

IGF2 Peptide-Based LYTACs for Targeted Degradation of Extracellular and Transmembrane Proteins

 , , , ,
, , , ,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
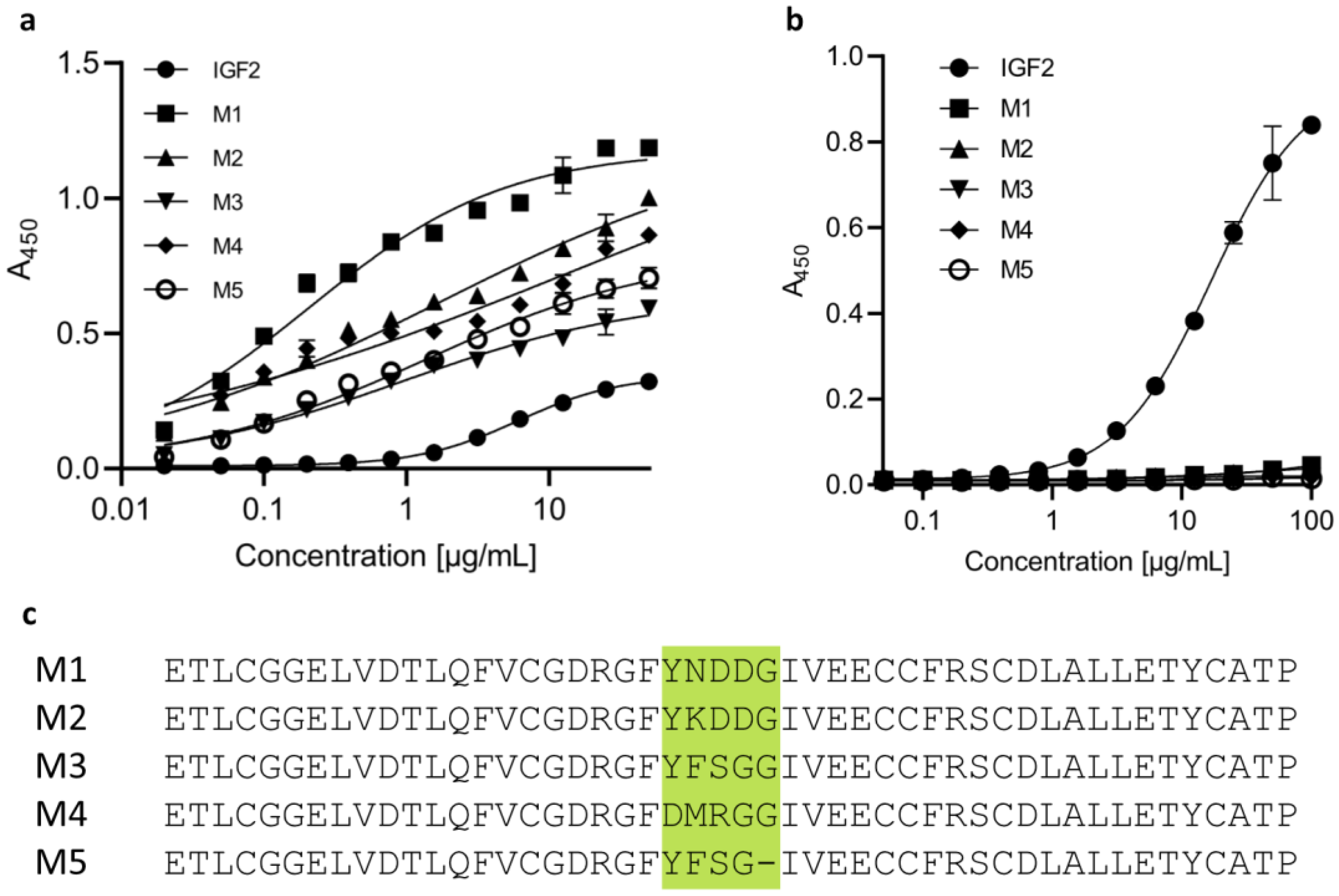
2.1. The Designed IGF2-Based Polypeptides Bind to IGF2R but Not to IGF1R
2.2. Design, Production, and Characterization of Bispecific Anti-PD-L1-Anti-IGF2R Compounds
2.3. Produced Bispecific Compounds Induce Internalization of Soluble PD-L1
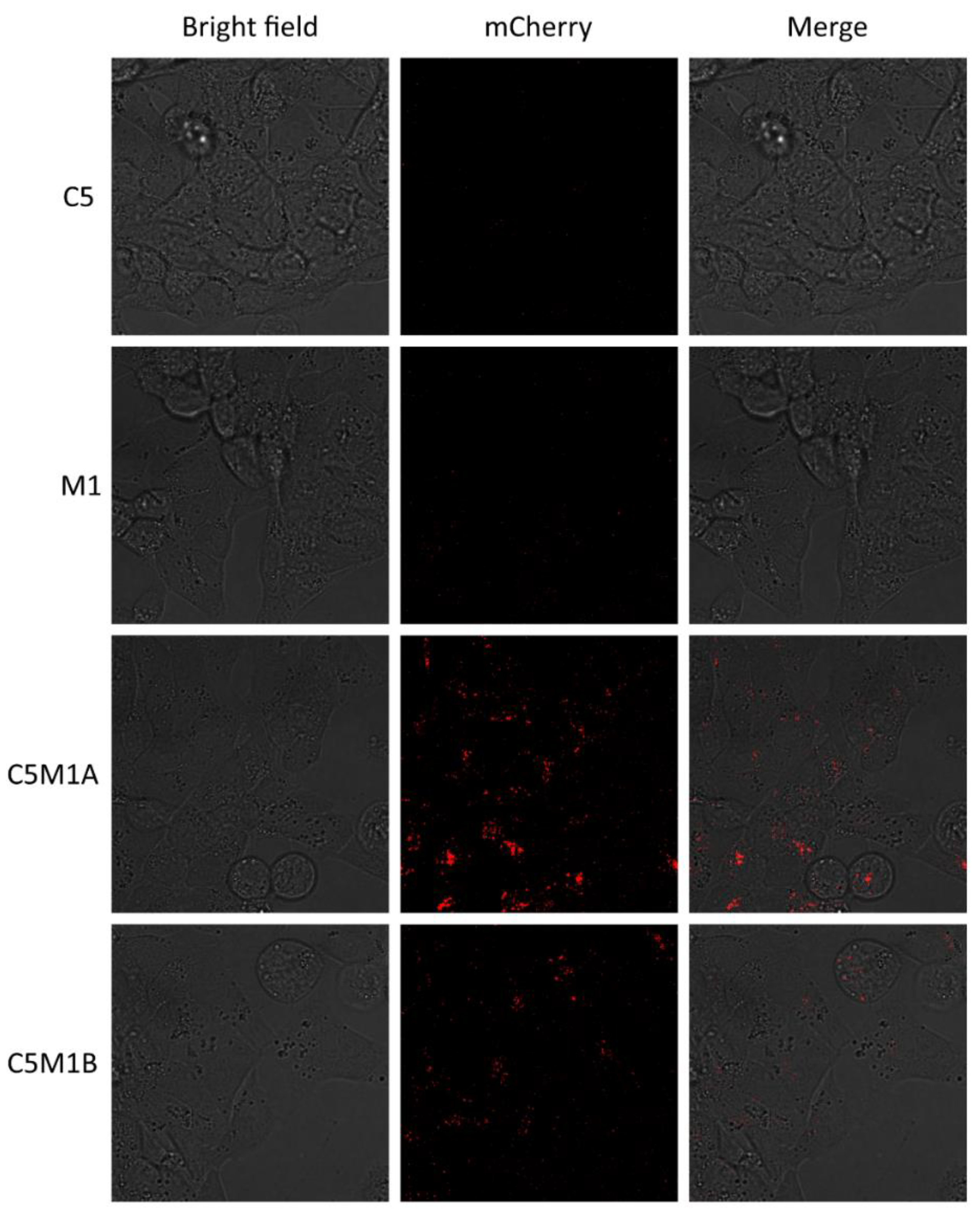
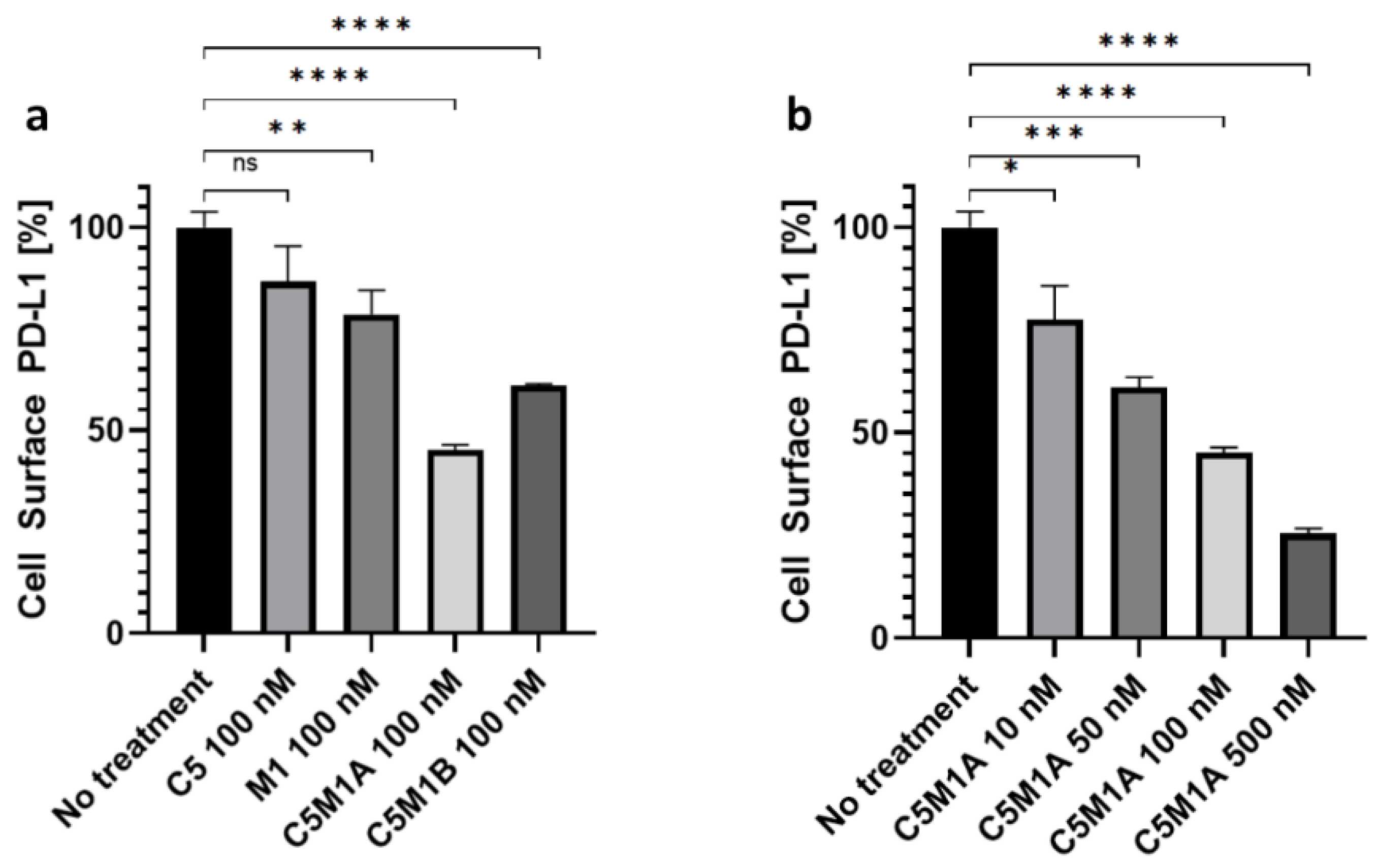
2.4. Bispecific Compounds Induce Internalization of Transmembrane PD-L1
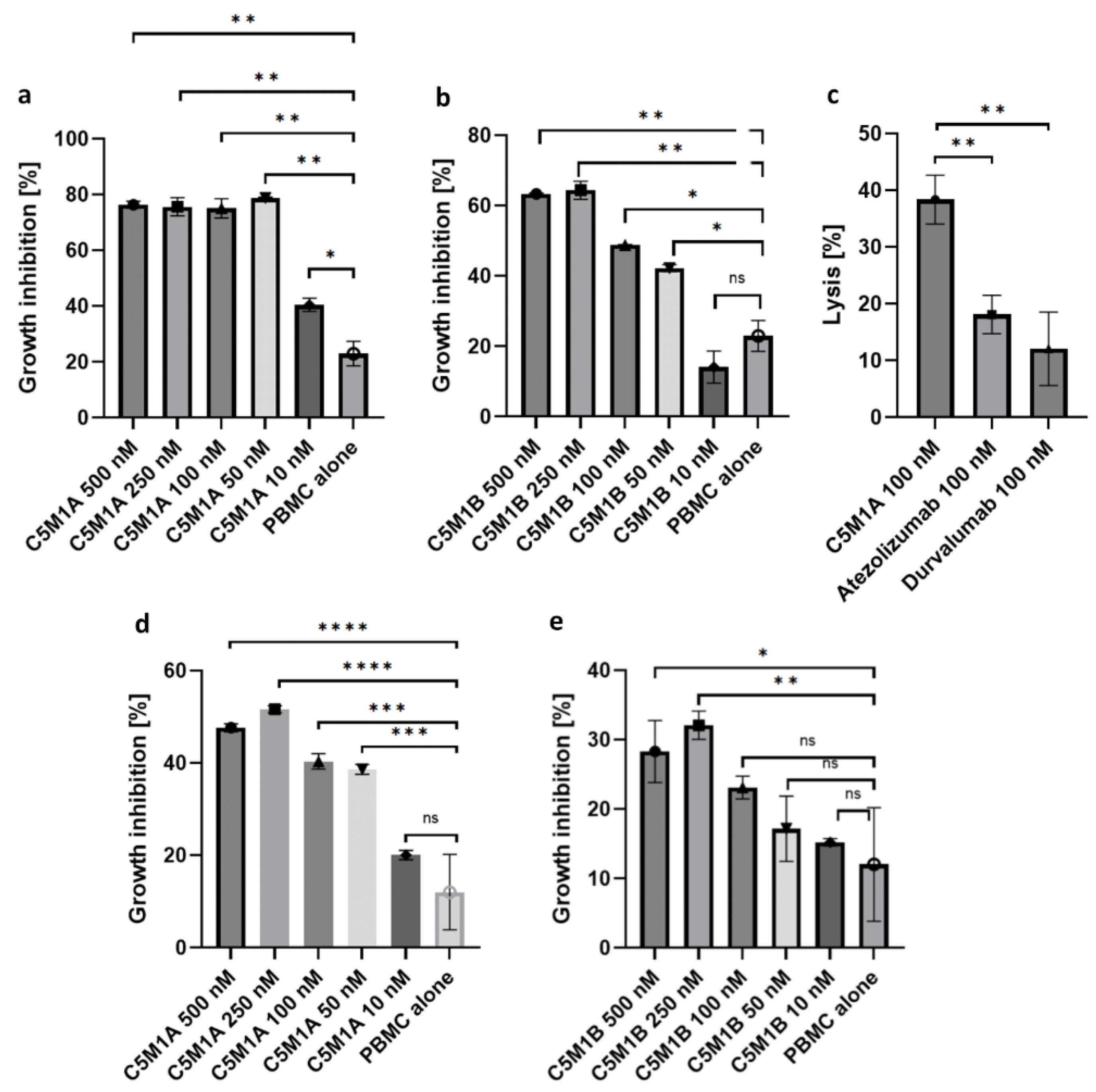
2.5. Bispecific Compounds Induce Tumor Cell Cytotoxicity When Incubated with PBMC
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Design of IGF2-Based Polypeptides with Abrogated IGF1R Binding
4.3. Stable Cell Lines Generation
4.4. Transient Protein Production in HEK293
4.5. Purification of Proteins
4.6. Indirect ELISA
4.7. Fluorescence Internalization Test
4.8. Flow Cytometry
4.9. PD-1/PD-L1 Blockade Bioassay
4.10. Human Tumor Cells Killing Assay
4.11. Cytotoxicity Test
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-Targeting Chimaeras for Degradation of Extracellular Proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs That Engage the Asialoglycoprotein Receptor for Targeted Protein Degradation. Nat. Chem. Biol. 2021, 17, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yun, Z.; Yuan, Z.; Tang, W. Targeted Degradation of Extracellular Secreted and Membrane Proteins. Trends Pharmacol. Sci. 2023, 44, 762–775. [Google Scholar] [CrossRef]
- Alabi, S.B.; Crews, C.M. Major Advances in Targeted Protein Degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, M.; Roux, A. Mechanisms of Clathrin-Mediated Endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Zhou, Y.; Teng, P.; Montgomery, N.T.; Li, X.; Tang, W. Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci. 2021, 7, 499–506. [Google Scholar] [CrossRef]
- Caianiello, D.F.; Zhang, M.; Ray, J.D.; Howell, R.A.; Swartzel, J.C.; Branham, E.M.J.; Chirkin, E.; Sabbasani, V.R.; Gong, A.Z.; McDonald, D.M.; et al. Bifunctional Small Molecules That Mediate the Degradation of Extracellular Proteins. Nat. Chem. Biol. 2021, 17, 947–953. [Google Scholar] [CrossRef]
- Zheng, J.; He, W.; Li, J.C.; Feng, X.; Li, Y.; Cheng, B.; Zhou, Y.; Li, M.; Liu, K.; Shao, X.; et al. Bifunctional Compounds as Molecular Degraders for Integrin-Facilitated Targeted Protein Degradation. J. Am. Chem. Soc. 2022, 144, 21831–21836. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lin, B.; Lu, Y.; Li, L.; Deng, K.; Zhang, S.; Zhang, H.; Yang, C.; Zhu, Z. Aptamer-LYTACs for Targeted Degradation of Extracellular and Membrane Proteins. Angew. Chem. Int. Ed. 2023, 62, e202218106. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Gao, Q.; Mao, M.; Zhang, C.; Yang, L.; Yang, Y.; Han, D. Bispecific Aptamer Chimeras Enable Targeted Protein Degradation on Cell Membranes. Angew. Chem. 2021, 133, 11367–11371. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; He, J.; Ou, C.; Donahue, T.C.; Muthana, M.M.; Su, L.; Wang, L.X. Site-Specific Chemoenzymatic Conjugation of High-Affinity M6P Glycan Ligands to Antibodies for Targeted Protein Degradation. ACS Chem. Biol. 2022, 17, 3013–3023. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.M.; Zhou, Y.; Teng, P.; Rault, L.N.; Liao, Y.; Tang, W. Development of Oligomeric Mannose-6-Phosphonate Conjugates for Targeted Protein Degradation. ACS Med. Chem. Lett. 2023, 14, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, H.; Tong, F.; Yun, C.; Xue, L.; Xu, J.; Jiang, X.; Cai, X. Harnessing the Lysosomal Sorting Signals of the Cation-Independent Mannose-6-Phosphate Receptor for Targeted Degradation of Membrane Proteins. J. Am. Chem. Soc. 2023, 145, 19107–19119. [Google Scholar] [CrossRef] [PubMed]
- Ahn, G.; Banik, S.M.; Bertozzi, C.R. Degradation from the Outside In: Targeting Extracellular and Membrane Proteins for Degradation through the Endolysosomal Pathway. Cell Chem. Biol. 2021, 28, 1072–1080. [Google Scholar] [CrossRef]
- Zhu, C.; Wang, W.; Wang, Y.; Zhang, Y.; Li, J. Dendronized DNA Chimeras Harness Scavenger Receptors to Degrade Cell Membrane Proteins. Angew. Chem. 2023, 62, e202300694. [Google Scholar] [CrossRef]
- Pance, K.; Gramespacher, J.A.; Byrnes, J.R.; Salangsang, F.; Serrano, J.-A.C.; Cotton, A.D.; Steri, V.; Wells, J.A. Modular Cytokine Receptor-Targeting Chimeras for Targeted Degradation of Cell Surface and Extracellular Proteins. Nat. Biotechnol. 2023, 41, 273–281. [Google Scholar] [CrossRef]
- Marei, H.; Tsai, W.-T.K.; Kee, Y.-S.; Ruiz, K.; He, J.; Cox, C.; Sun, T.; Penikalapati, S.; Dwivedi, P.; Choi, M.; et al. Antibody Targeting of E3 Ubiquitin Ligases for Receptor Degradation. Nature 2022, 610, 182–189. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Yu, L.; Huang, X.; Wang, X.; Han, D.; Yang, Y.; Liu, Z. Covalent LYTAC Enabled by DNA Aptamers for Immune Checkpoint Degradation Therapy. J. Am. Chem. Soc. 2023, 145, 1–16. [Google Scholar] [CrossRef]
- Howell, R.; Wang, R.; McDonald, D.; Spiegel, D.A. Bifunctional Molecules That Induce Targeted Degradation and Transcytosis of Extracellular Proteins in Brain Cells. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Loppinet, E.; Besser, H.A.; Lee, C.; Zhang, W.; Cui, B.; Khosla, C. Targeted Lysosomal Degradation of Secreted and Cell Surface Proteins through the LRP-1 Pathway. J. Am. Chem. Soc. 2023, 145, 18705–18710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Duque-Jimenez, J.; Brixi, G.; Facchinetti, F.; Rhee, K.; Feng, W.W.; Jänne, P.A.; Zhou, X. Transferrin Receptor Targeting Chimeras (TransTACs) for Membrane Protein Degradation. BioRxiv 2023. [Google Scholar] [CrossRef]
- Brown, J.; Esnouf, R.M.; Jones, M.A.; Linnell, J.; Harlos, K.; Hassan, A.B.; Jones, E.Y. Structure of a Functional IGF2R Fragment Determined from the Anomalous Scattering of Sulfur. EMBO J. 2002, 21, 1054–1062. [Google Scholar] [CrossRef]
- Brown, J.; Delaine, C.; Zaccheo, O.J.; Siebold, C.; Gilbert, R.J.; van Boxel, G.; Denley, A.; Wallace, J.C.; Hassan, A.B.; Forbes, B.E.; et al. Structure and Functional Analysis of the IGF-II/IGF2R Interaction. EMBO J. 2008, 27, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Prata, M.J.; Alves, S. Mannose-6-Phosphate Pathway: A Review on Its Role in Lysosomal Function and Dysfunction. Mol. Genet. Metab. 2012, 105, 542–550. [Google Scholar] [CrossRef]
- Gary-Bobo, M.; Nirde, P.; Jeanjean, A.; Morere, A.; Garcia, M. Mannose 6-Phosphate Receptor Targeting and Its Applications in Human Diseases. Curr. Med. Chem. 2007, 14, 2945–2953. [Google Scholar] [CrossRef]
- Bochel, A.J.; Williams, C.; McCoy, A.J.; Hoppe, H.-J.; Winter, A.J.; Nicholls, R.D.; Harlos, K.; Jones, E.Y.; Berger, I.; Hassan, A.B.; et al. Structure of the Human Cation-Independent Mannose 6-Phosphate/IGF2 Receptor Domains 7–11 Uncovers the Mannose 6-Phosphate Binding Site of Domain 9. Structure 2020, 28, 1300–1312. [Google Scholar] [CrossRef]
- Dahms, N.M.; Hancock, M.K. P-Type Lectins. Biochim. Biophys. Acta Gen. Subj. 2002, 1572, 317–340. [Google Scholar] [CrossRef]
- Ghobrial, G.; Araujo, L.; Jinwala, F.; Li, S.; Lee, L.Y. The Structure and Biological Function of CREG. Front. Cell Dev. Biol. 2018, 6, 1–7. [Google Scholar] [CrossRef]
- Sacher, M.; Di Bacco, A.; Lunin, V.V.; Zheng, Y.; Wagner, J.; Gill, G.; Cygler, M. The Crystal Structure of CREG, a Secreted Glycoprotein Involved in Cellular Growth and Differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 18326–18331. [Google Scholar] [CrossRef]
- Song, X.; Lasanajak, Y.; Olson, L.J.; Boonen, M.; Dahms, N.M.; Kornfeld, S.; Cummings, R.D.; Smith, D.F. Glycan Microarray Analysis of P-Type Lectins Reveals Distinct Phosphomannose Glycan Recognition. J. Biol. Chem. 2009, 284, 35201–35214. [Google Scholar] [CrossRef] [PubMed]
- Zaccheo, O.J.; Prince, S.N.; Miller, D.M.; Williams, C.; Kemp, C.F.; Brown, J.; Jones, E.Y.; Catto, L.E.; Crump, M.P.; Hassan, A.B. Kinetics of Insulin-like Growth Factor II (IGF-II) Interaction with Domain 11 of the Human IGF-II/Mannose 6-Phosphate Receptor: Function of CD and AB Loop Solvent-Exposed Residues. J. Mol. Biol. 2006, 359, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-Phosphate Receptors: New Twists in the Tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Rensen, P.C.N.; Sliedregt, L.A.J.M.; Ferns, M.; Kieviet, E.; van Rossenberg, S.M.W.; van Leeuwen, S.H.; van Berkel, T.J.C.; Biessen, E.A.L. Determination of the Upper Size Limit for Uptake and Processing of Ligands by the Asialoglycoprotein Receptor on Hepatocytesin Vitroandin Vivo. J. Biol. Chem. 2001, 276, 37577–37584. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative Molecular Formats and Therapeutic Applications for Bispecific Antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Brown, K.A.; Seol, H.; Pillai, D.K.; Sankoorikal, B.J.; Formolo, C.A.; Mac, J.T.; Edwards, N.J.; Rose, M.C.; Hathout, Y. The Human Secretome Atlas Initiative: Implications in Health and Disease Conditions. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2454–2461. [Google Scholar] [CrossRef]
- de Goeij, B.E.C.G.; Vink, T.; ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody–Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef]
- DeVay, R.M.; Delaria, K.; Zhu, G.; Holz, C.; Foletti, D.; Sutton, J.; Bolton, G.; Dushin, R.; Bee, C.; Pons, J.; et al. Improved Lysosomal Trafficking Can Modulate the Potency of Antibody Drug Conjugates. Bioconjug. Chem. 2017, 28, 1102–1114. [Google Scholar] [CrossRef]
- Zhu, G.; Chen, X. Aptamer-Based Targeted Therapy. Adv. Drug Deliv. Rev. 2018, 134, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Brody, E.N.; Gold, L. Aptamers as Therapeutic and Diagnostic Agents. Rev. Mol. Biotechnol. 2000, 74, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.W.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. Chapter Nineteen—Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Meth. Enzymol. 2011, 487, 545–574. [Google Scholar]
- Nivón, L.G.; Moretti, R.; Baker, D. A Pareto-Optimal Refinement Method for Protein Design Scaffolds. PLoS ONE 2013, 8, e59004. [Google Scholar] [CrossRef]
- Huang, P.-S.; Ban, Y.-E.A.; Richter, F.; Andre, I.; Vernon, R.; Schief, W.R.; Baker, D. RosettaRemodel: A Generalized Framework for Flexible Backbone Protein Design. PLoS ONE 2011, 6, e24109. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikitiuk, M.; Barczyński, J.; Bielski, P.; Arciniega, M.; Tyrcha, U.; Hec, A.; Lipińska, A.D.; Rychłowski, M.; Holak, T.A.; Sitar, T. IGF2 Peptide-Based LYTACs for Targeted Degradation of Extracellular and Transmembrane Proteins. Molecules 2023, 28, 7519. https://doi.org/10.3390/molecules28227519
Mikitiuk M, Barczyński J, Bielski P, Arciniega M, Tyrcha U, Hec A, Lipińska AD, Rychłowski M, Holak TA, Sitar T. IGF2 Peptide-Based LYTACs for Targeted Degradation of Extracellular and Transmembrane Proteins. Molecules. 2023; 28(22):7519. https://doi.org/10.3390/molecules28227519
Chicago/Turabian StyleMikitiuk, Michał, Jan Barczyński, Przemysław Bielski, Marcelino Arciniega, Urszula Tyrcha, Aleksandra Hec, Andrea D. Lipińska, Michał Rychłowski, Tad A. Holak, and Tomasz Sitar. 2023. "IGF2 Peptide-Based LYTACs for Targeted Degradation of Extracellular and Transmembrane Proteins" Molecules 28, no. 22: 7519. https://doi.org/10.3390/molecules28227519
APA StyleMikitiuk, M., Barczyński, J., Bielski, P., Arciniega, M., Tyrcha, U., Hec, A., Lipińska, A. D., Rychłowski, M., Holak, T. A., & Sitar, T. (2023). IGF2 Peptide-Based LYTACs for Targeted Degradation of Extracellular and Transmembrane Proteins. Molecules, 28(22), 7519. https://doi.org/10.3390/molecules28227519