Bromobutane (eq) | Time (h) | Temperature (°C) | Yield (%) | Chemical Purity (%) |
|---|---|---|---|---|---|---|---|
| 1 | DMF | K2CO3 | 1.05 | 3 | 75 | 82 | 87.43 |
| 2 | EtOH | K2CO3 | 1.05 | 3 | 75 | 64 | 78.60 |
| 3 | i-PrOH | K2CO3 | 1.05 | 3 | 75 | 64 | 72.30 |
| 4 | MeCN | K2CO3 | 1.05 | 3 | 75 | 58 | 70.05 |
| 5 | THF | K2CO3 | 1.05 | 3 | 75 | 60 | 73.18 |
| 6 | EtOH | Na2CO3 | 1.05 | 3 | 75 | 66 | 79.01 |
| 7 | EtOH | DIPEA | 1.05 | 3 | 75 | 49 | 62.36 |
| 8 | EtOH | K3PO4 | 1.05 | 3 | 75 | 57 | 75.12 |
| 9 | EtOH | NaOH | 1.05 | 3 | 75 | 25 | 46.52 |
| 10 | EtOH | Na2CO3 | 1.25 | 3 | 75 | 73 | 87.29 |
| 11 | EtOH | Na2CO3 | 1.50 | 3 | 75 | 78 | 91.63 |
| 12 | EtOH | Na2CO3 | 1.75 | 3 | 75 | 80 | 93.45 |
| 13 | EtOH | Na2CO3 | 2.00 | 3 | 75 | 80 | 95.59 |
| 14 | EtOH | Na2CO3 | 1.50 | 5 | 75 | 93 | 99.12 |
| 15 | EtOH | Na2CO3 | 1.50 | 8 | 75 | 93 | 98.97 |
| 16 | EtOH | Na2CO3 | 1.50 | 12 | 75 | 93 | 98.85 |
| 17 | EtOH | Na2CO3 | 1.50 | 5 | 40 | 69 | 83.54 |
| 18 | EtOH | Na2CO3 | 1.50 | 5 | 100 | 89 | 98.94 |
| 19 | DMF | Na2CO3 | 1.50 | 5 | 75 | 85 | 85.94 |
| Entry | Solvent | Yield (%) | Chemical Purity (%) | ee (%) |
|---|---|---|---|---|
| 1 | Water | 85 | 98.48 | 97.12 |
| 2 | i-BuOH | 57 | 99.52 | 99.12 |
| 3 | i-PrOH | 82 | 99.90 | 99.30 |
| 4 | EtOH | 73 | 99.87 | 98.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Q.; Gan, H.; Li, C.; Gui, G.; Wang, J.; Zha, X. The Optimization of the Synthesis Process and the Identification of Levobupivacaine Hydrochloride. Molecules 2023, 28, 7482. https://doi.org/10.3390/molecules28227482
Yan Q, Gan H, Li C, Gui G, Wang J, Zha X. The Optimization of the Synthesis Process and the Identification of Levobupivacaine Hydrochloride. Molecules. 2023; 28(22):7482. https://doi.org/10.3390/molecules28227482
Chicago/Turabian StyleYan, Qiuming, Houjun Gan, Chunzheng Li, Gang Gui, Jianbo Wang, and Xiaoming Zha. 2023. "The Optimization of the Synthesis Process and the Identification of Levobupivacaine Hydrochloride" Molecules 28, no. 22: 7482. https://doi.org/10.3390/molecules28227482