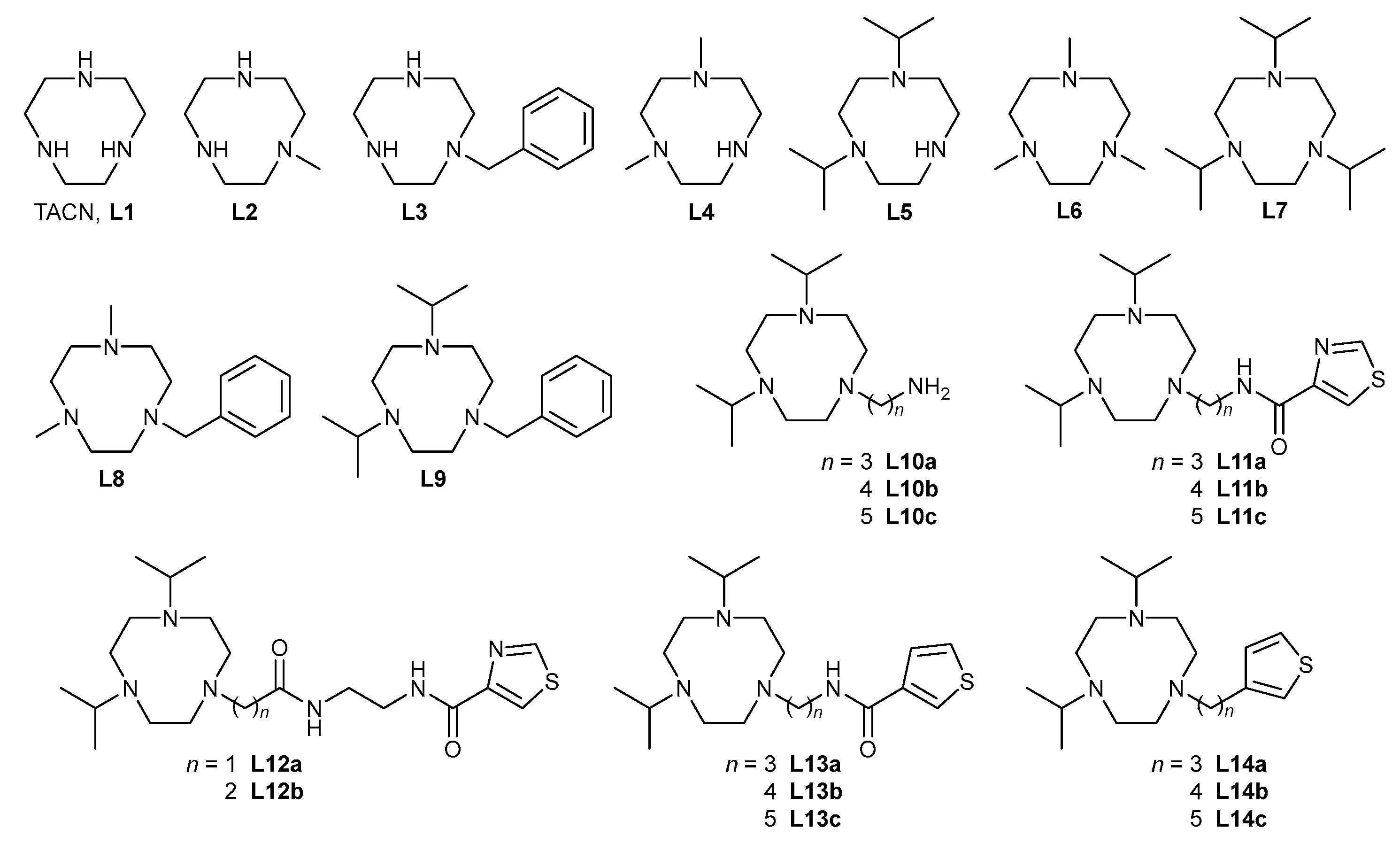
4.2. Synthesis
4.2.1. 1,4,7-Trimethyl-1,4,7-triazacyclononane (L6)
Compound
L6 was prepared using a modified version of the procedure reported in [
30].
Compound L1 (200 mg, 1.55 mmol) was dissolved in 37% aq. formaldehyde (8 mL, 108 mmol, 70 equiv.). To the solution, formic acid (2 mL, 53 mmol, 34 equiv.) was added, and the mixture was heated to reflux for 24 h. The mixture was evaporated to dryness in vacuo, and the residue was taken in 15 mL of 10% aq. NaOH. The product was extracted with CHCl3 (3 × 20 mL), and the organic phases were combined and dried over Na2SO4. The drying agent was filtered off, and volatiles were evaporated in vacuo, affording 232 mg (87%) of L6 in the form of a colourless oil.
NMR (CDCl3): 1H: δ 2.36 (s, 9H, CH3); 2.65 (s, 12H, CH2). 13C{1H}: δ 46.76 (3C, CH3); 57.06 (6C, CH2).
MS-ESI (+): 172.3 ([M + H]+, calc. 172.2).
4.2.2. 1,4,7-Tri(iso-propyl)-1,4,7-triazacyclononane (L7)
The compound L1 (1.00 g, 7.74 mmol) was dissolved in MeCN (50 mL). Potassium carbonate (6.41 g, 46.4 mmol, 6 equiv.) and iso-propyl bromide (3.14 g, 25.5 mmol, 3.3 equiv.) were added, and the mixture was stirred under a condenser at 60 °C for 24 h. The solids were filtered off using a S4 frit, and the filtrate was evaporated in vacuo. The residue was dissolved in CHCl3 (20 mL) and extracted with water (2 × 20 mL). The organic fraction was dried over Na2SO4, filtered and evaporated, yielding L7 as a colourless oil (1.43 g, 72%).
NMR (CDCl3): 1H δ 0.96 (d, 3JHH = 6.5, 18H, CH3); 2.63 (s, 12H, CH2); 2.86 (sept, 3JHH = 6.4, 3H, CH). 13C{1H}: δ 18.33 (6C, CH3); 52.73 (6C, CH2); 54.30 (3C, CH).
MS-ESI (+): 256.4 ([M + H]+, calc. 256.3).
4.2.3. 1-Benzyl-4,7-dimethyl-1,4,7-triazacyclononane (L8)
Compound L8 was prepared and isolated using a procedure analogous to the procedure described above for L6. Starting with 3.00 g (13.7 mmol) of L3, 37% aq. formaldehyde (50 mL, 670 mmol, 49 equiv.) and formic acid (20 mL, 530 mmol, 39 equiv.), product L8 was obtained in the form of a yellowish oil. Yield: 3.23 g (95%).
NMR (CDCl3): 1H: δ 2.35 (s, 6H, CH3); 2.66 (m, 4H, CH2(cycle)); 2.73 (m, 4H, CH2(cycle)); 2.82 (s, 4H, CH2(cycle)); 3.66 (s, 2H, CH2Ph); 7.30 (m, 5H, arom.). 13C{1H}: δ 46.61 (2C); 56.04 (2C); 56.82 (2C); 57.09 (2C); 63.42 (1C, CH2Ph); 126.75 (1C), 129.08 (2C), 129.12 (2C) and 140.25 (1C), all arom.
MS-ESI (+): 248.3 ([M + H]+, calc. 248.4).
4.2.4. 1-Benzyl-4,7-di(iso-propyl)-1,4,7-triazacyclononane (L9)
Compound L9 was prepared and isolated using a procedure analogous to the procedure described above for L7. L3∙2HCl∙H2O (2.00 g, 6.44 mmol) was suspended in MeCN (50 mL), and K2CO3 (2.67 g, 19.3 mmol, 3 equiv.) was added. To the stirred suspension, iso-propyl bromide (3.96 g, 32.2 mmol, 5 equiv.) was added, and the mixture was stirred at 60 °C overnight. After a work-up similar to L7, product L9 was obtained in the form of a yellowish oil. Yield: 1.93 g (98%).
NMR (CDCl3): 1H: δ 0.97 (d, 3JHH = 6.4, 12H, CH3); 2.50–2.72 (m, 8H, CH2(cycle)); 2.81–2.97 (m, 6H, CH2(cycle) + CH); 3.67 (s, 2H, CH2Ph); 7.30 (m, 5H, arom.). 13C{1H}: δ 18.42 (4C, CH3); 52.41, 52.76 and 54.79 (all 2C, CH2(cycle)); 55.37 (2C, CH); 62.21 (1C, CH2Ph); 126.63 (1C), 128.13 (2C), 129.02 (2C) and 140.52 (1C), all arom.
MS-ESI (+): 304.2 ([M + H]+, calc. 304.3).
4.2.5. 1,4-Dimethyl-1,4,7-triazacyclononane (L4) and 1,4-di(iso-propyl)-1,4,7-triazacyclononane (L5)
Compound L8 or L9 (2.00 g) was dissolved in a mixture of methanol (30 mL), water (5 mL) and acetic acid (10 mL). A Pd/C catalyst (15% wt., 200 mg) was added, and the reaction mixture was stirred overnight at room temperature under a hydrogen atmosphere at atmospheric pressure (balloon). The suspension was filtered, and the filtrate was evaporated to dryness and than co-evaporated with water (3 × 20 mL). The residue was then taken into 20 mL of 10% aq. NaOH, and the product (L4 or L5, respectively) was extracted using CHCl3 (3 × 30 mL). Organic fractions were combined and dried over Na2SO4. The drying agent was filtered off, and the volatiles were evaporated in vacuo, providing products in the form of light-yellow oils.
L4: Yield: 1.08 g (85%).
NMR (CDCl3): 1H: δ 2.64 (m, 4H, CH2(cycle)); 2.50 (m, 8H, CH2(cycle)); 2.37 ppm (s, 6H, CH3). 13C{1H}: δ 45.46, 46.22, 53.40 and 54.45 (all 2C, CH2(cycle) and CH3).
MS-ESI (+): 158.2 ([M + H]+, calc. 158.3).
L5: Yield: 1.16 g (84%).
NMR (CDCl3): 1H: δ 1.00 (d, 3JHH = 6.7, 12H, CH3); 2.48 (s, 4H, CH2(cycle)); 2.58 (m, 4H, CH2(cycle)), 2.68 (m, 4H, CH2(cycle)); 2.86 (sept, 3JHH = 6.7, 2H, CH). 13C{1H}: δ 18.81 (4C, CH3); 47.05, 47.78 and 48.99 (all 2C, CH2(cycle)); 53.05 (s, 2C, CH).
MS-ESI (+): 214.3 ([M + H]+, calc. 214.2).
4.2.6. 1-[ω-(Phthalimido)-ALKYL]-4,7-di(iso-propyl)-1,4,7-triazacyclononane (alkyl: 2a—propyl, 2b—butyl, 2c—pentyl)
General procedure: the compound L5 was dissolved in MeCN; 30 mL per 1 g of the starting compound was used. Then, K2CO3 (3 equiv.) and alkylating agent 1a–c (1.1 equiv.) were added, and the mixture was stirred at 60 °C for 2 days. Then, the mixture was filtered through an S4 frit and the filtrate was evaporated to dryness. The residue was dissolved in CHCl3 (30 mL) and extracted using water (2 × 20 mL). The organic fraction was dried over Na2SO4 and evaporated, yielding a bright yellow oil which was purified using flash chromatography on reverse-phase silica gel (GM1). After combining and evaporating the fractions containing the product, compounds were isolated as oily materials containing non-stoichiometrical amounts of TFA, which prevented the exact calculation of the yields (but no solvent remains were present, according to NMR spectroscopy).
2a: Starting from 0.45 g (2.1 mmol) of L5, the product 2a·xTFA was obtained in a yield of 0.63 g.
NMR (D2O): 1H: δ 1.38 (d, 3JHH = 6.5, 12H, CH3); 1.89–2.02 (m, 2H, CH2CH2CH2); 2.82–2.89 (m, 4H, CH2(cycle)); 3.03 (sept, 3JHH = 6.4, 2H, CH); 3.10 (br s, 2H, CH2CH2CH2); 3.24 (br s, 2H, CH2CH2CH2); 3.54–3.80 (m, 8H, CH2(cycle)); 7.82 (m, 4H, arom.). 13C{1H}: δ 14.91 (4C, CH3); 17.18, 22.66 and 35.67 (all 1C, CH2CH2CH2); 45.25, 47.39, 51.65 and 59.85 (all 2C, CH2(cycle) and CH); 123.34, 131.21 and 134.76, (all 2C, arom.); 170.63 (2C, CO).
MS-ESI (+): 401.5 ([M + H]+, calc. 401.3).
2b: Starting from 0.48 g (2.2 mmol) of L5, the product 2b·xTFA was obtained in a yield of 0.71 g.
NMR (D2O): 1H: δ 1.35 (d, 3JHH = 7.2, 12H, CH3); 1.53–1.71 (m, 4H, CH2CH2CH2CH2); 2.83–2.91 (m, 2H, CH2CH2CH2CH2); 2.99–3.17 (m, 4H, CH2(cycle)); 3.24 (sept, 3JHH = 6.9, 2H, CH); 3.34–3.49 (m, 2H, CH2CH2CH2CH2); 3.56 (s, 4H, CH2(cycle)); 3.66 (m, 4H CH2(cycle)); 7.79 (m, 4H, arom.). 13C{1H}: δ 20.19 (4C, CH3); 22.17, 26.17, 42.27 and 50.35 (all 1C, CH2CH2CH2CH2); 52.26, 53.08, 59.61 and 64.55 (all 2C, CH2(cycle) and CH); 128.32, 136.16 and 139.81 (all 2C, arom.); 175.75 (2C, CO).
MS-ESI (+): 415.4 ([M + H]+, calc. 415.3).
2c: Starting from 0.66 g (3.1 mmol) of L5, the product 2c·xTFA was obtained in a yield of 0.94 g.
NMR (D2O): 1H: δ 1.35 (d, 3JHH = 6.1, 12H, CH3); 1.63–1.71 (m, 6H, CH2CH2CH2CH2CH2); 2.83–2.95 (m, 2H, CH2CH2CH2CH2CH2); 3.07–3.29 (m, 6H, CH2(cycle) and CH2CH2CH2CH2CH2), 3.41 (sept, 3JHH = 6.1, 2H, CH); 3.50 (s, 4H, CH2(cycle)); 3.59–3.65 (m, 4H CH2(cycle)); 7.79 (m, 4H, arom.). 13C{1H}: δ 18.59 (4C, CH3); 19.93, 25.97, 26.49, 40.46 and 48.50 (all 1C, CH2CH2CH2CH2CH2); 49.88, 51.41, 58.50 and 62.07 (all 2C, CH2(cycle) and CH); 126.27, 134.23 and 137.73 (all 2C, arom.); 173.79 (2C, CO).
MS-ESI (+): 429.5 ([M + H]+, calc. 429.3).
4.2.7. 1-[ω-(Amino)-alkyl]-4,7-di(iso-propyl)-1,4,7-triazacyclononane (alkyl: L10a—propyl, L10b—butyl, L10c—pentyl)
General procedure: the chosen compound 2a–c (in the form of non-stoichiometric trifluoroacetate) was dissolved in a mixture of EtOH (10 mL) and hydrazine monohydrate (80%, 10 mL), and the reaction mixture was refluxed overnight. The solvents were evaporated, the solid residue was dissolved in EtOH and filtered to remove most of phtalhydrazide formed during deprotection and the filtrate was evaporated to dryness. However, there was still some phtalhydrazide present in the residue; therefore, it was further hydrolysed. The residue was dissolved in EtOH (20 mL), and an excess of solid NaOH (1 g) was added. The mixture was heated to reflux overnight. Volatiles were evaporated, and the residue was dissolved in water and extracted using DCM (3 × 30 mL). Organic fractions were combined, dried over Na2SO4, filtered and evaporated, yielding L10a–c, respectively, as viscous, light-yellow oils.
L10a: Starting from 0.63 g of 2a∙xTFA, product L10a was obtained in a yield of 0.37 g (65% to L5 over two steps).
NMR (CDCl3): 1H: δ 0.97 (d, 3JHH = 6.6, 12H, CH3); 1.63 (p, 3JHH = 6.8, 2H, CH2CH2CH2); 2.56–2.61 (m, 6H, CH2(cycle) and CH2CH2CH2); 2.65–2.72 (m, 4H, CH2(cycle)); 2.72–2.79 (m, 2H, CH2CH2CH2); 2.81–2.86 (m, 4H, CH2(cycle)); 2.91 (sept, 3JHH = 6.5, 2H, CH). 13C{1H}: δ 18.31 (4C, CH3); 31.69 (2C, CH2CH2CH2); 40.80, 52.39, 52.47, 54.70, 55.38 and 55.97 (CH2CH2CH2, CH2(cycle) and CH).
MS-ESI (+): 271.4 ([M + H]+, calc. 271.3).
L10b: Starting from 0.71 g of 2b∙xTFA, product L10b was obtained in a yield of 0.44 g (69% to L5 over two steps).
NMR (CDCl3): 1H: δ 1.00 (d, 3JHH = 6.7, 12H, CH3); 1.34–1.50 (m, 2H, CH2CH2CH2CH2); 1.50–1.65 (m, 2H, CH2CH2CH2CH2); 2.62 (t, 3JHH = 7.1, 2H, CH2CH2CH2CH2); 2.69–2.80 (m, 6H, CH2(cycle) and CH2CH2CH2CH2); 2.80–2.87 (m, 4H, CH2(cycle)); 2.89 (s, 4H, CH2(cycle)); 3.24 (sept, 3JHH = 6.6, 2H, CH). 13C{1H}: δ 18.41 (4C, CH3); 25.04 and 31.57 (all 1C, CH2CH2CH2CH2); 42.07, 51.89, 52.04, 54.64, 54.79 and 57.83 (CH2CH2CH2CH2, CH2(cycle) and CH).
MS-ESI (+): 285.2 ([M + H]+, calc. 285.3).
L10c: Starting from 0.94 g of 2c∙xTFA, product L10c was obtained in a yield of 0.63 g (68% to L5 over two steps).
NMR (CDCl3): 1H: δ 0.99 (d, 3JHH = 6.7, 12H, CH3); 1.32 (p, 3JHH = 7.7, 2H, CH2CH2CH2CH2CH2); 1.43–1.53 (m, 4H, CH2CH2CH2CH2CH2); 2.46–2.56 (m, 2H, CH2CH2CH2CH2CH2); 2.56–2.64 (m, 4H, CH2(cycle)); 2.64–2.76 (m, 6H, CH2(cycle) and CH2CH2CH2CH2CH2); 2.83–2.90 (m, 4H, CH2(cycle)); 2.93 (sept, 3JHH = 6.6, 2H, CH). 13C{1H}: δ 18.30 (4C, CH3); 24.16, 25.98, 29.85 and 30.40 (all 1C, CH2CH2CH2CH2CH2); 40.78, 46.80, 49.82, 53.80 and 55.45 (CH2CH2CH2CH2CH2, CH2(cycle) and CH).
MS-ESI (+): 299.4 ([M + H]+, calc. 299.3).
4.2.8. 1-[ω-(Thiazole-4-ylcarboxamido)alkyl]-4,7-di(iso-propyl)-1,4,7-triazacyclononane (alkyl: L11a—propyl, L11b—butyl, L11c—pentyl)
General procedure: the chosen amine L10a–c (1 equiv.) was dissolved in DMF (20 mL). To this solution, Et3N (1 equiv.) was added. In another flask, to a prepared solution of thiazole-4-carboxylic acid (1.5 equiv.) and Et3N (2 equiv.) in DMF (20 mL), HATU (1.5 equiv.) was added. After 5 min, the solution containing amine L10a–c was added dropwise to the flask containing the thiazole-4-carboxylic acid–HATU solution, and the resulting mixture was stirred at room temperature overnight. The reaction mixture was evaporated to dryness in vacuo, and the residue was purified using column chromatography on silica gel (EtOH:conc. aq. NH3 5:1). Product-containing fractions were combined and evaporated, yielding the ligands L11a–c as light yellow oils.
L11a: Starting from 0.3 g (1.11 mmol) of L10a, product L11a was obtained in a yield of 0.05 g (12%).
NMR (CDCl3): 1H: δ 1.03 (d, 3JHH = 6.6, 12H, CH3); 1.83–1.95 (m, 2H, CH2CH2CH2); 2.67 (br s, 4H, CH2(cycle)); 2.82 (br s, 6H CH2(cycle) and CH2CH2CH2); 3.00 (sept, 3JHH = 6.7, 2H, CH); 3.09 (br s, 4H, CH2(cycle)); 3.56 (q, 3JHH = 6.4, 2H, CH2NHCO); 8.26 (d, 4JHH = 2.1, 1H, CCHS); 8.29 (s, 1H, NH); 8.76 (d, 4JHH = 2.1, 1H, NCHS). 13C{1H}: δ 18.25 (4C, CH3); 18.66, 26.49 and 37.74 (all 1C, CH2CH2CH2); 50.18, 52.53 and 54.20 (very br, CH2(cycle)); 54.63 (2C, CH); 123.05, 151.40 and 152.66 (all 1C, thiazole); 161.32 (1C, CO).
MS-ESI (+): 382.4 ([M + H]+, calc. 382.3).
L11b: Starting from 0.13 g (0.5 mmol) of L10b, product L11b was obtained in a yield of 0.03 g (17%).
NMR: 1H (CD3OD): δ 1.03 (d, 3JHH = 6.6, 12H, CH3); 1.50–1.74 (m, 4H, CH2CH2CH2CH2); 2.54–2.64 (m, 2H, CH2CH2CH2CH2); 2.68 (s, 4H, CH2(cycle)); 2.71-2.80 (m, 4H, CH2(cycle)); 3.00 (sept, 3JHH = 6.8, 2H, CH); 3.44 (t, JHH = 6.3, 2H, CH2NHCO); 8.23 (d, 4JHH = 2.0, 1H, CCHS); 9.00 (d, 4JHH = 2.0, 1H, NCHS). 13C{1H} (CDCl3): δ 18.32 (4C, CH3); 23.73 and 27.12 (all 1C, CH2CH2CH2CH2); 38.77, 46.36, 46.71, 49.62, 53.69 and 55.17 (CH2CH2CH2CH2, CH2(cycle) and CH); 123.23, 151.41 and 153.14 (all 1C, thiazole); 161.37 (1C, CO).
MS-ESI (+): 396.4 ([M + H]+, calc. 396.3).
L11c: Starting from 0.30 g (1.01 mmol) of L10c, product L11c was obtained in a yield of 0.12 g (29%).
NMR (CD3OD): 1H: δ 1.02 (d, 3JHH = 6.5, 12H, CH3); 1.36–1.46 (m, 2H, CH2CH2CH2CH2CH2); 1.50–1.59 (m, 2H, CH2CH2CH2CH2CH2); 1.66 (p, 3JHH = 7.2, 2H, CH2CH2CH2CH2CH2); 2.55 (t, 3JHH = 7.6, 2H, CH2CH2CH2CH2CH2); 2.64 (s, 4H, CH2(cycle)), 2.69–2.77 (m, 4H, CH2(cycle)), 2.89–2.99 (m, 6H, CH2(cycle) and CH); 3.41 (t, 3JHH = 7.0, 2H, CH2NHCO); 8.22 (d, 4JHH = 2.0, 1H, CCHS); 8.99 (d, 4JHH = 2.2, 1H, NCHS). 13C{1H}: 18.48 (4C, CH3); 25.89, 28.01, 30.43 and 40.23 (all 1C, CH2CH2CH2CH2CH2); 50.35, 50.80 and 52.95 (all very br s, CH2(cycle)), 55.43 and 57.81 (CH2CH2CH2CH2CH2 and CH); 124.61, 152.07 and 155.36 (all 1C, thiazole); 163.44 (1C, CO).
MS-ESI (+): 410.4 ([M + H]+, calc. 410.3).
4.2.9. N-(2-Aminoethyl)-4-thiazolecarboxamide (5)
To a stirring solution of methyl-4-thiazolecarboxylate 4 (0.8 g, 5.6 mmol) in MeOH (30 mL), ethylenediamine (1 equiv., 0.34 g) was added, and the mixture was refluxed overnight. The solvent was evaporated, and the crude product containing both the monomeric product and its dimer was purified via column chromatography on silica gel, using EtOH/NH3 20/1 (v/v) as a mobile phase. Fractions containing the product were combined and evaporated, yielding 0.32 g (34%) of 5 in the form of a yellow oil.
NMR (CDCl3): 1H: δ 1.69 (s, 2H, NH2); 2.93 (t, 3JHH = 6.0, 2H, CH2), 3.50 (q, 3JHH = 6.0, 2H, CH2), 7.77 (s, 1H, NH), 8.16 (d, 4JHH = 2.1, 1H, CCHS), 8.74 (d, 4JHH = 2.2, 1H, NCHS). 13C{1H}: δ 41.63 and 42.24 (all 1C, CH2CH2); 123.24, 151.23 and 152.83 (all 1C, thiazole); 161.40 (1C, CO).
MS-ESI: (+) 144.1 ([M + H]+, calc. 144.0).
4.2.10. N-[2-(2-Chloroacetyl)amidoethyl]-4-thiazolecarboxamide (6a) and N-[2-(3-chloropropionyl)amidoethyl]-4-thiazolecarboxamide (6b)
To a stirring solution of N-(2-aminoethyl)-4-thiazolecarboxamide 5 and Et3N (1.2 equiv.) in dry DCM, chloroacetyl chloride (1.2 equiv.) in dry DCM was added dropwise. During the addition, the mixture was cooled to 0 °C in an ice–water bath. After the addition, the mixture was allowed to warm up to room temperature and was left stirring overnight. Volatiles were evaporated, and the resulting reaction mixture was purified using column chromatography on silica gel, using MeOH:conc. aq. NH3 20:1 (6a) or ethyl acetate/methanol 3:1 (6b) as mobile phases. Fractions containing products were combined and evaporated, yielding compound 6a in form of a yellow powder and compound 6b as a yellow oil.
6a: Starting from 0.24 g of 5, product 6a was obtained in a yield of 0.13 g (37%).
NMR (CDCl3): 1H: δ 3.57 (m, 2H, CH2CH2); 3.66 (m, 2H, CH2CH2); 4.05 (s, 2H, CH2Cl); 7.31 (s, 1H, NH); 7.79 (s, 1H, NH); 8.21 (d, 4JHH = 2.1, 1H, CCHS), 8.78 (d, 4JHH = 2.1, 1H, NCHS); 13C{1H}: δ 39.25, 40.91 and 42.65 (all 1C, CH2); 123.67, 150.52 and 152.96 (all 1C, thiazole); 162.10 and 166.93 (all 1C, CO).
MS-ESI: (+) 247.7 ([M + H]+, calc. 248.0).
6b: Starting from 0.19 g of 5, product 6a was obtained in a yield of 0.18 g (62%) at 70% purity, according to 1H NMR.
NMR (CDCl3): 1H δ 2.66 (t, 3JHH = 6.6, 2H, CH2CO); 3.51–3.75 (m, 4H, NCH2CH2N); 3.80 (t, 3JHH = 6.5, 2H, CH2Cl); 6.55 (s, 1H, NH); 7.80 (s, 1H, NH); 8.20 (d, 4JHH = 2.1, CCHS), 8.79 (d, 4JHH = 2.1, 1H, NCHS). 13C{1H}: δ 39.37, 39.71, 40.22 and 40.98 (all 1C, CH2); 123.66, 150.73 and 153.05 (all 1C, thiazole); 162.52 and 170.18 (all 1C, CO).
MS-ESI: (+) 261.7 ([M + H]+, calc. 262.0).
4.2.11. {N-[N-(Thiazol-4-yl-carbonyl)-2-amidoethyl]amidocarbonylmethyl}-4,7-di(iso-propyl)-1,4,7-triazacyclononane (L12a) and (2-{N-[N-(thiazol-4-yl-carbonyl)-2-amidoethyl]amidocarbonyl}ethyl)-4,7-di(iso-propyl)-1,4,7-triazacyclononane (L12b)
To a stirred suspension of L5 (ca 0.1 g, 1 equiv.) and K2CO3 (4 equiv.) in MeCN (30 mL), alkylating agent 6a or 6b (1.1 equiv.) was added, and the mixture was heated to reflux overnight (the alkylating reagent 6 dissolved at a higher temperature). After filtration through S4 frit, the filtrate was evaporated to dryness. Work-up for L12a: The residue was dissolved in DCM and extracted with water (3 × 15 mL). The organic fraction was dried over Na2SO4, filtered and evaporated. Product L12a was obtained in the form of light brown oil. Work-up for L12b: The reaction mixture was purified using reversed-phase flash chromatography (GM2) and the fraction containing the product was evaporated, yielding L12b·xTFA in the form of a light yellow oil.
L12a: Starting from 0.1 g (0.5 mmol) of L5, product L12a was obtained in a yield of 0.09 g (47%).
NMR (CDCl3): 1H: δ 0.92 (d, 3JHH = 6.6, 12H, CH3); 2.53–2.64 (m, 12H, CH2(cycle)); 2.76–2.85 (sept, 3JHH = 6.5, 2H, CH); 3.26 (s, 2H, NCH2CO); 3.46–3.53 (m, 2H, CONHCH2); 3.59 (q, JHH = 5.9, 2H, CONHCH2); 7.99 (s, 1H, NH), 8.14 (d, 4JHH = 2.1, 1H, CCHS); 8.75 (d, 4JHH = 2.2, 1H, NCHS); 9.71 (s, 1H, NH). 13C{1H}: δ 18.33 (4C, CH3); 38.70, 40.27, 49.96 and 54.90 (all 2C, CH2(cycle) and CH); 55.36 (1C, CH2CO); 59.48 and 61.59 (all 1C, CONHCH2CH2NHCO); 122.99, 151.38 and 152.77 (all 1C, thiazole); 161.37 and 174.49 (all 1C, CO).
MS-ESI: (+) 425.4 ([M + H]+, calc.425.3).
L12b: Starting from 0.13 g (0.6 mmol) of L5, product L12b·xTFA was obtained in a yield of 0.22 g.
NMR (D2O): 1H (80 °C): δ 1.87 (d, 3JHH = 6.6, 12H, CH3); 3.03 (t, 3JHH = 6.9, 2H, NCH2CH2CO); 3.50 (t, 3JHH = 6.9, 2H, NCH2CH2CO); 3.57 (br s, 4H, CH2(cycle)); 3.78–3.95 (br m, 4H, CH2(cycle)); 4.05–4.09 (m, 2H, NHCH2CH2NH); 4.09–4.13 (m, 2H, NHCH2CH2NH); 4.15–4.24 (m, 4H, CH2(cycle) and CH); 8.82 (d, 4JHH = 2.0, 1H, CCHS); 9.59 (d, 4JHH = 2.0, 1H, NCHS). 13C{1H} (25 °C): δ 15.63 (4C, CH3); 16.57, 31.28, 38.81, 38.82, 46.48 and 46.87 (CH2(cycle), CH and NCH2CH2CO); 49.35 and 60.00 (all 1C, CONHCH2CH2NHCO); 125.09, 148.87 and 155.91 (all 1C, thiazole); 163.45 and 176.26 (all 1C, CO).
MS-ESI: (+) 439.5 ([M + H]+, calc. 439.3).
4.2.12. 1-[ω-(Thiophene-3-ylcarboxamido)alkyl]-4,7-di(iso-propyl)-1,4,7-triazacyclononane (Alkyl: L13a—propyl, L13b—butyl, L13c—pentyl)
General procedure: 3-thiophenecarboxylic acid was dissolved in SOCl2 (excess), and the mixture was heated to reflux for 3 h. Excess SOCl2 was evaporated, and the 3-thiophenecarbonyl chloride (7) formed was used in the next step without purification. To a mixture of 10a–c (1 equiv.) and Et3N (3 equiv.) in DCM, 3-thiophenecarbonyl chloride (7) (1.1 equiv.) was added, and the mixture was left stirring at room temperature overnight. Then, the solvent was evaporated, and the product was purified using reversed-phase flash chromatography (GM2). Product-containing fractions were combined and evaporated, yielding L13a–c·xTFA as light-yellow oils.
L13a: Starting from 82 mg (0.3 mmol) of L10a, 24 mg of product L13a was obtained.
NMR (CDCl3): 1H: δ 1.35 (d, 3JHH = 6.5, 12H, CH3); 1.89–1.99 (m, 2H, CH2CH2CH2); 2.94 (m, 2H, CH2CH2CH2); 3.01–3.52 (br m, 14H, CH2(cycle) and CH2CH2CH2); 3.60 (p, 3JHH = 6.6, 2H, CH); 7.28 (s, 1H, NH); 7.32 (dd, 3JHH = 5.1, 4JHH = 2.9, 1H, SCHCH); 7.51 (dd, 3JHH = 5.1, 4JHH = 1.2, 1H, SCHCH); 8.06 (dd, 4JHH = 2.9 and 1.2, 1H, SCHC). 13C{1H}: δ 16.77 (4C, CH3); 17.01 (very br, 1C, CH2CH2CH2); 25.08 (1C, CH2CH2CH2); 37.43, 46.53, 47.12, 49.09 and 59.45 (all very br, CH2CH2CH2, CH2(cycle) and CH); 126.47, 129.19, 136.67 and 160.84 (all 1C, thiophene); and 164.22 (1C, CO).
MS-ESI: (+) 381.5 ([L13a + H]+, calc. 381.3).
L13b: Starting from 57 mg (0.2 mmol) of L10b, 21 mg of product L13b·xTFA was obtained.
NMR (D2O): 1H: δ 1.33 (d, 3JHH = 6.6, 12H, CH3); 1.58–1.70 (m, 4H, CH2CH2CH2CH2); 2.92–2.86 (m, 2H, CH2CH2CH2CH2); 3.03–3.29 (br m, 6H, CH2(cycle) and CH2CH2CH2CH2); 3.34–3.45 (m, 4H, CH2(cycle)); 3.53 (s, 4H, CH2(cycle)); 3.64 (sept, 3JHH = 6.6, 2H, CH); 7.43 (dd, 3JHH = 5.1, 4JHH = 1.4, 1H, SCHCH); 7.53 (dd, 3JHH = 5.1, 4JHH = 2.9, 1H, SCHCH); 8.02 (dd, 4JHH = 3.0 and 1.3, 1H, SCHC). 13C{1H}: δ 19.46 (4C, CH3); 21.29 and 25.39 (all 1C, CH2CH2CH2CH2); 43.13, 49.56, 52.43, 59.27 and 63.63 (CH2CH2CH2CH2, CH2(cycle) and CH); 130.02, 131.72, 133.76 and 140.11 (all 1C, thiophene); 170.22 (1C, CO).
MS-ESI: (+) 395.5 ([L13b + H]+, calc. 395.3).
L13c: Starting from 130 mg (0.4 mmol) of L10c, 85 mg of product L13c was obtained.
NMR (D2O): 1H: δ 1.30 (d, 3JHH = 6.6, 12H, CH3); 1.35–1.41 (m, 2H, CH2CH2CH2CH2CH2); 1.56–1.72 (m, 4H, CH2CH2CH2CH2CH2); 2.95–3.46 (br m, 14H, CH2(cycle) + CH); 3.47–3.58 (m, 4H, CH2CH2CH2CH2CH2); 6.74 (s, 1H, NH); 7.32 (d, 3JHH = 5.1, 1H, SCHCH); 7.46 (d, 3JHH = 5.1, 1H, SCHCH); 7.97 (br s, 1H, SCHC). 13C{1H}: δ 16.38 and 17.49 (4C, CH3); 23.88, 24.34 and 28.77 (all 1C, CH2CH2CH2CH2CH2); 40.00, 46.28, 47.53, 49.23, 56.42 and 59.69 (CH2CH2CH2CH2CH2, CH2(cycle) and CH); 126.52, 128.08, 130.12 and 136.65 (all 1C, thiophene); 166.57 (1C, CO).
MS-ESI: (+) 409.5 ([L13c + H]+, calc. 409.3).
4.2.13. 1,4-bis(tert-Butyloxycarbonyl)-1,4,7-triazonane (8)
Compound
8 was prepared using a modified procedure previously published in [
24]. To a stirred solution of TACN (1.50 g, 11.6 mmol) and Et
3N (2.27 mL, 16.3 mmol, 1.4 equiv.) in anhydrous CHCl
3 (20 mL) in an ice bath, Boc
2O (4.31 g, 19.7 mmol, 1.7 equiv.) dissolved in anhydrous CHCl
3 (10 mL) was added dropwise over the course of 1 h. The mixture was then slowly warmed to room temperature and left stirring overnight. The solvent was removed under reduced pressure to yield a viscid, colourless oil which was purified via silica gel chromatography (DCM:MeOH 15:1) to produce pure product
8 as a colourless oil (2.12 g, 67%). The characterisation data are identical to those found in [
24].
4.2.14. 3-(3-Bromopropyl)thiophene (9a) and 3-(5-bromopentyl)thiophene (9c)
General procedure: compounds
9a and
9c were prepared from 3-bromothiophene in a procedure analogous to the procedure described for compound
9b [
27], using corresponding di-bromoalkyl reagents. The reaction mixtures were purified via column chromatography using hexane:EtOAc 100:3 as a mobile phase (TLC visualisation: vaniline), affording
9a–
c in the form of colourless liquids.
9a Starting from 6.0 g (37 mmol) of 3-bromothiophene, 2.2 g of 9a was obtained after purification (27%).
NMR (CDCl3): 1H: δ 2.10–2.18 (m, 2H, CH2CH2CH2); 2.75–2.83 (m, 2H, CH2CH2CH2); 3.38 (t, JHH = 6.6, 2H, CH2CH2CH2); 6.93 (dd, 3JHH = 5.1, 4JHH = 1.3, 1H, SCHCH); 6.97 (dd, 4JHH = 3.1 and 1.3, 1H, SCHC); 7.25 (dd, 3JHH = 5.1, 4JHH = 3.1, SCHCH). 13C{1H}: δ 28.49, 33.24 and 33.38 (all 1C, CH2CH2CH2); 120.90, 125.75, 128.15 and 140.77 (all 1C, thiophene).
9b: Starting from 6.0 g (37 mmol) of 3-bromothiophene, 3.5 g of
9b was obtained after purification (44%). Characterisation data are identical with literature [
27].
9c: Starting from 6.0 g (37 mmol) of 3-bromothiophene, 3.9 g of 9c was obtained after purification (45%).
NMR (CDCl3): 1H: δ 1.42–1.54 (m, 2H, CH2CH2CH2CH2CH2); 1.59–1.70 (m, 2H, CH2CH2CH2CH2CH2); 1.82–1.94 (m, 2H, CH2CH2CH2CH2CH2); 2.60–2.68 (m, 2H, CH2CH2CH2CH2CH2); 3.40 (t, 3JHH = 6.8, 2H, CH2CH2CH2CH2CH2); 6.94–6.90 (m, 2H, SCHCH and SCHC); 7.24 (dd, 3JHH = 4.7, 4JHH = 3.1, 1H, SCHCH). 13C{1H}: δ 27.94, 29.81, 30.16, 32.74 and 33.89 (all 1C, CH2CH2CH2CH2CH2); 120.13, 125.38, 128.27 and 142.71 (all 1C, thiophene).
4.2.15. 1-[ω-(Thiophene-3-yl)-alkyl]-4,7-bis(tert-butyloxycarbonyl)-1,4,7-triazacyclononane (alkyl: 10a—propyl, 10b—butyl, 10c—pentyl)
General procedure: To a suspension of di-protected macrocycle 8 (ca 1 g) and K2CO3 (3 equiv.) in MeCN (30 mL), alkylating agent 9a–c (1.1 equiv.) was added. The mixture was heated to 60 °C and left stirring overnight. Then the mixture was filtered through an S4 frit and evaporated. The residue was purified via chromatography, using: 10a,b on silica gel and hexane/ethylacetate 4.5:1 as a mobile phase. Product-containing fractions were combined and evaporated, yielding products 10a,b as colourless oils. 10c was purified via flash chromatography (GM1), yielding 10c as a yellow oil in trifluoroacetate form.
10a: Starting from 0.75 g (2.3 mmol) of 8, product 10a was obtained after purification in a yield of 0.75 g (73%).
NMR (CDCl3): 1H: δ 1.45 (s, 18H, CH3); 1.69–1.87 (m, 2H, CH2CH2CH2); 2.41–2.77 (m, 8H, CH2CH2CH2 and CH2(cycle)); 3.14–3.33 (m, 4H, CH2(cycle)); 3.37–3.56 (m, 4H, CH2(cycle)); 6.93–6.99 (m, 2H, SCHCH and SCHC); 7.18–7.27 (m, 1H, SCHCH). 13C{1H}: δ 28.67 (6C, CH3); 49.63, 50.62, 53.65, 54.00, 56.32 and 56.44 (CH2CH2CH2 and CH2(cycle)); 79.43 (2C, C(CH3)3; 119.99, 125.25, 128.21 and 142.11 (all 1C, thiophene); 155.41 (2C, CO).
MS-ESI: (+) 454.5 ([M + H]+, calc. 454.3).
10b: Starting from 1.28 g (3.9 mmol) of 8, product 10b was obtained after purification in a yield of 0.98 g (54%).
NMR (CDCl3): 1H: δ 1.48 (s, 18H, CH3); 1.55–1.70 (m, 4H, CH2CH2CH2CH2); 2.35–2.76 (br m, 8H, CH2CH2CH2CH2 and CH2(cycle)); 3.12–3.32 (m, 4H, CH2(cycle)); 3.38–3.52 (br m, 4H, CH2(cycle)); 6.93–6.99 (m, 2H, SCHCH and SCHC); 7.21–7.25 (m, 1H, SCHCH). 13C{1H}: δ 28.49 (6C, CH3); 28.68 and 30.31 (all 1C, CH2CH2CH2CH2); 49.92, 50.77, 51.00, 54.11, 56.79 (CH2CH2CH2CH2 and CH2(cycle)); 79.54 (2C, C(CH3)3; 120.11, 125.32, 128.32 and 141.92 (all 1C, thiophene); 155.69 (2C, CO).
MS-ESI: (+) 468.5 ([M + H]+, calc. 468.3).
10c: Starting from 0.45 g (1.4 mmol) of 8, product 10c·xTFA was obtained after purification in a yield of 0.4 g.
NMR (CD3OD): 1H: δ 1.51 (s, 18H, CH3); 1.65–1.85 (m, 4H, CH2CH2CH2CH2CH2); 2.68 (t, JHH = 7.5, 2H, CH2CH2CH2CH2CH2); 3.32–3.83 (br m, 16H, CH2CH2CH2CH2CH2 and CH2(cycle)); 6.96 (dd, 3JHH = 5.0, 4JHH = 1.3, 1H, SCHCH); 7.05 (dd, 4JHH = 2.9 and 1.3, 1H, SCHC); 7.32 (dd, 3JHH = 5.0, 4JHH = 2.9, 1H, SCHCH). 13C{1H}: δ 25.15 and 27.06 (all 1C, CH2CH2CH2CH2CH2) 28.60 (6C, CH3); 30.71, 31.01, 45.16, 52.11, 52.81 and 57.56 (CH2CH2CH2CH2CH2 and CH2(cycle)); 82.92 (2C, C(CH3)3; 121.20, 126.32, 129.09 and 143.54 (all 1C, thiophene); 157.24 (2C, CO).
MS-ESI: (+) 482.5 ([M + H]+, calc. 482.3).
4.2.16. 1-[ω-(Thiophene-3-yl)-alkyl]-1,4,7-triazacyclononane (alkyl: 11a—propyl, 11b—butyl, 11c—pentyl)
General procedure: compound 10a–c·xTFA was suffused with 6M HCl and left stirring at room temperature overnight. During this time, the originally insoluble oil dissolved as product 11a–c was formed. The solution was evaporated to dryness, yielding 11a–c·xHCl as a white or slightly yellow powder.
11a: Starting from 0.75 g (1.7 mmol) of 10a, product 11a·xHCl was obtained in a yield of 0.55 g.
NMR (D2O): 1H: δ 1.90–2.00 (m, 2H, CH2CH2CH2); 2.69 (t, 3JHH = 7.4, 2H, CH2CH2CH2); 2.88–2.95 (m, 2H, CH2CH2CH2); 3.17 (dd, 3JHH = 6.8 and 4.9, 4H, CH2(cycle)); 3.34 (dd, 3JHH = 6.8 and 4.9, 4H, CH2(cycle)); 3.51 (s, 4H, CH2(cycle)); 7.08 (dd, 3JHH = 5.0, 4JHH = 1.3, 1H, SCHCH); 7.15 (dd, 4JHH = 2.9 and 1.3, 1H, SCHC); 7.43 (dd, 3JHH = 5.0, 4JHH = 2.9, 1H, SCHCH). 13C{1H}: δ 25.48 and 27.52 (all 1C, CH2CH2CH2); 42.55, 43.35, 48.58 and 55.62 (all 2C, CH2(cycle) and CH2CH2CH2); 121.62, 126.95, 129.00 and 142.56 (all 1C, thiophene).
MS-ESI: (+) 254.3 ([M + H]+, calc. 254.2).
11b: Starting from 0.36 g (0.8 mmol) of 10b, product 11b·xHCl was obtained in a yield of 0.26 g.
NMR (D2O): 1H: δ 1.64 (m, 4H, CH2CH2CH2CH2); 2.70 (t, 3JHH = 6.7, 2H, CH2CH2CH2CH2); 2.94 (t, 3JHH = 7.6, 2H, CH2CH2CH2CH2); 3.16 (t, 3JHH = 5.9, 4H, CH2(cycle)); 3.30 (t, 3JHH = 5.8, 4H, CH2(cycle)); 3.44 (s, 4H, CH2(cycle)); 7.07 (dd, 3JHH = 5.0, 4JHH = 1.3, 1H, SCHCH); 7.13 (dd, 4JHH = 3.2 and 1.3, 1H, SCHC); 7.42 (dd, 3JHH = 5.0, 4JHH = 3.2, 1H, SCHCH). 13C{1H}: δ 23.97, 27.83 and 29.61 (all 1C, CH2CH2CH2CH2); 42.73, 43.37, 49.05 and 56.25 (CH2CH2CH2CH2 and CH2(cycle)); 121.37, 126.79, 129.14 and 143.50 (all 1C, thiophene).
MS-ESI: (+) 268.3 ([M + H]+, calc. 268.2).
11c: Starting from 0.4 g (0.8 mmol) of 10c, product 11c·xHCl was obtained in a yield of 0.28 g.
NMR (D2O): 1H: δ 1.28–1.39 (m, 2H, CH2CH2CH2CH2CH2); 1.60–1.71 (m, 6H, CH2CH2CH2CH2CH2); 2.67 (t, 3JHH = 7.4, 2H, CH2CH2CH2CH2CH2); 3.22 (dd, 3JHH = 6.8 and 4.7, 4H, CH2(cycle)); 3.32 (dd, 3JHH = 6.8 and 4.7, 4H, CH2(cycle)); 3.42 (s, 4H, CH2(cycle)); 7.06 (dd, 3JHH = 4.9, 4JHH = 1.2, 1H, SCHCH); 7.11 (dd, 4JHH = 3.0 and 1.2, 1H, SCHC); 7.40 (dd, 3JHH = 4.9, 4JHH = 3.0, 1H, SCHCH). 13C{1H}: δ 24.64, 26.74, 30.09 and 30.12 (all 1C, CH2CH2CH2CH2CH2); 42.98, 43.40, 49.49 and 57.00 (CH2CH2CH2CH2CH2 and CH2(cycle)); 121.42, 126.81, 129.55 and 144.19 (all 1C, thiophene).
MS-ESI: (+) 282.4 ([M + H]+, calc. 282.2).
4.2.17. 1-[ω-(Thiophene-3-yl)-alkyl]-4,7-bis(iso-propyl)-1,4,7-triazacyclononane (alkyl: L14a—propyl, L14b—butyl, L14c—pentyl)
General procedure: compound 11a–c·xHCl and K2CO3 (excess) were suspended in MeCN (30 mL) and stirred at room temperature for 1 h in order to convert the hydrochloride into a free amine base. Then, iso-propyl bromide (5 equiv.) was added, and the mixture was heated to 60 °C and left stirring overnight. The reaction progress was followed by HPLC-MS, and the alkylating agent was occasionally added in order to maximise the yield of the reaction. After the alkylation was complete according to HPLC-MS, the mixture was filtered through S4 frit and evaporated. The residue was redissolved in MeCN and purified using flash chromatography on reverse-phase silica gel (GM1). Product-containing fractions were combined and evaporated, yielding final ligands L14a–c·xTFA as colourless or light-yellow oils.
L14a: Starting from 0.55 g of 11a·xHCl, product L14a·xTFA was obtained in a yield of 0.46 g.
NMR (D2O): 1H: δ 1.35 (d, 3JHH = 6.3, 12H, CH3); 1.89–1.99 (m, 2H, CH2CH2CH2); 2.68 (t, 3JHH = 7.3, 2H, CH2CH2CH2); 2.84–2.92 (m, 2H, CH2CH2CH2); 3.06–3.45 (br m, 8H, CH2(cycle)); 3.51 (s, 4H, CH2(cycle)); 3.64 (sept, 3JHH = 6.6, 2H, CH); 7.07 (dd, 3JHH = 5.0, 4JHH = 1.5, 1H, SCHCHC); 7.14 (dd, 4JHH = 3.3 and 1.3, 1H, SCHC); 7.43 (dd, 3JHH = 5.0, 4JHH = 3.3, 1H, SCHCH). 13C{1H}: δ 16.07, 17.67 (4C, CH3); 25.04, 27.44, 46.07, 47.71, 48.84, 55.29 and 59.92 (all 1C, CH2(cycle), CH2CH2CH2 and CH); 121.65, 127.02, 128.93 and 142.45 (all 1C, thiophene).
MS-ESI: (+) 338.3 ([M +H]+, calc. 338.3).
L14b: Starting from 0.37 of 11b·xHCl, product L14b·xTFA was obtained in a yield of 0.31 g.
NMR (D2O): 1H: δ 1.32 (d, 3JHH = 6.6, 12H, CH3); 1.57–1.67 (m, 4H, CH2CH2CH2CH2); 2.70 (t, JHH = 6.6, 2H, CH2CH2CH2CH2)); 2.86–2.95 (m, 2H, CH2CH2CH2CH2); 3.08–3.41 (br m, 8H, CH2(cycle)); 3.45 (s, 4H, CH2(cycle)); 3.59 (sept, 3JHH = 6.6, 2H, CH); 7.06 (dd, 3JHH = 4.9, 4JHH = 1.3, 1H, SCHCH); 7.11 (dd, 4JHH = 2.9 and 1.1, 1H, SCHC); 7.41 (dd, 3JHH = 4.9, 4JHH = 3.0, 1H, SCHCH). 13C{1H}: δ 16.30,17.55 (4C, CH3); 23.81, 27.63, 29.58, 46.19, 47.52, 49.30, 56.44 and 59.64 (all 1C, CH2(cycle), CH2CH2CH2CH2 and CH); 121.41, 126.79, 129.11 and 143.35 (all 1C, thiophene).
MS-ESI: 352.4 (+) ([M + H]+, calc. 352.3).
L14c: Starting from 0.42 of 11c·xHCl, product L14c·xTFA was obtained in a yield of 0.34 g.
NMR (CD3OD): 1H: δ 1.30 (d, 3JHH = 7.1, 12H, CH3); 1.55–1.67 (m, 4H, CH2CH2CH2CH2CH2); 2.62 (t, 3JHH = 7.6, 2H, CH2CH2CH2CH2CH2), 2.83–2.91 (m, 4H, CH2CH2CH2CH2CH2); 3.09 (br s, 2H, CH2(cycle)); 3.17–3.39 (br m, 8H, CH2(cycle)); 3.39–3.54 (br m, 4H, CH2(cycle)); 3.59 (sept, 3JHH = 6.6, 2H, CH); 6.91–6.94 (m, 2H, SCHCH and SCHC); 7.24 (d, 3JHH = 4.2, 1H, SCHCH). 13C{1H}: δ 17.72 (4C, CH3); 27.74, 30.97, 31.39; 46.77, 47.19,49.28, 50.01 (all very br), 57.30; 121.02, 126.22, 129.09 and 143.83 (all 1C, thiophene).
MS-ESI: (+) 366.4 ([M + H]+, calc. 366.3).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}