

Identification of Dual-Target Inhibitors for Epidermal Growth Factor Receptor and AKT: Virtual Screening Based on Structure and Molecular Dynamics Study


Abstract
:1. Introduction
2. Results and Discussion
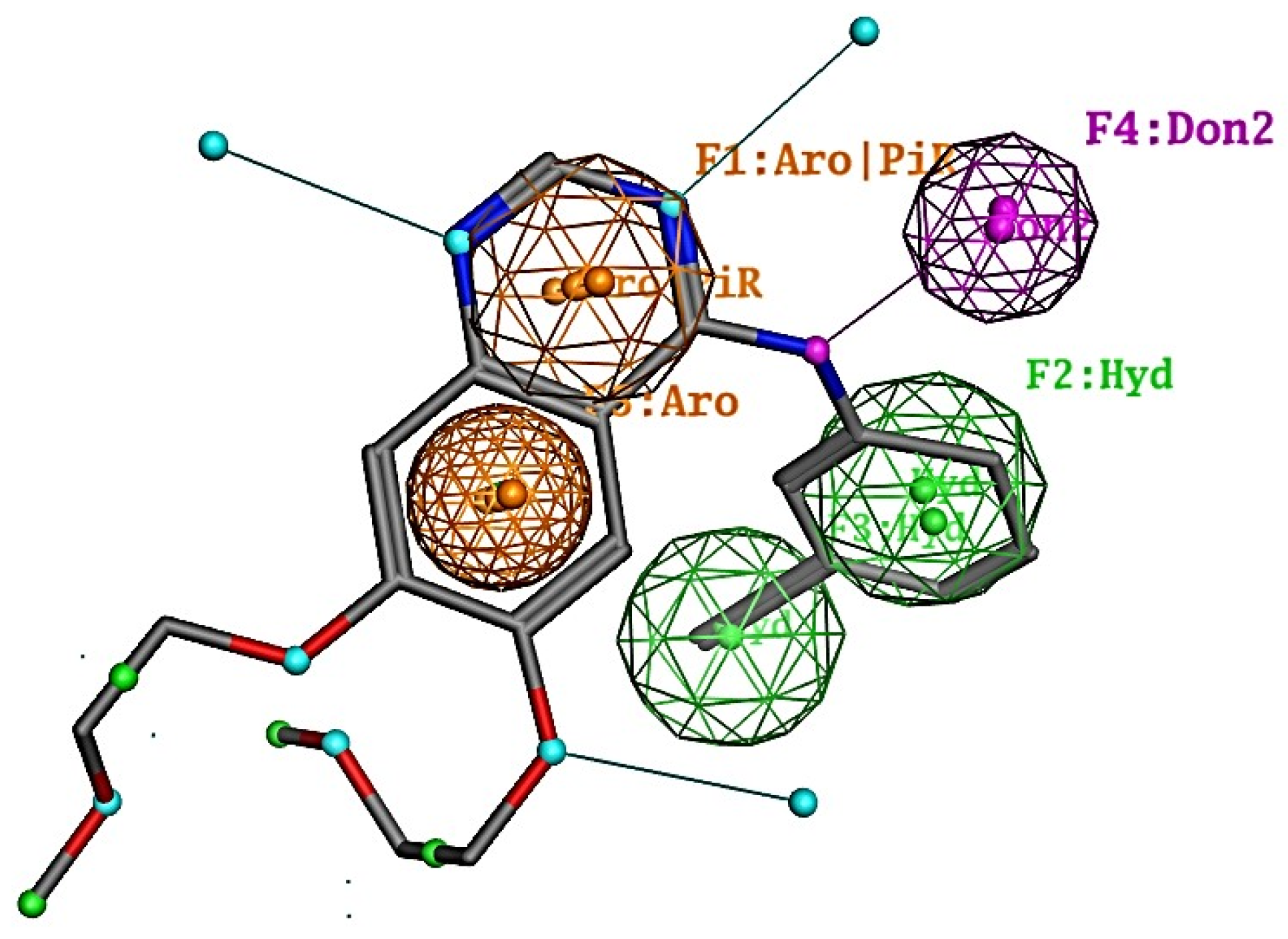
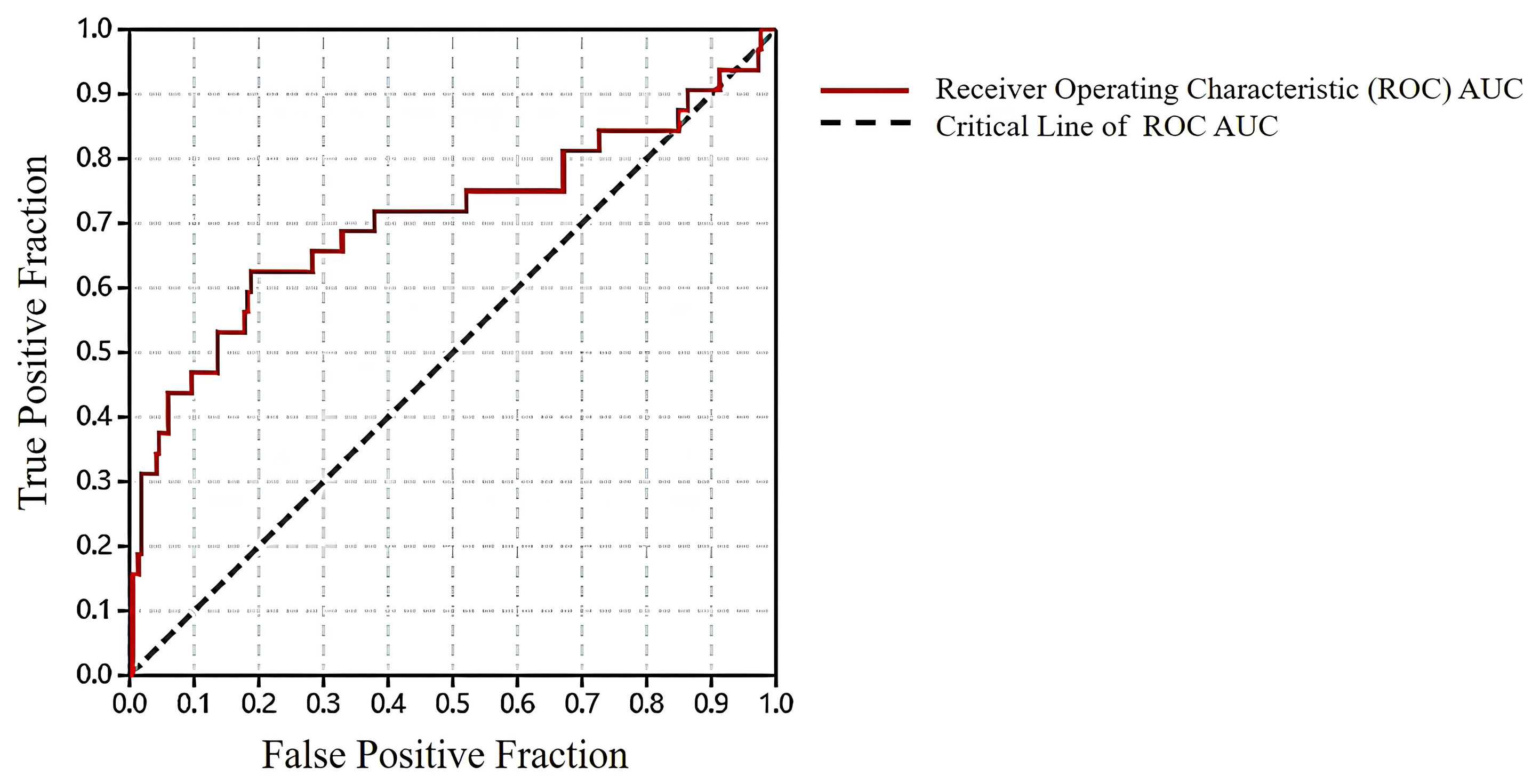
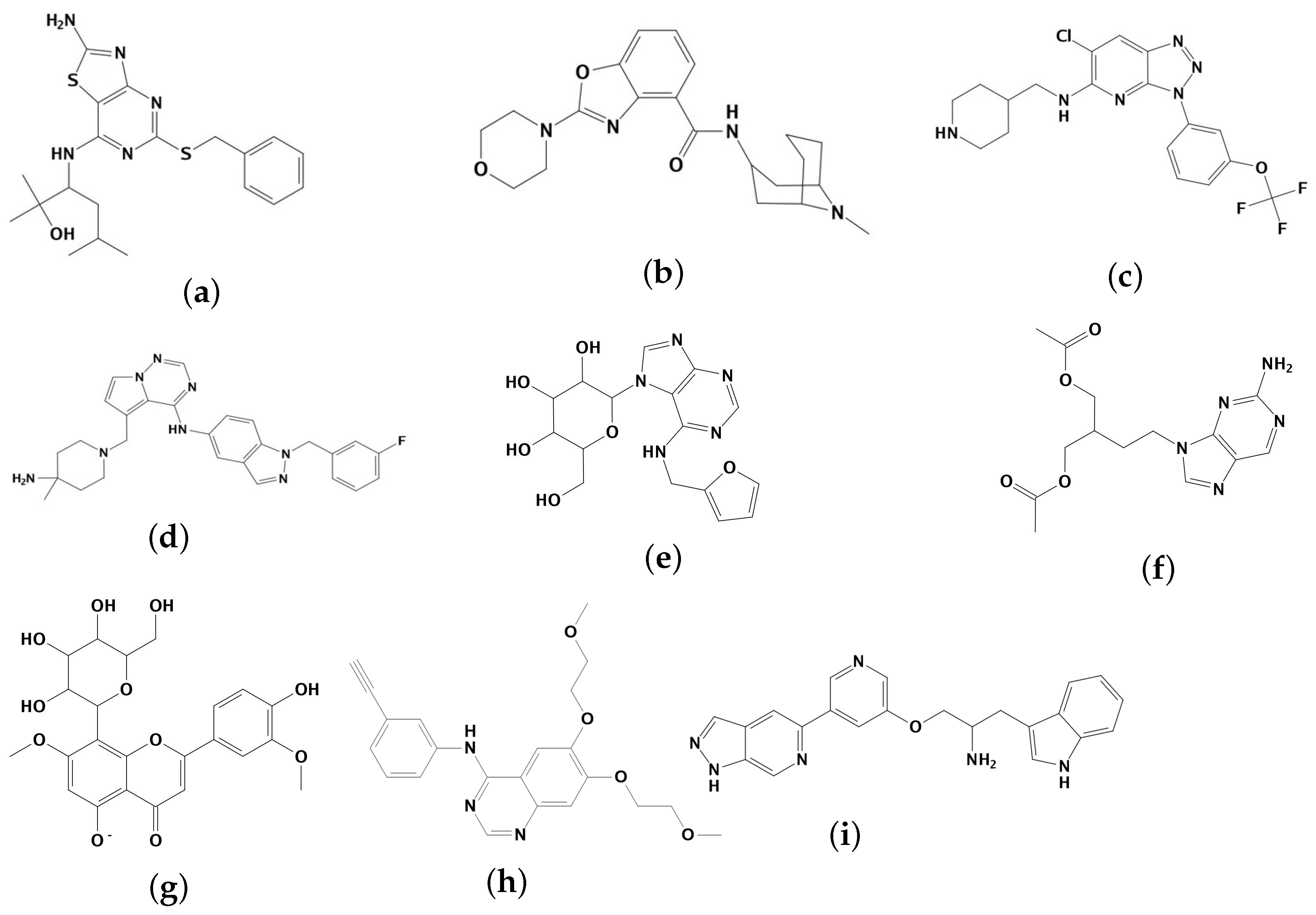
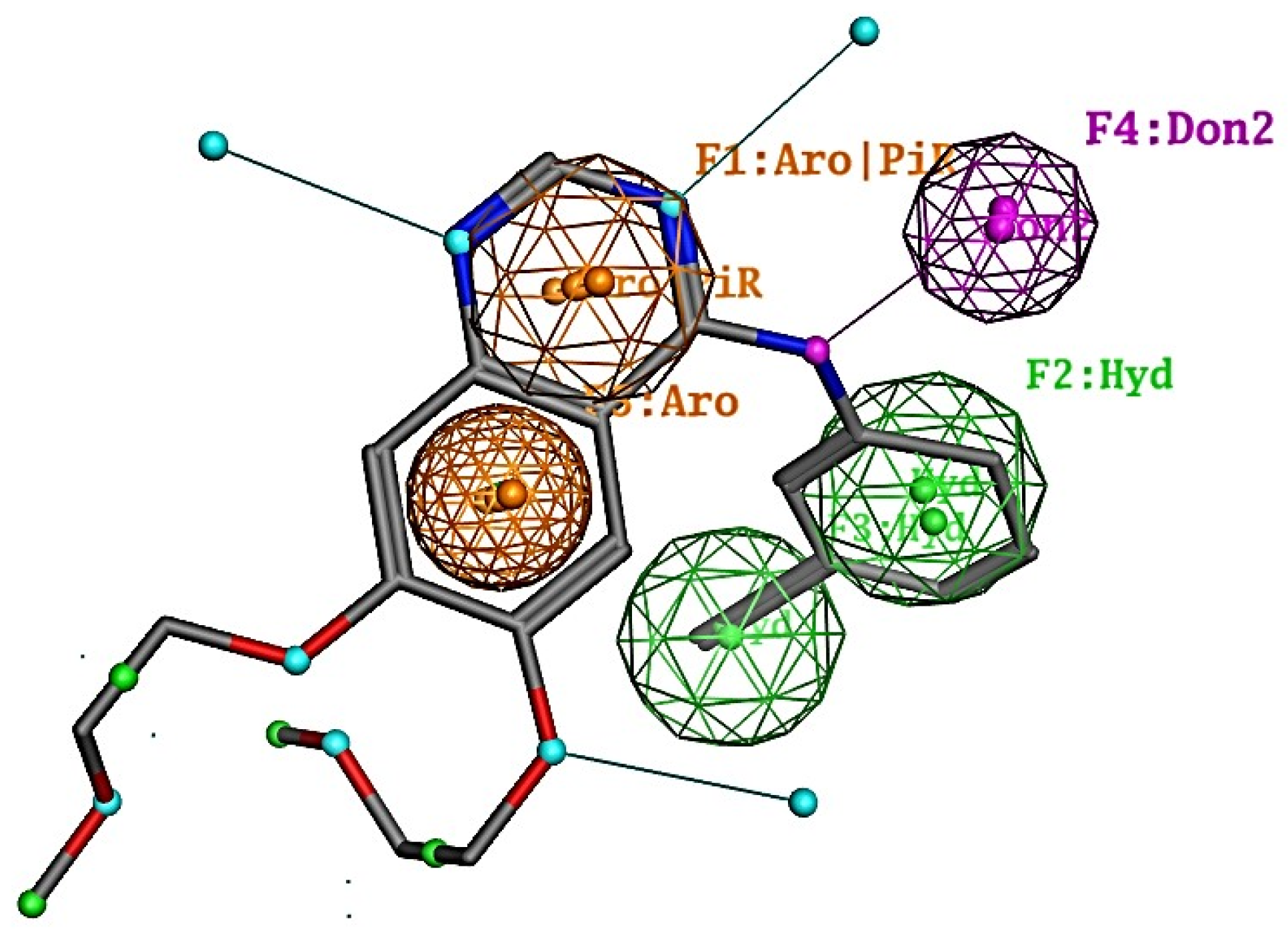
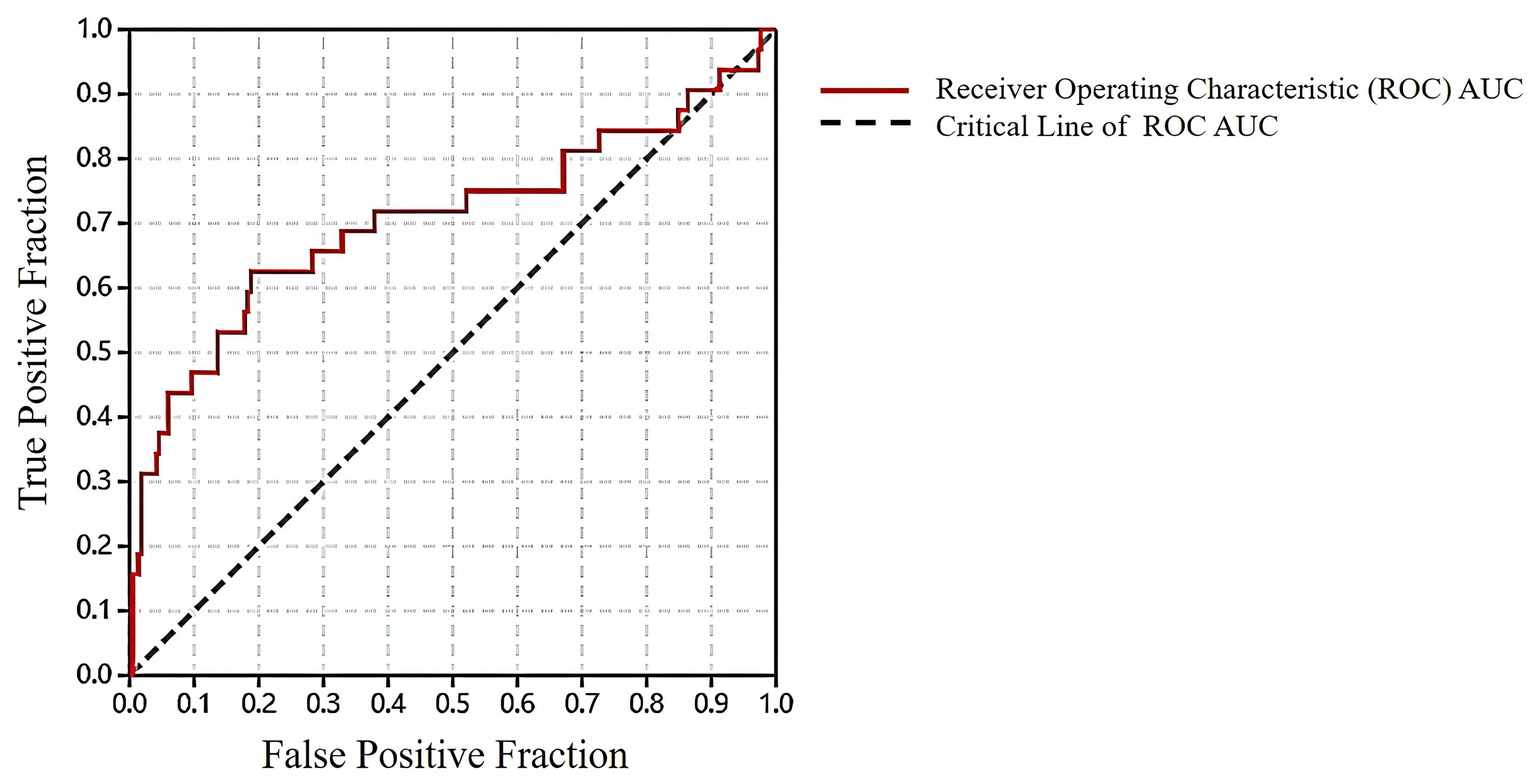
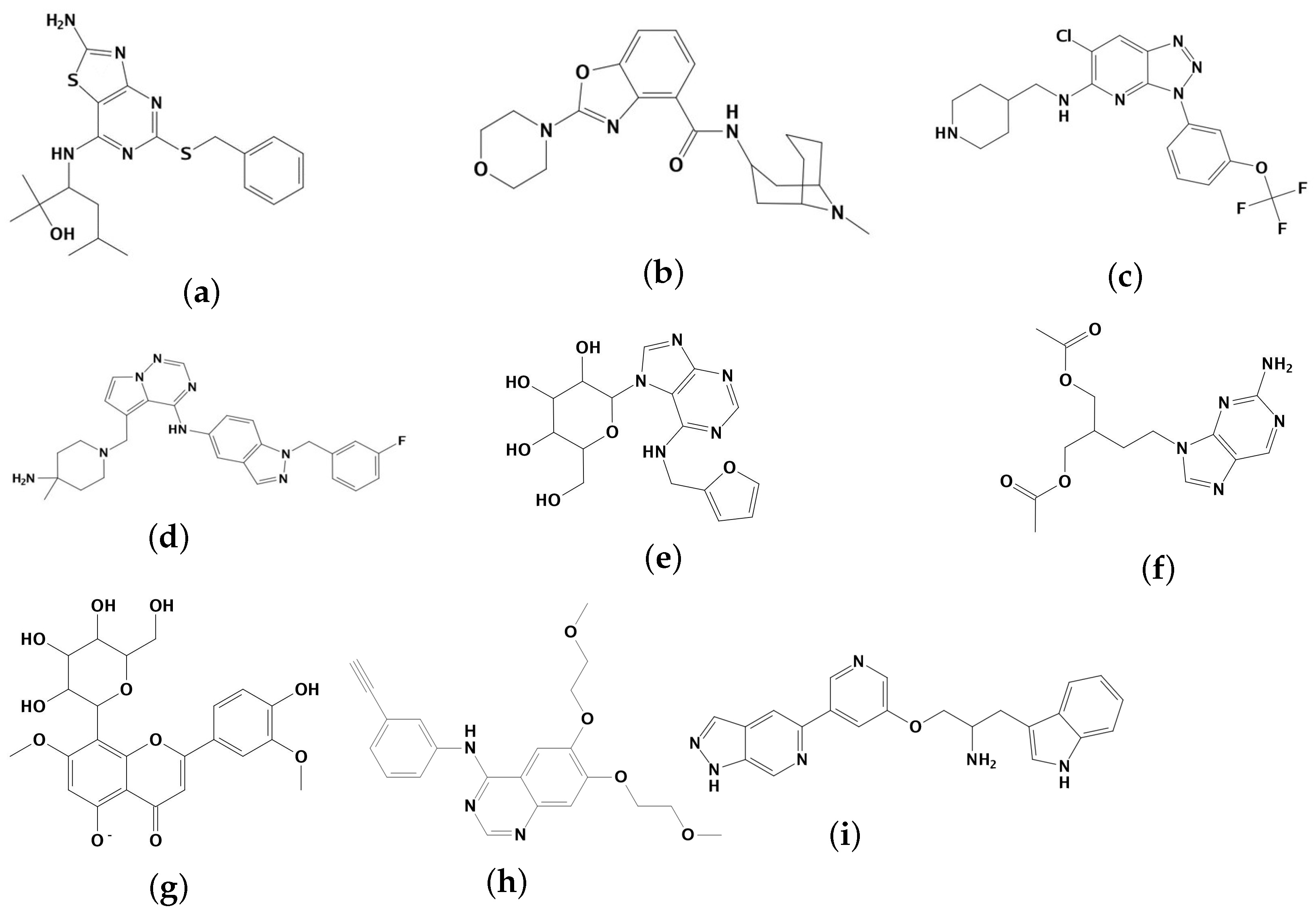
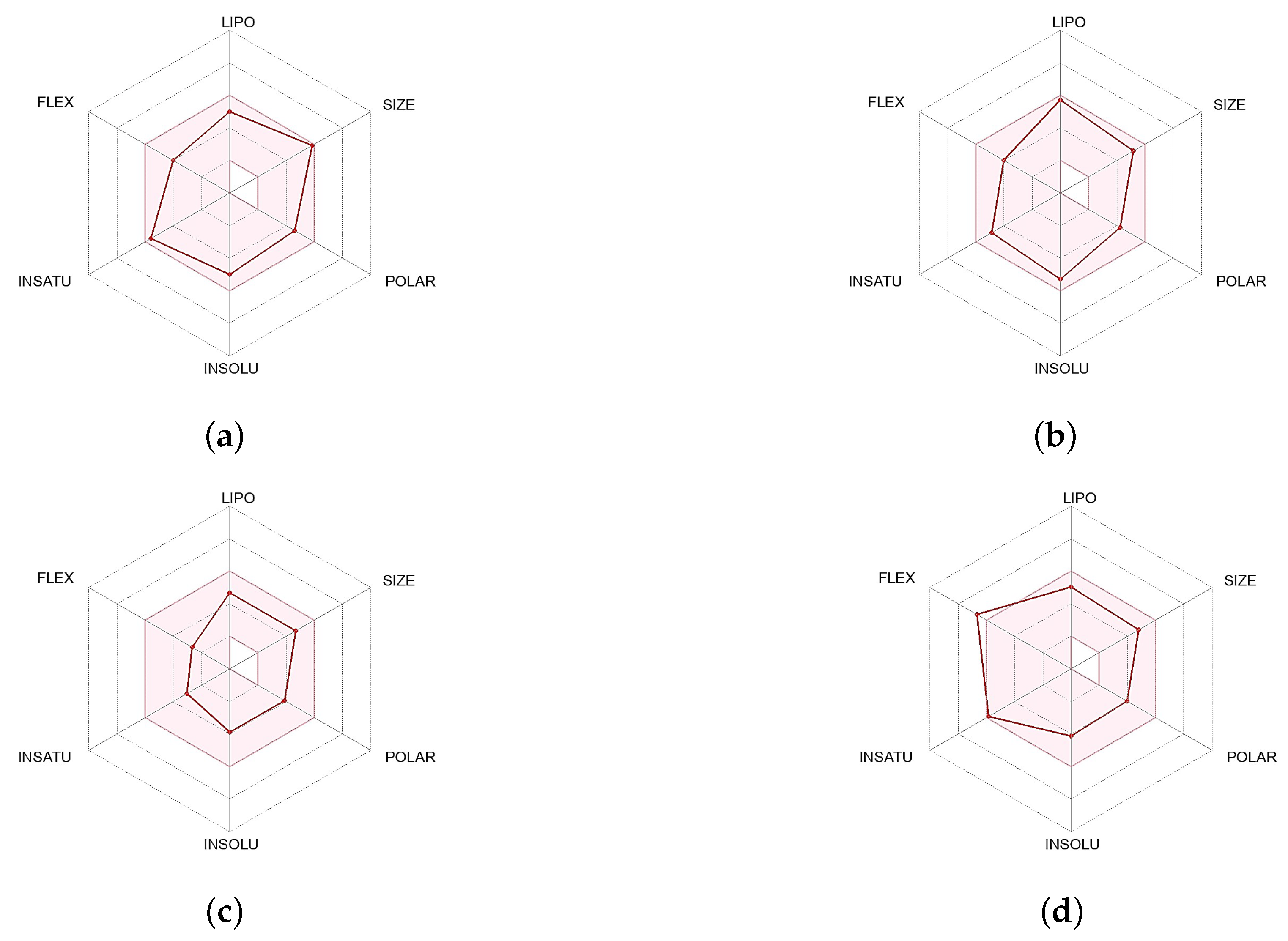
2.1. Results of Pharmacophore and Molecular Docking Screening
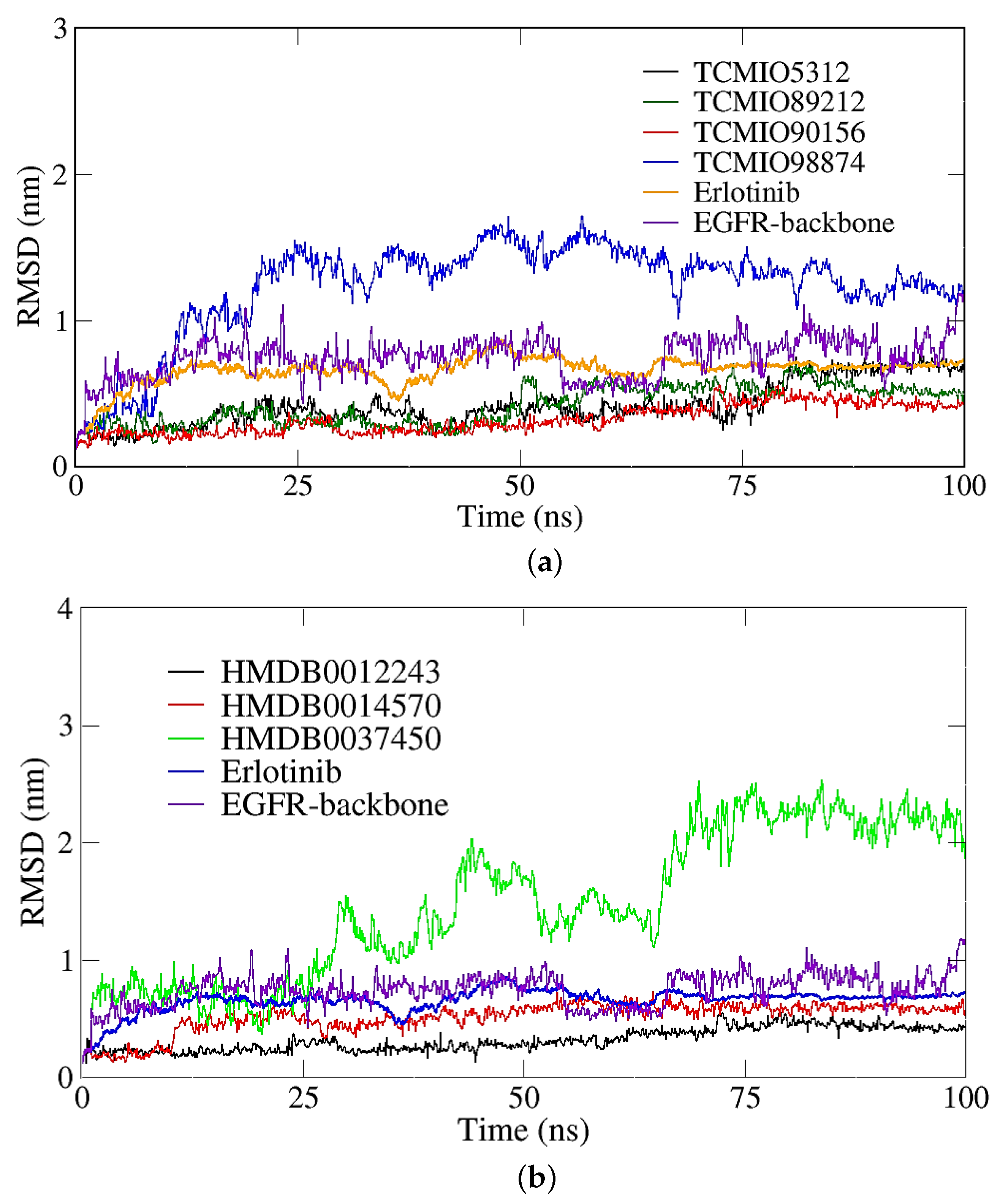
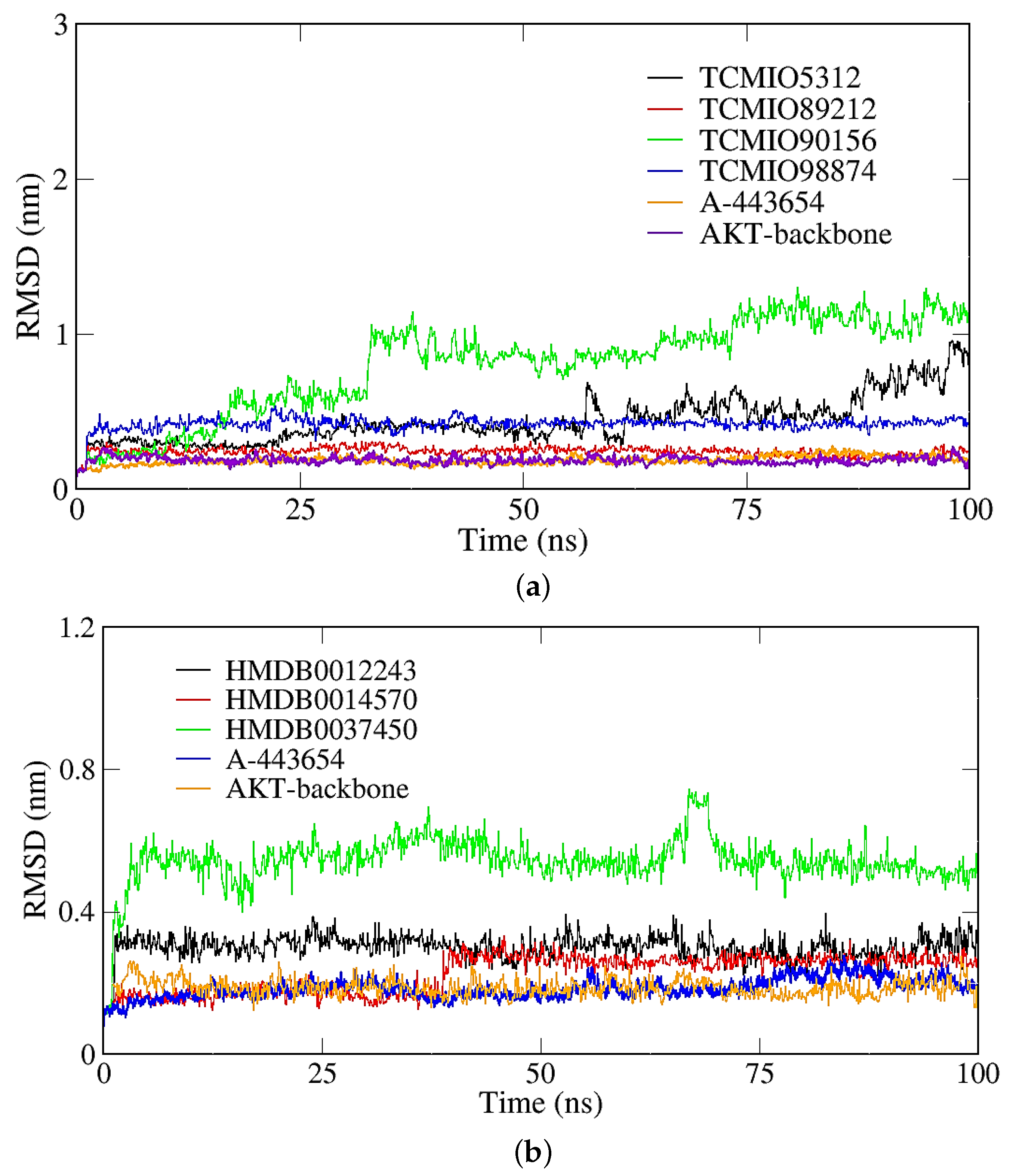
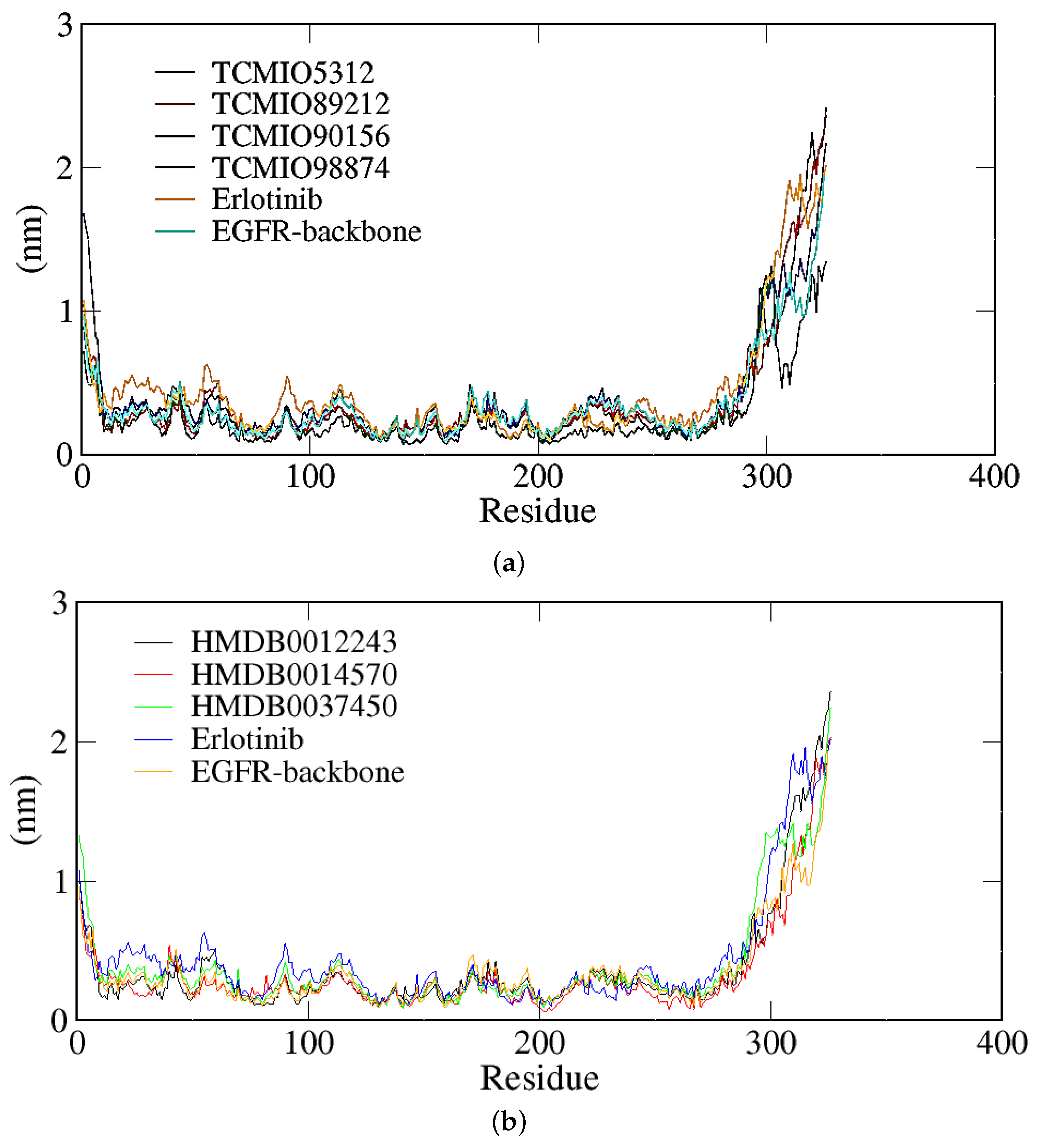
2.2. MD Simulation Results
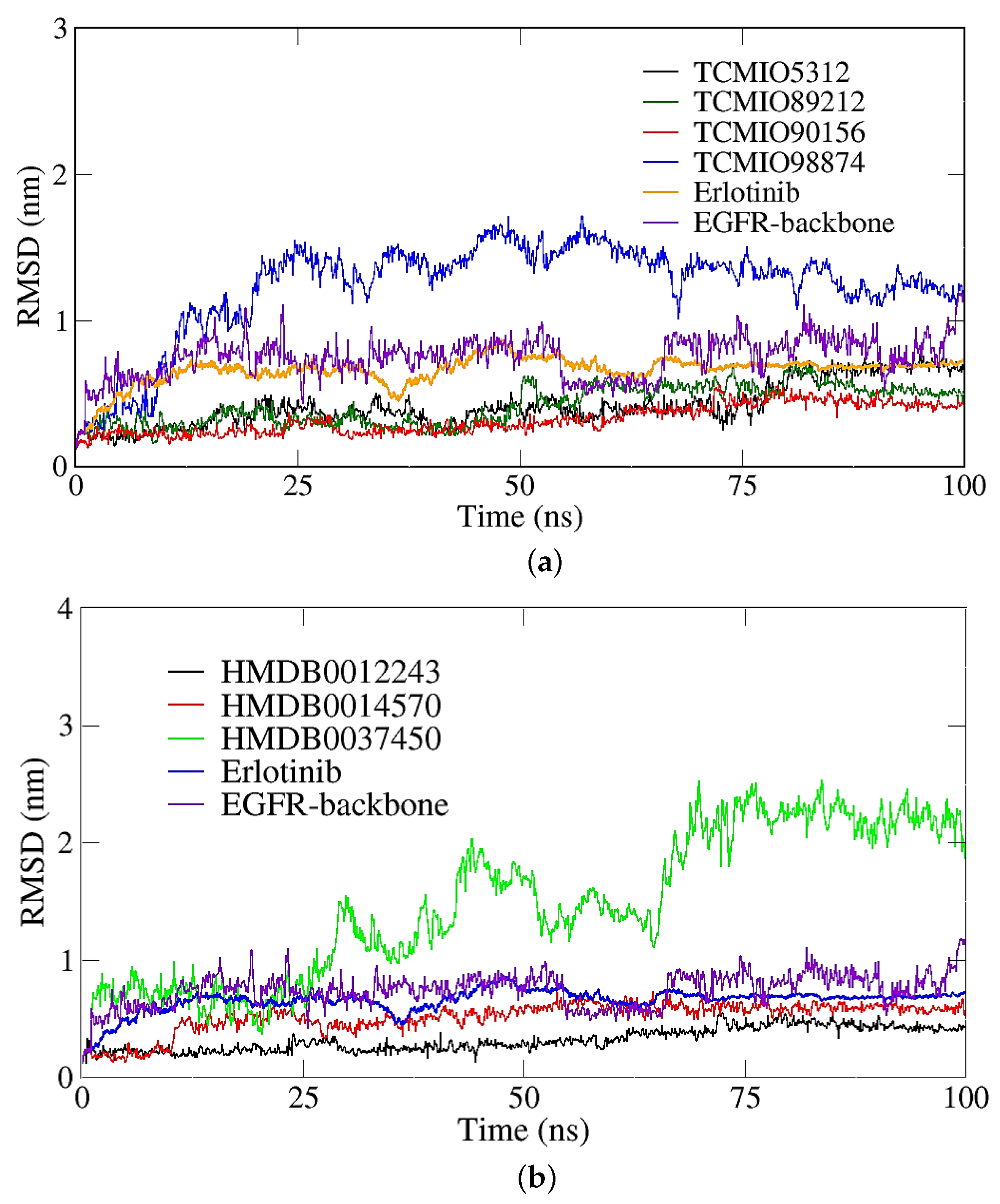
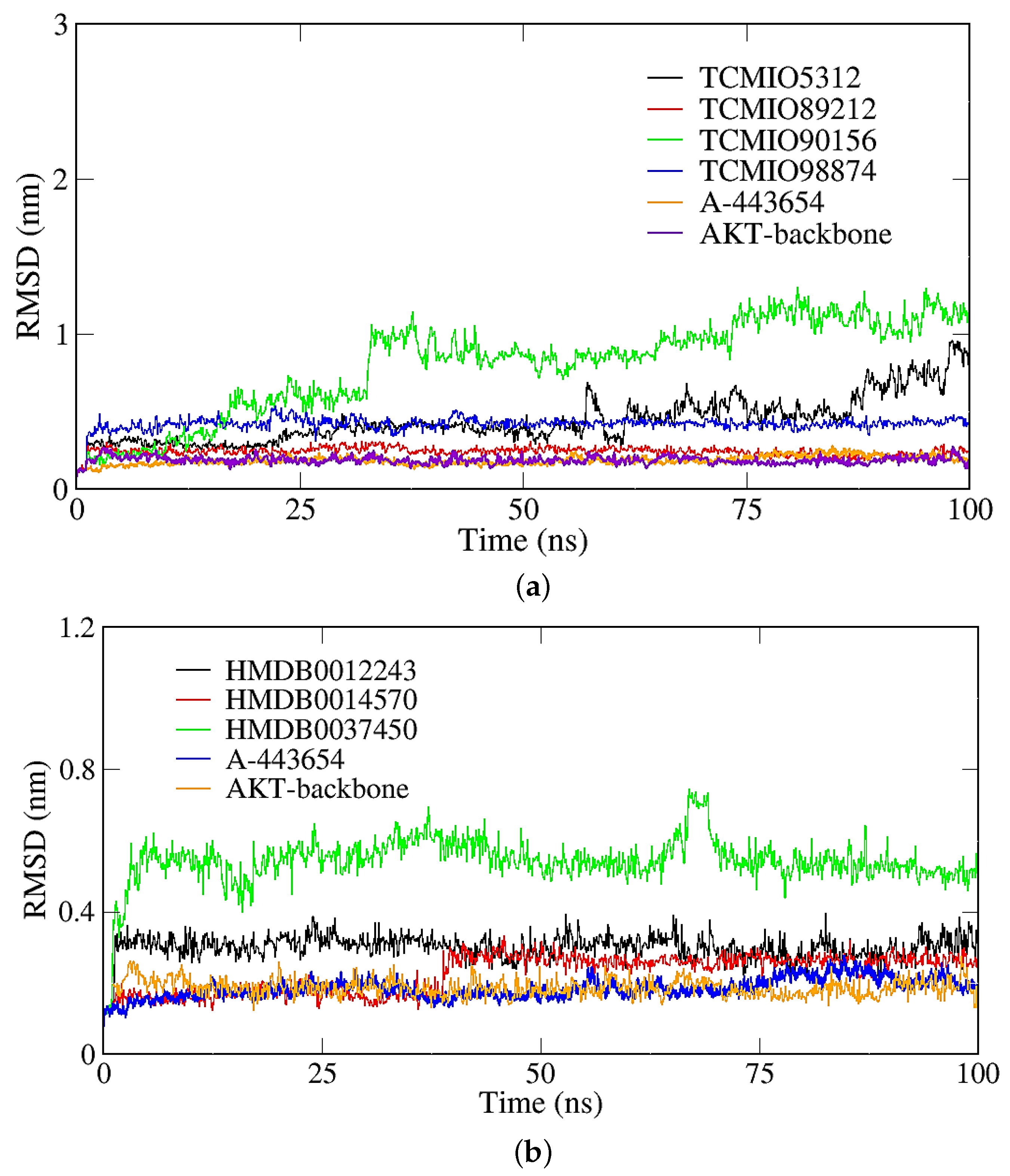
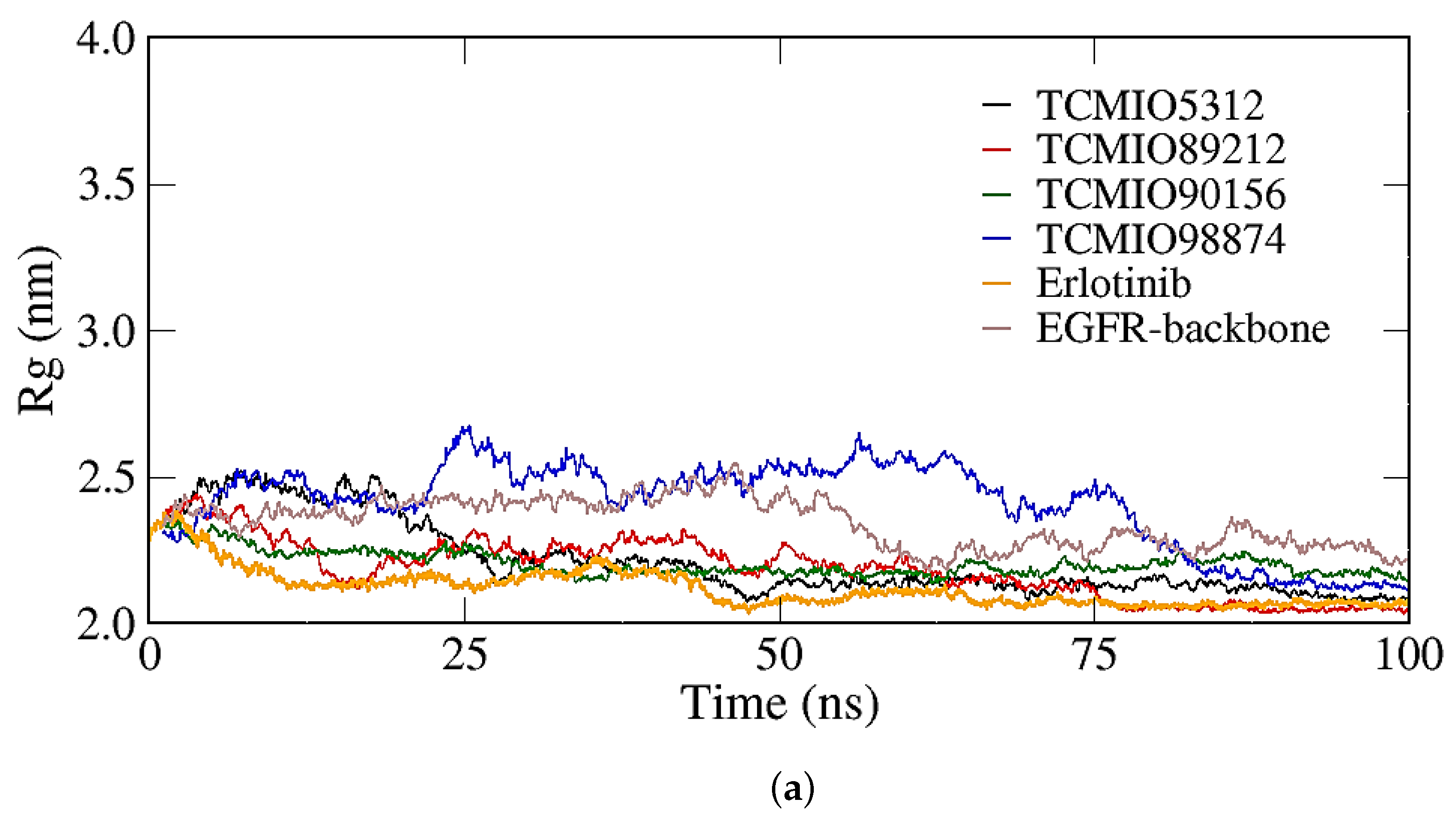
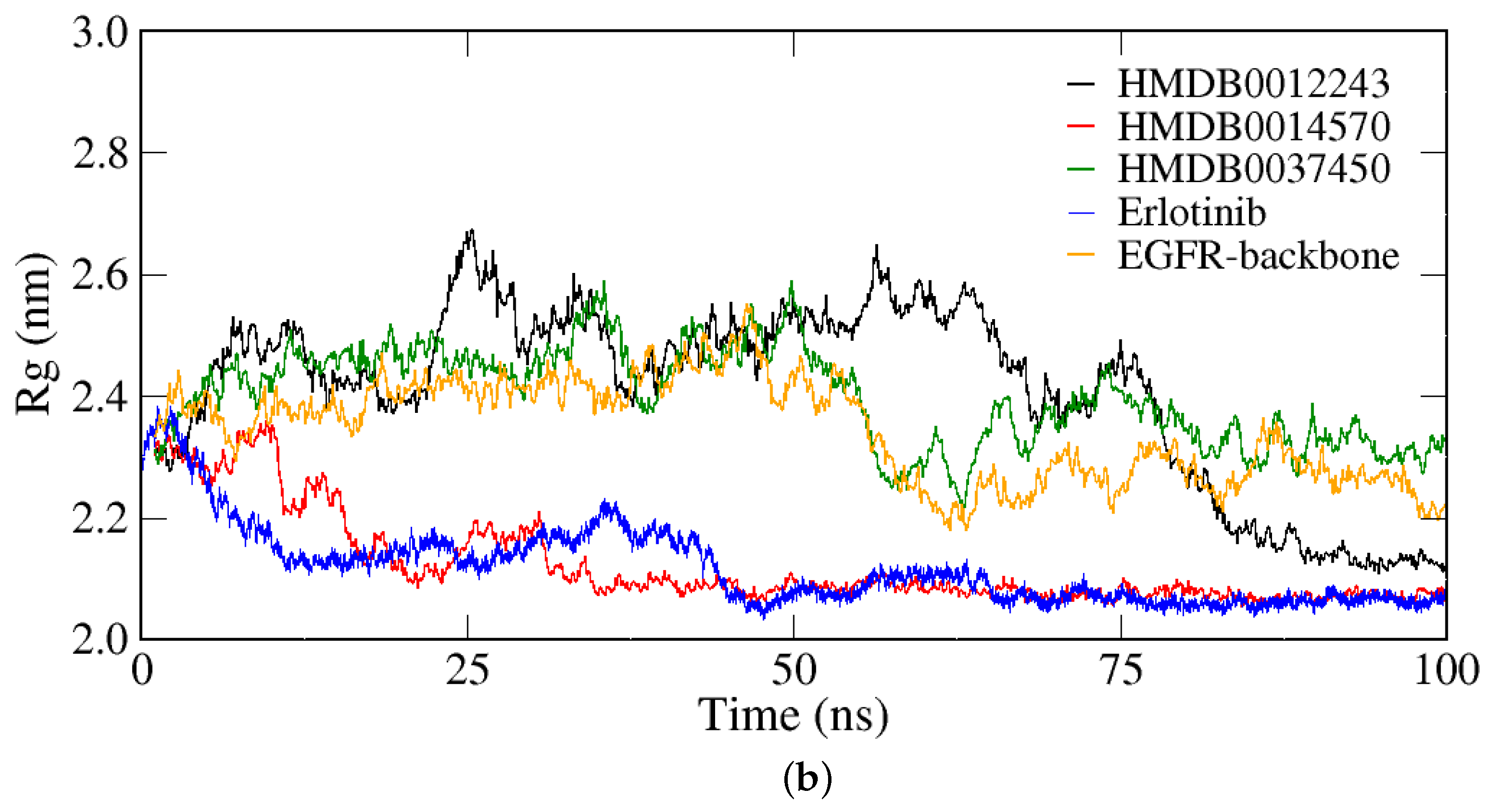
2.2.1. RMSD, RMSF, and Rg Analyses
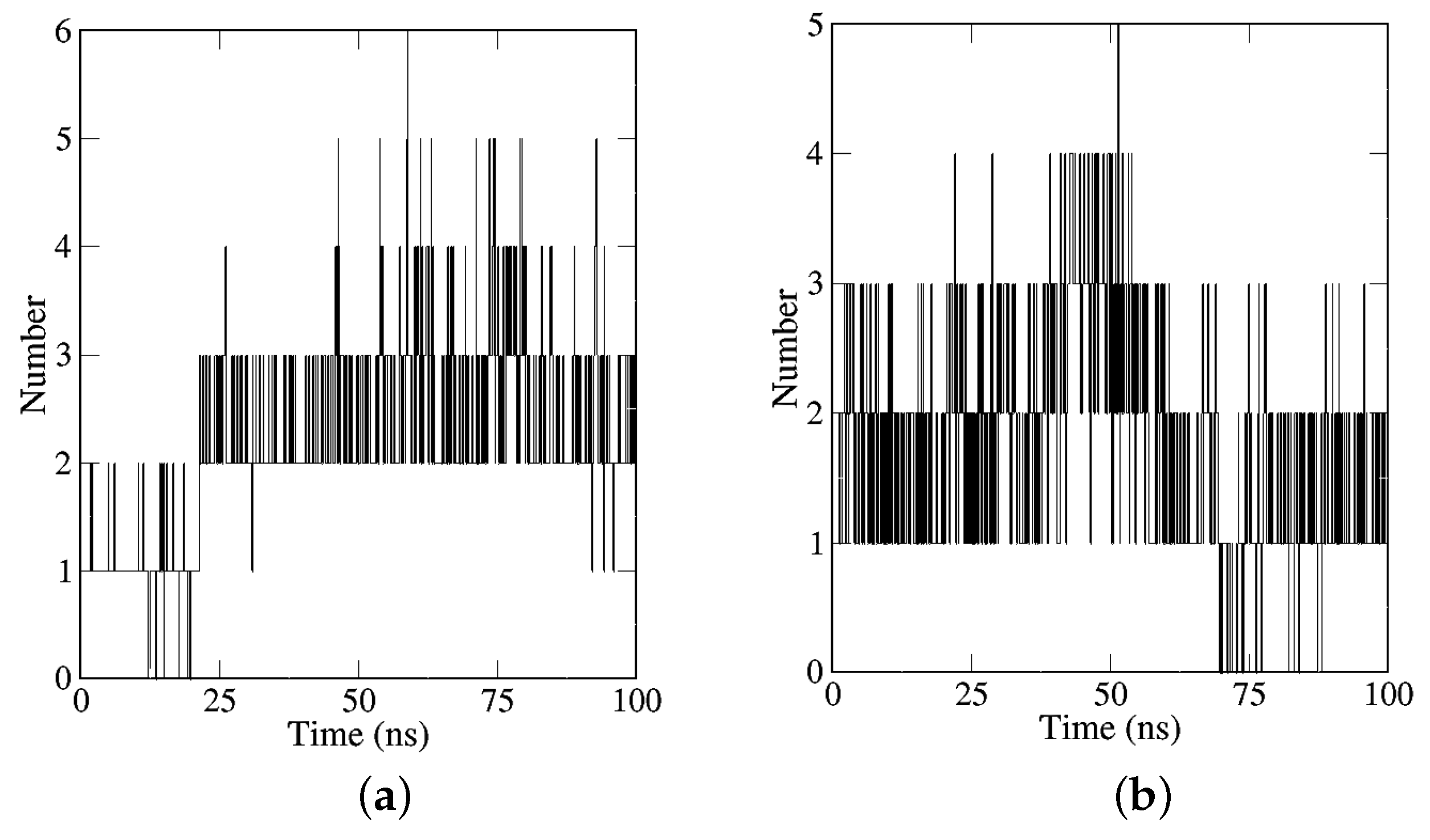
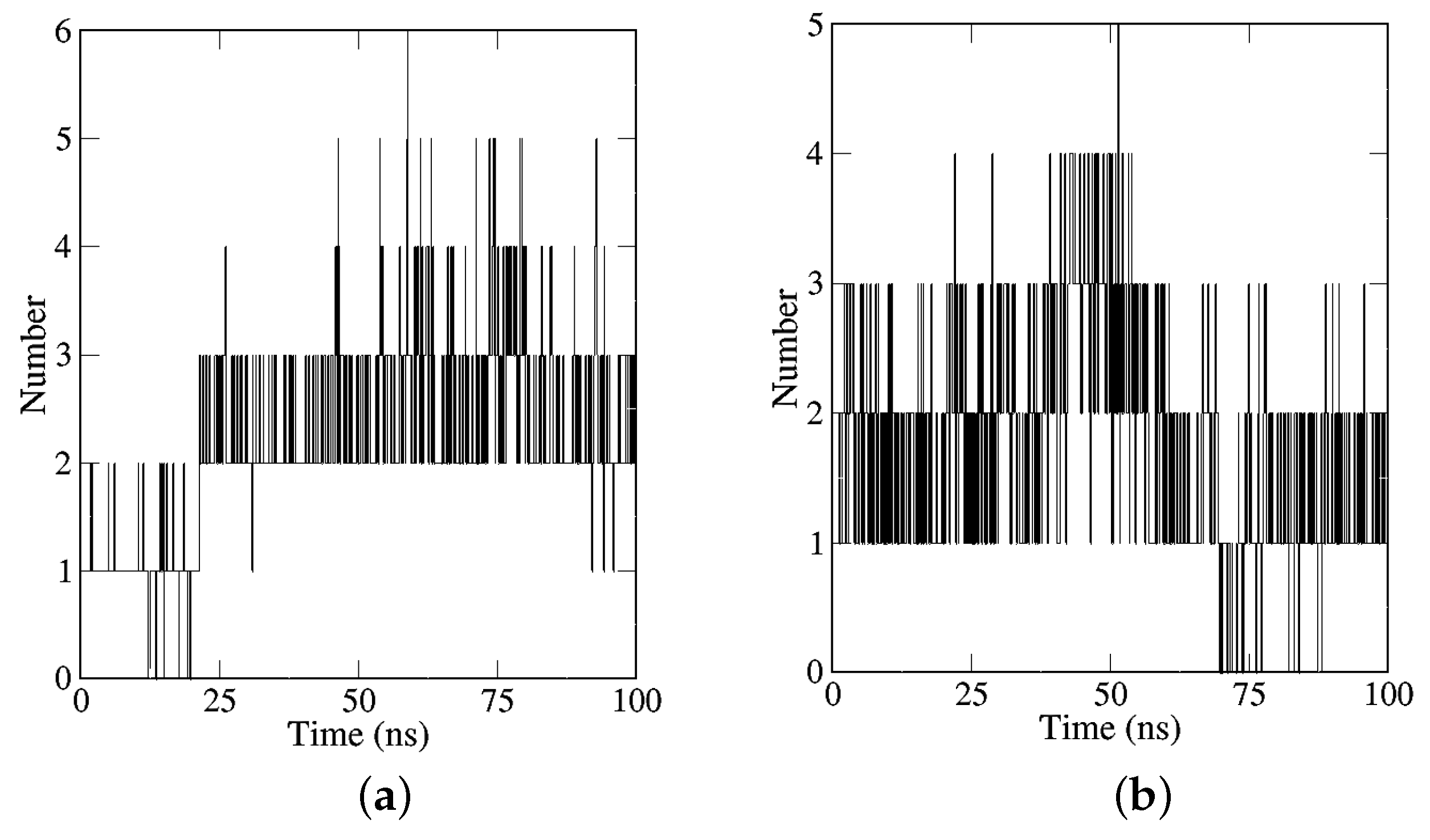
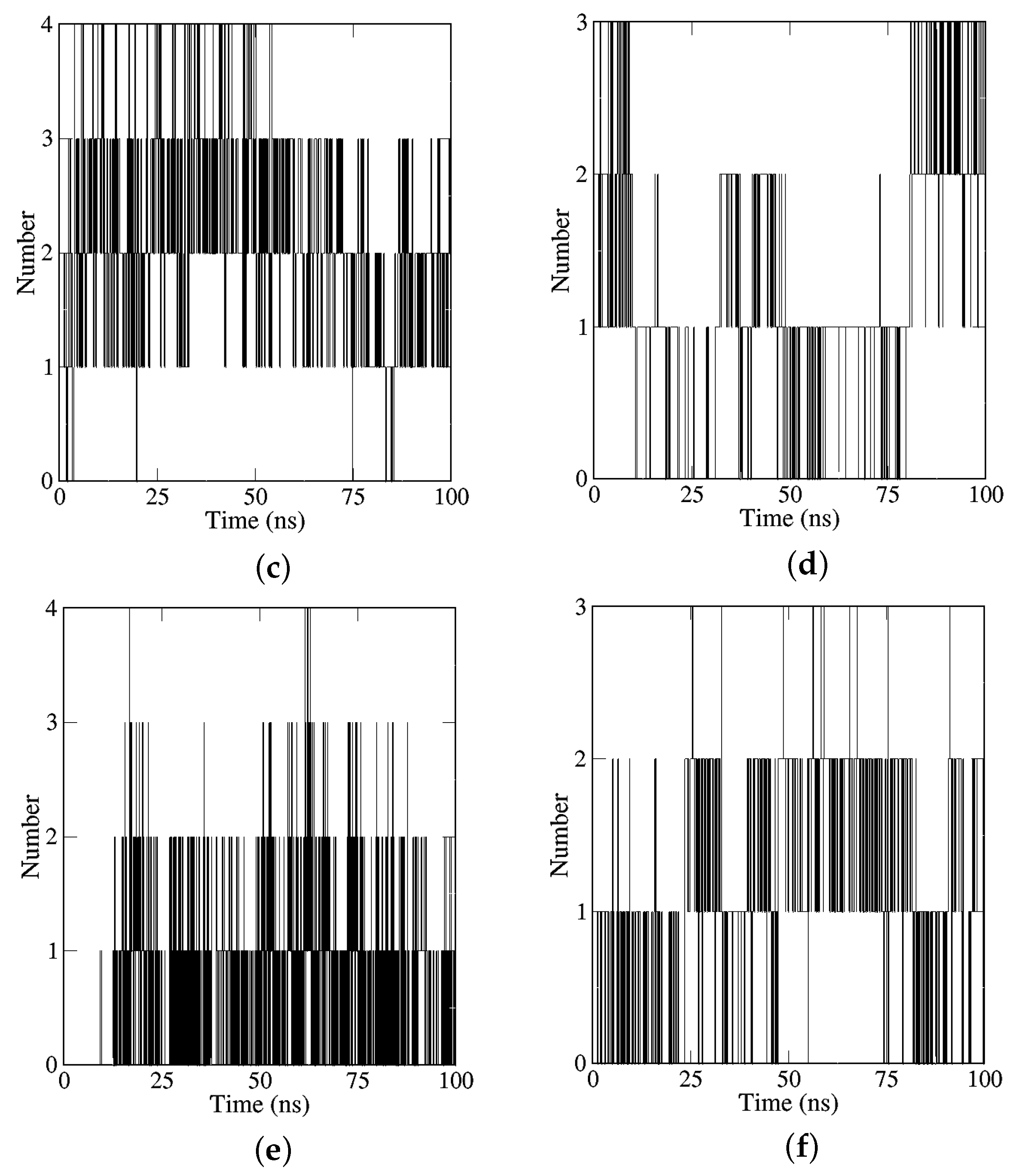
2.2.2. Analyses of H-Bonds
2.2.3. Binding Free Energy Analyses
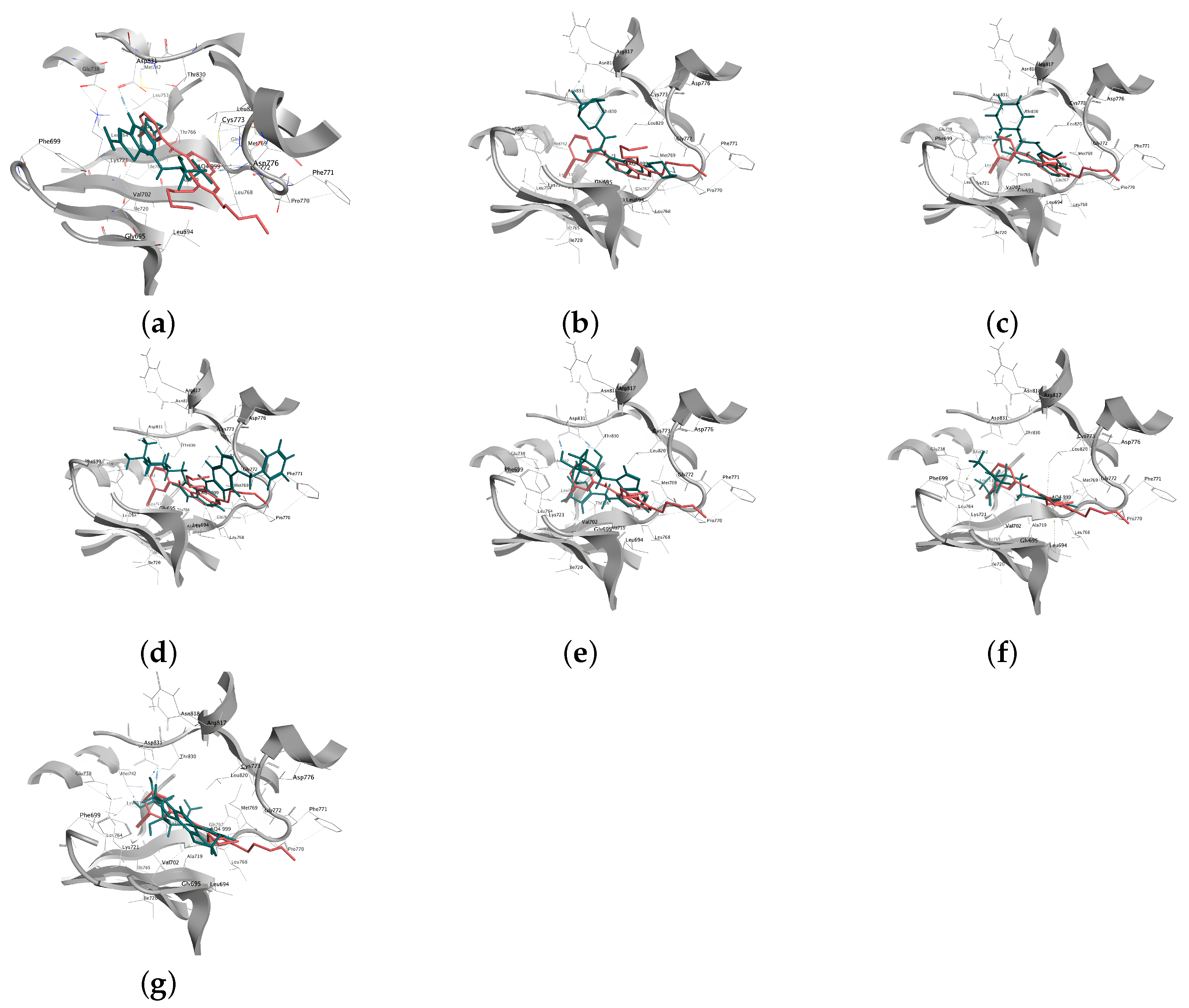
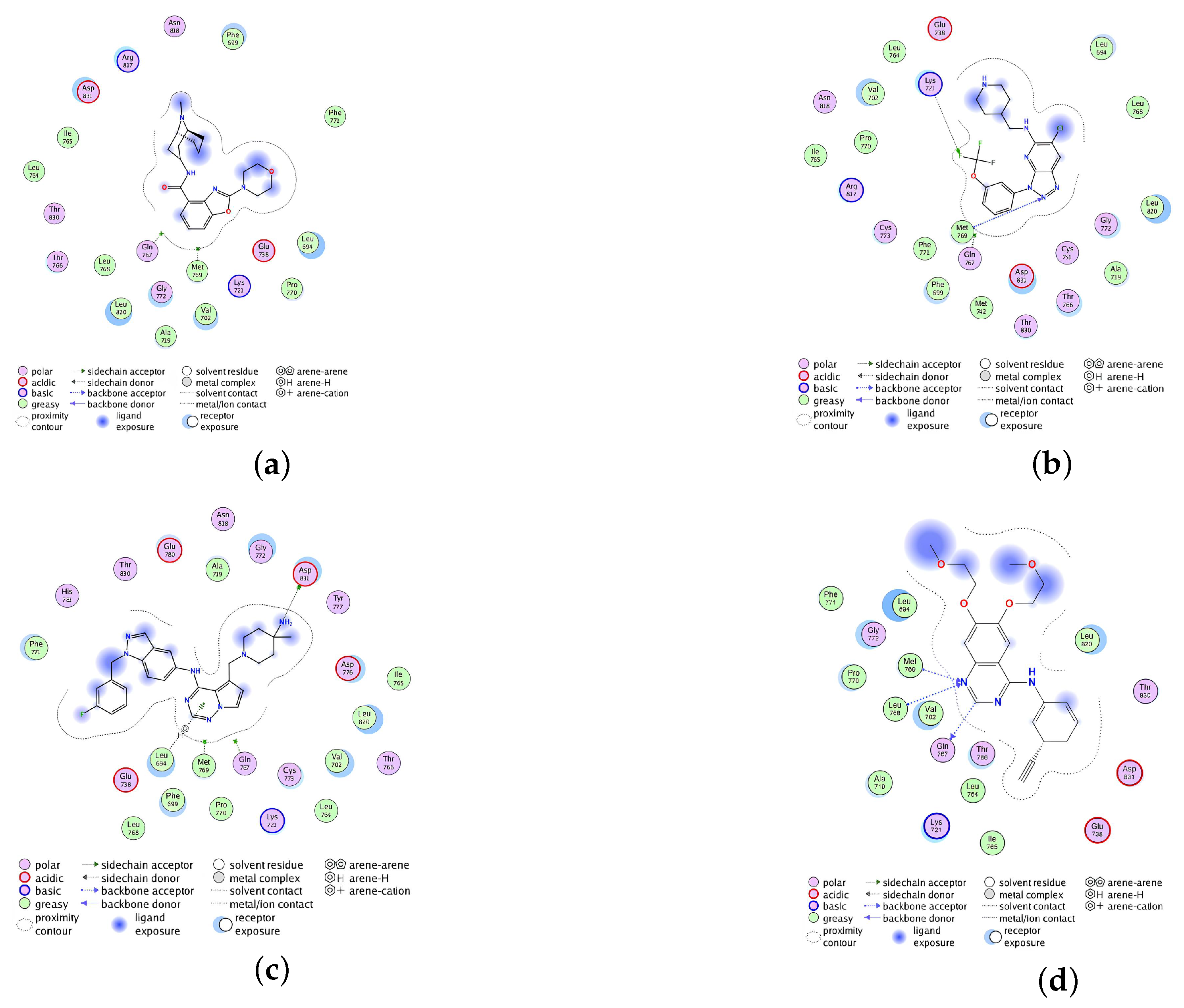
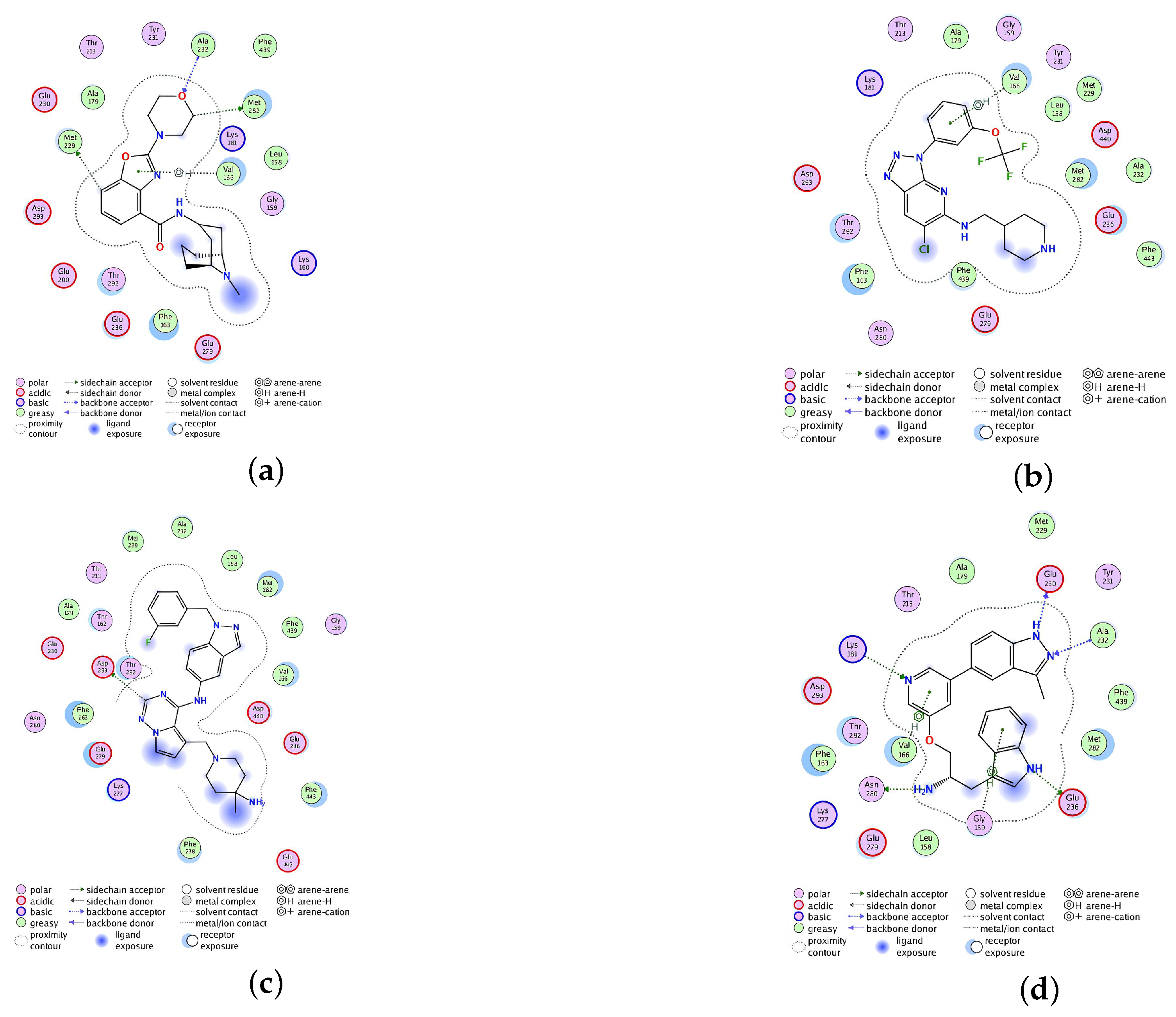
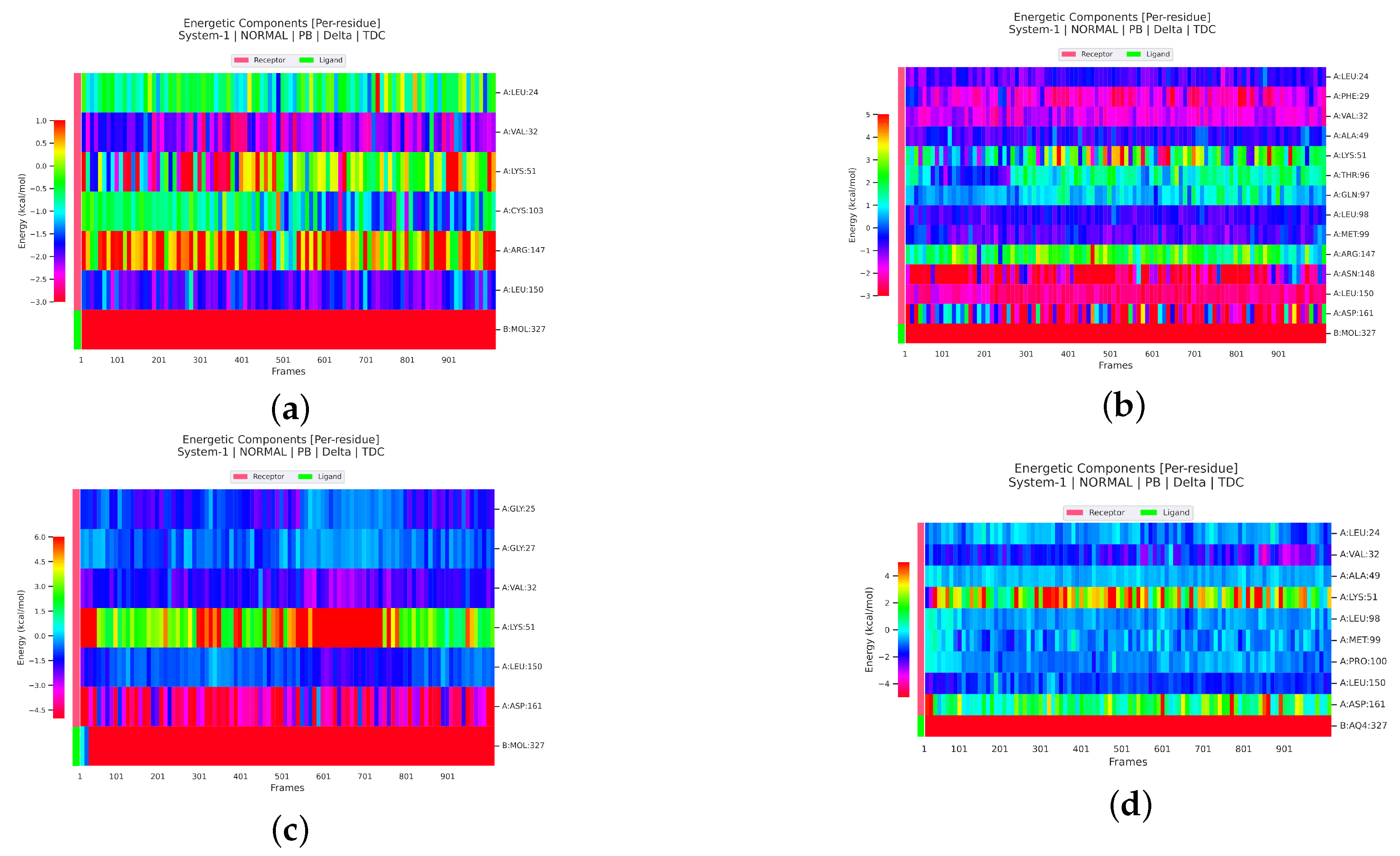
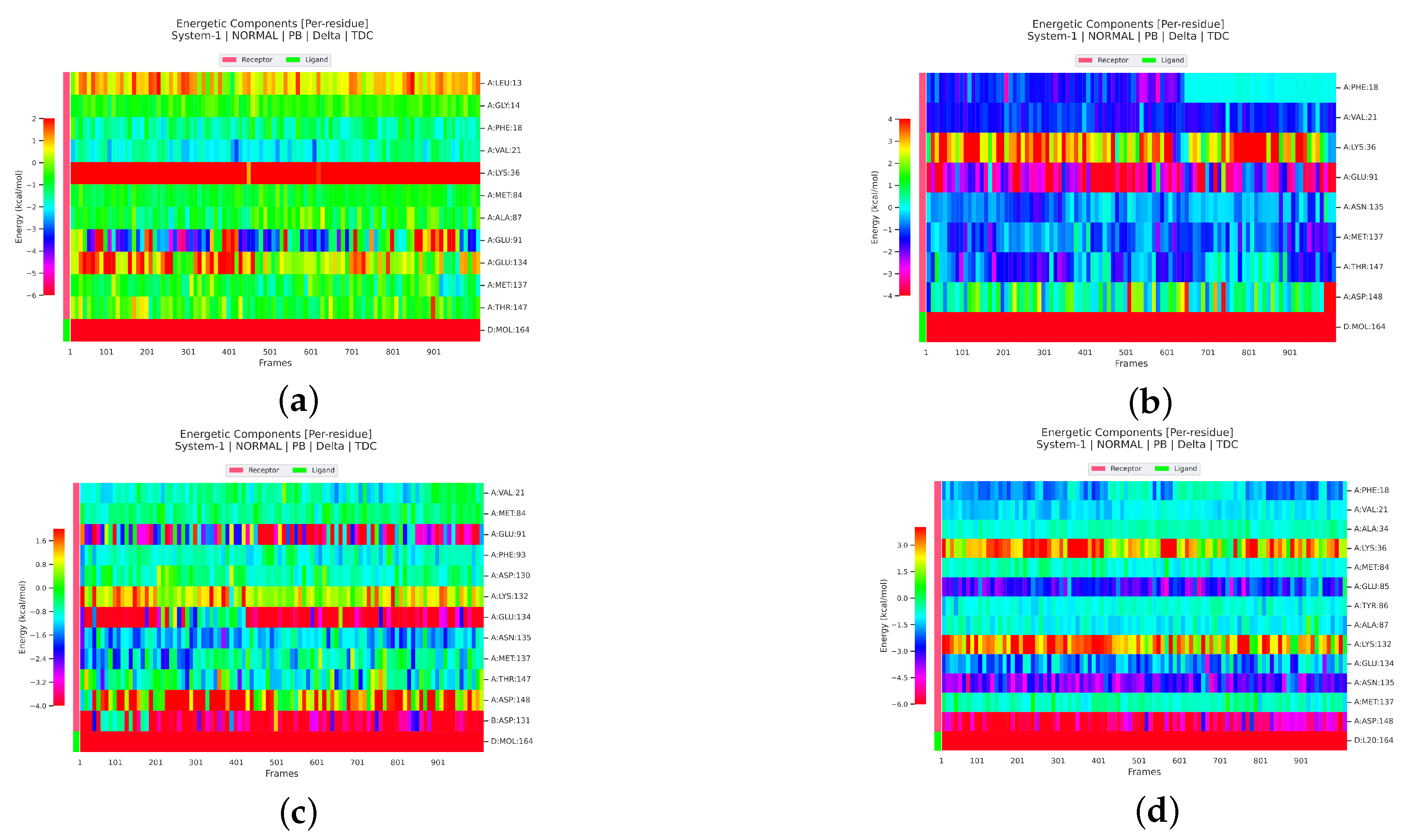
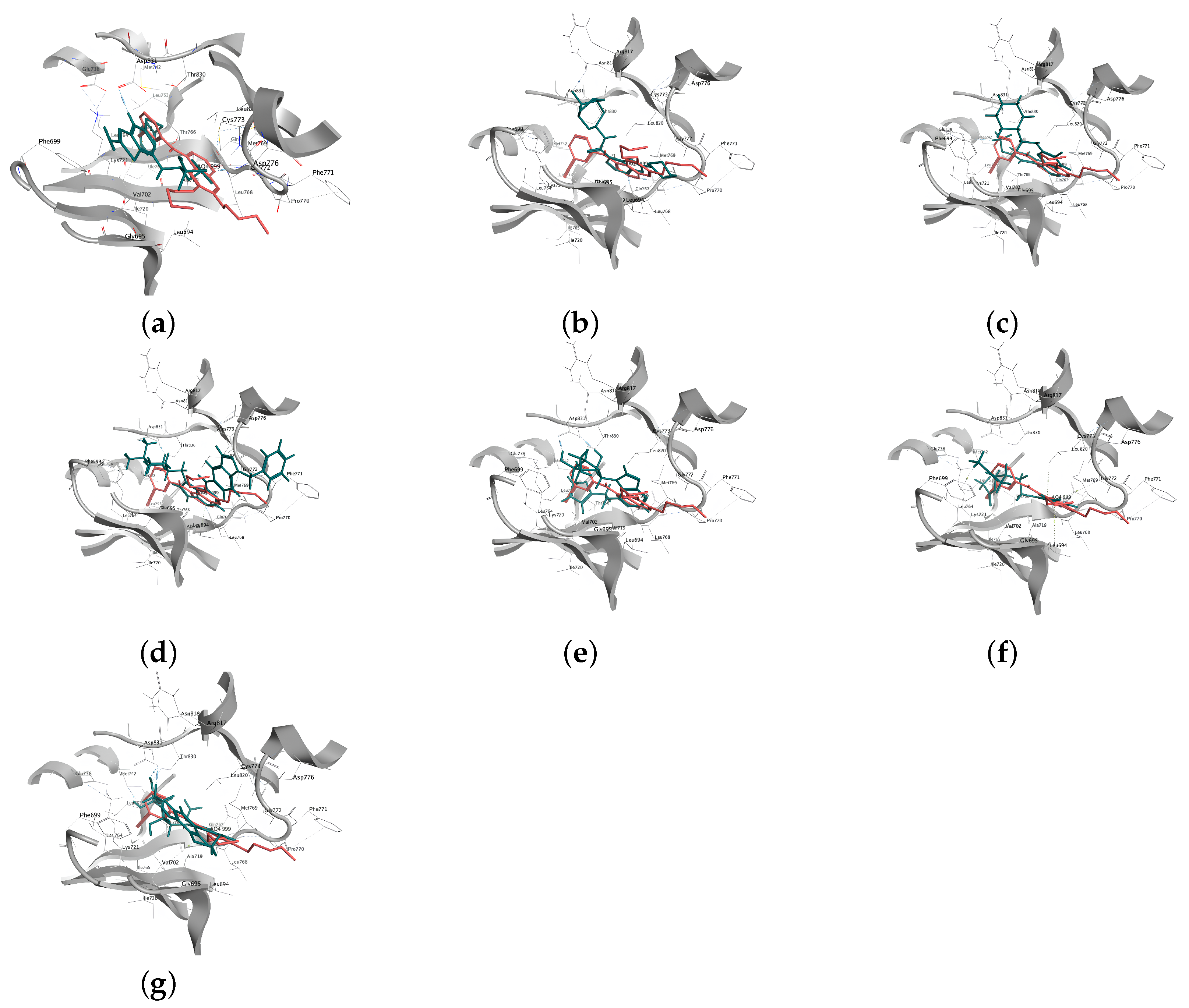
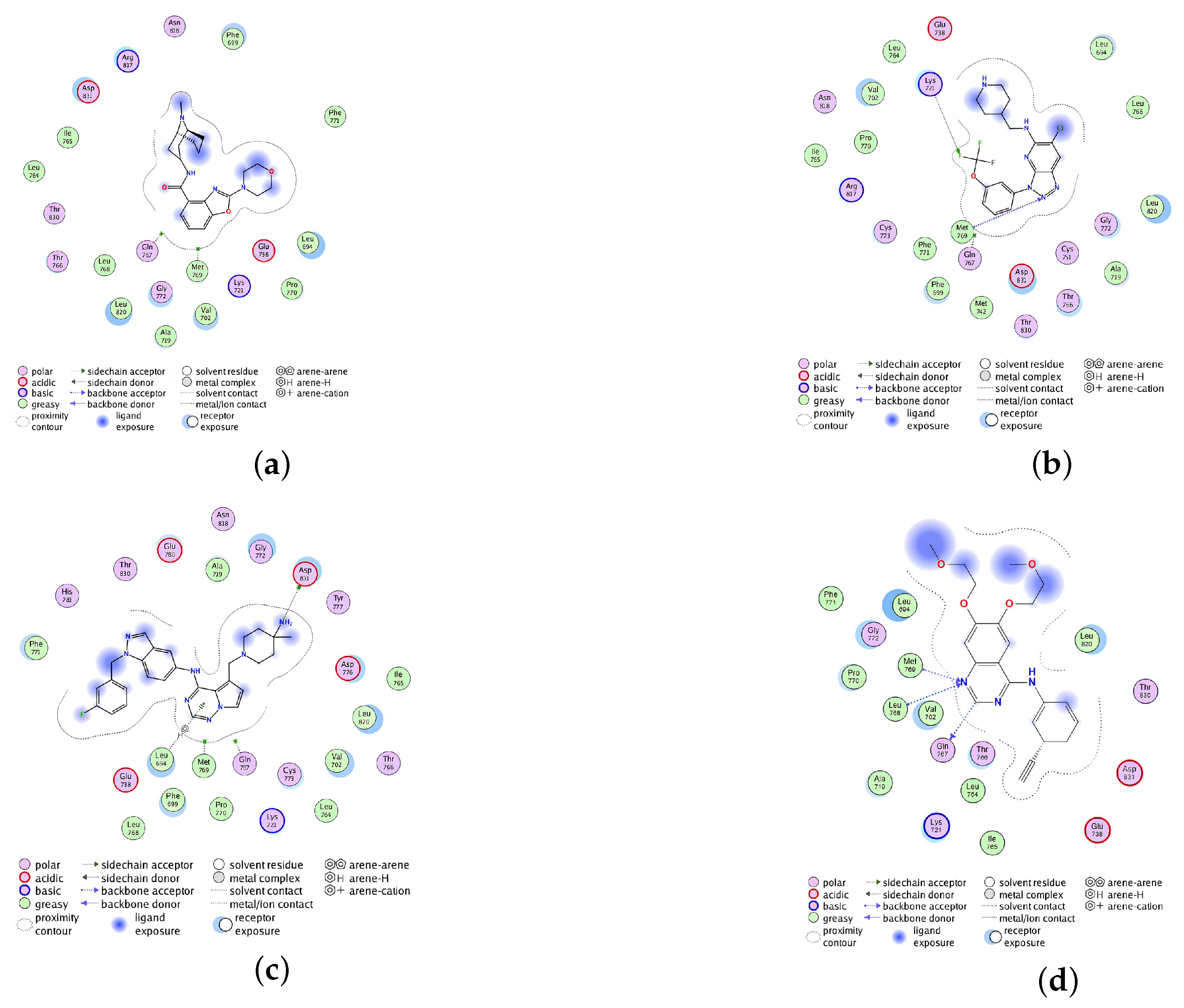
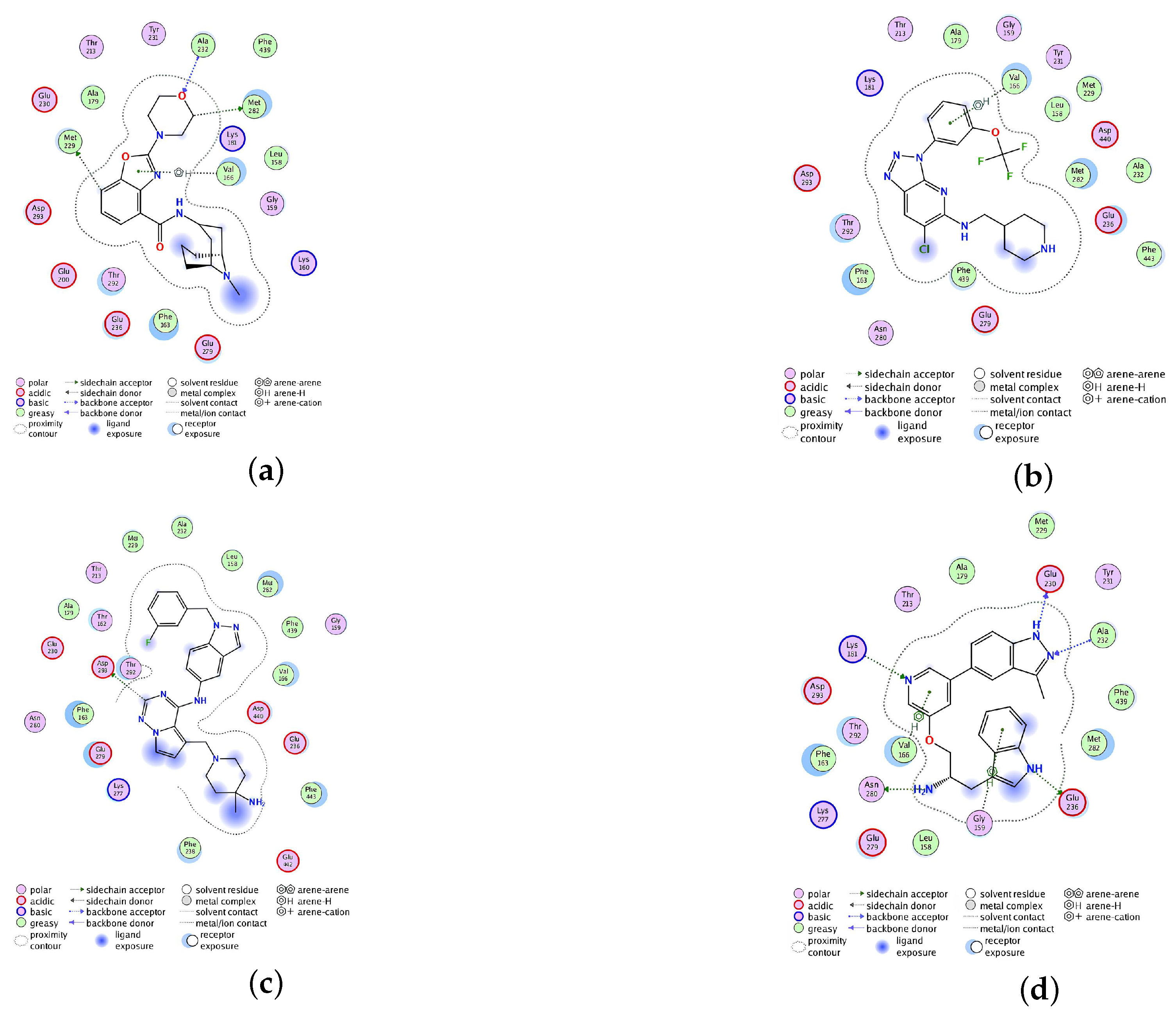
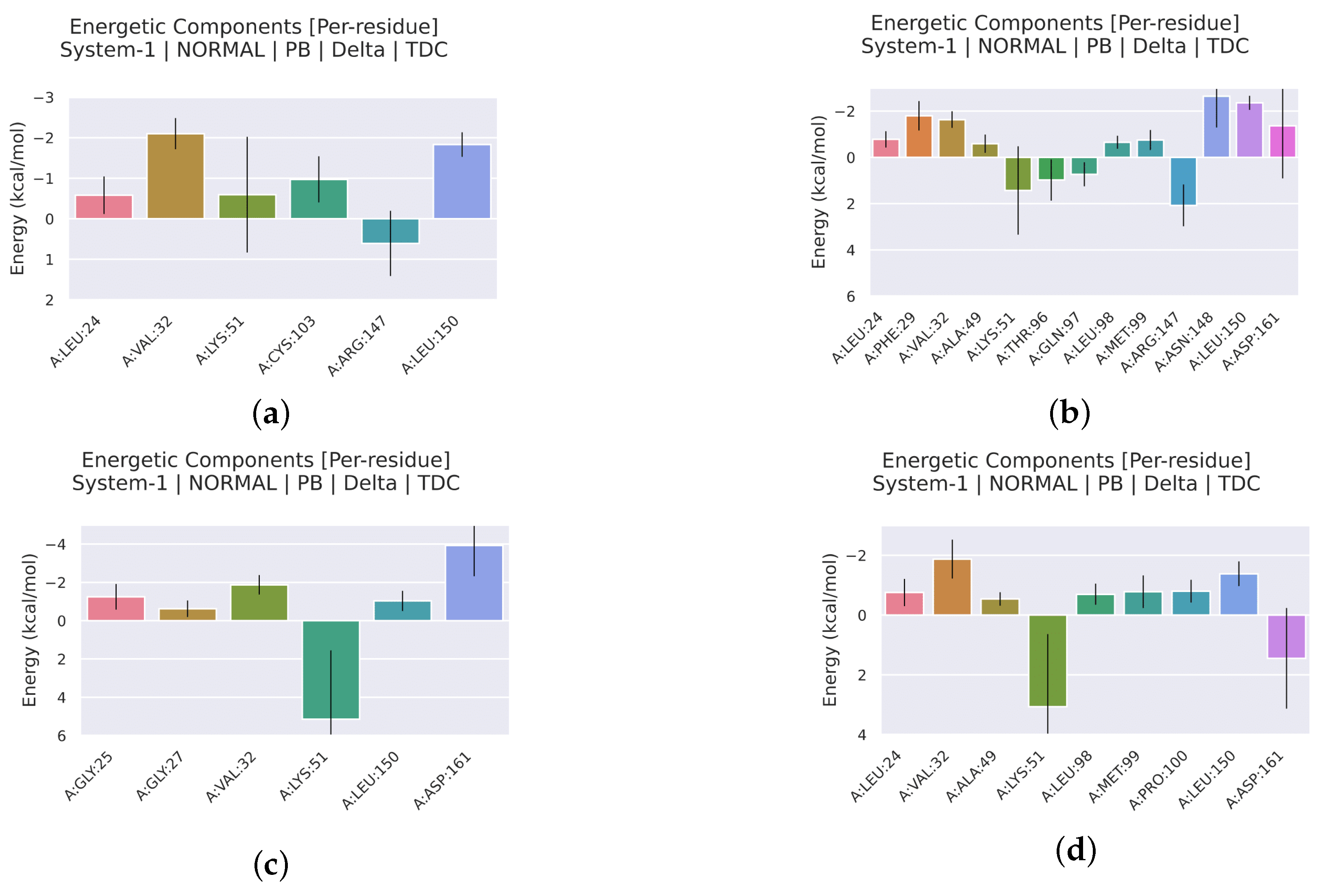
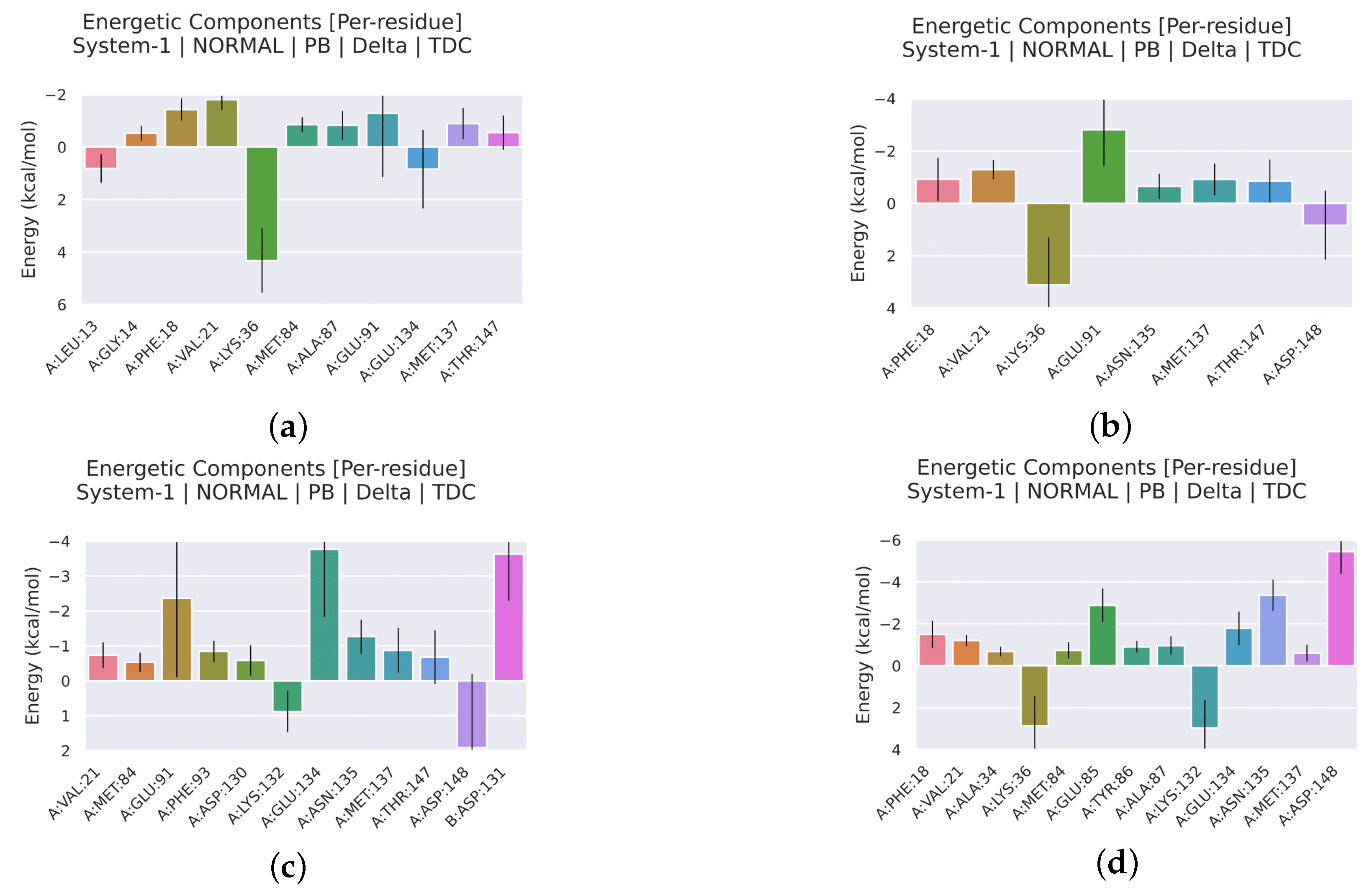
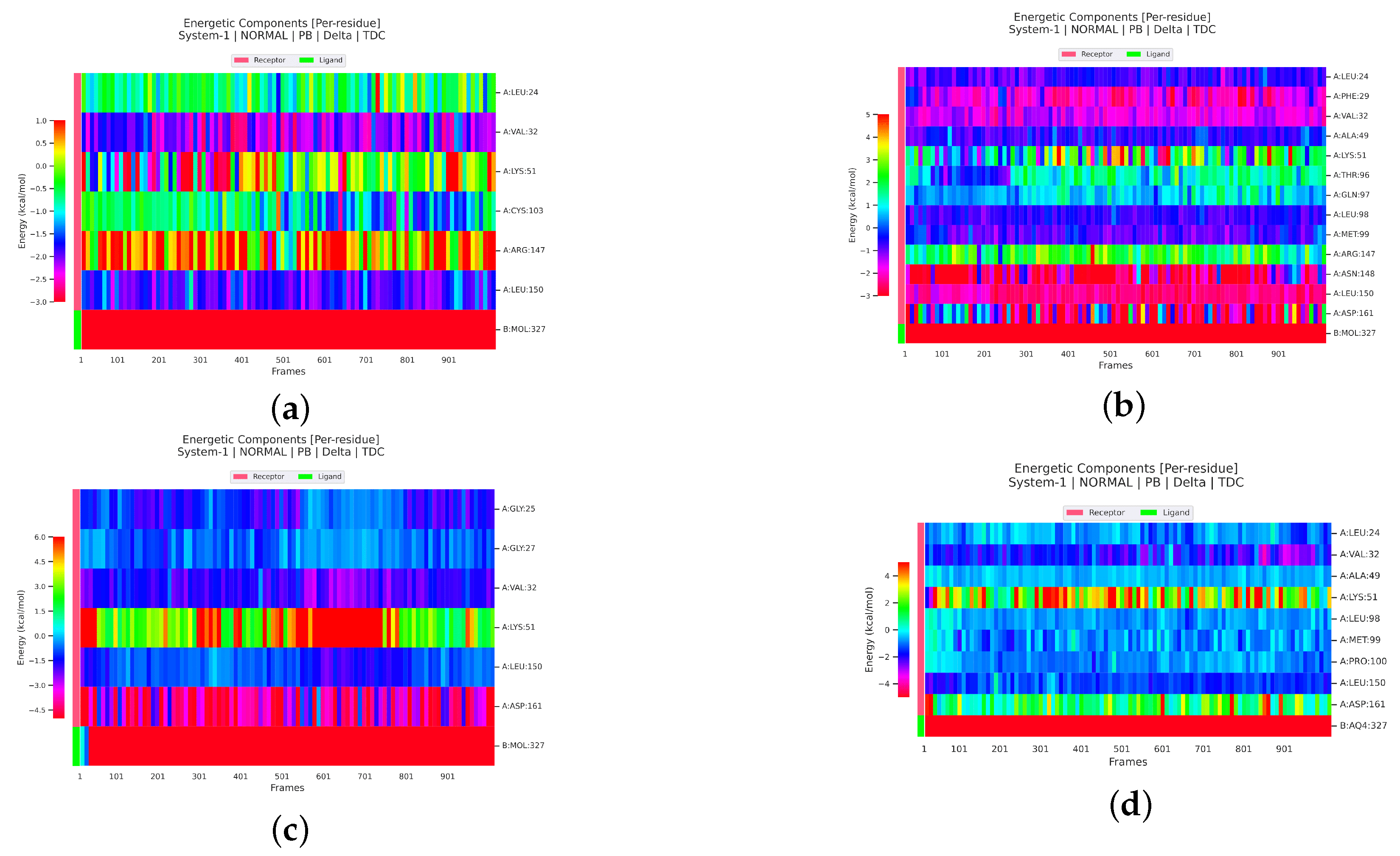
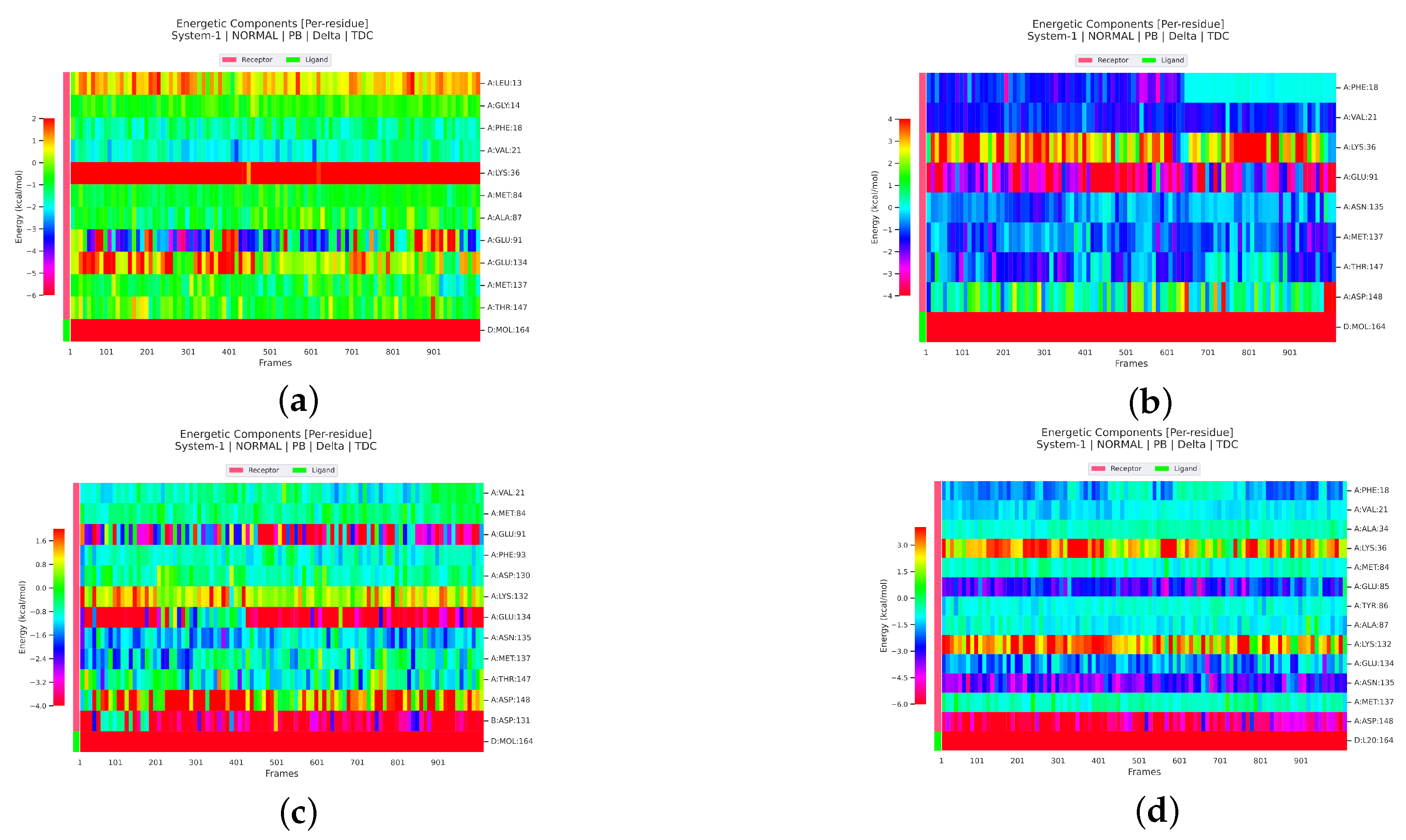
2.2.4. Analysis of the Interaction between Ligands and Proteins
2.2.5. ADMET Profiling
3. Screening Methods
3.1. Pharmacophore and Molecular Docking
3.2. Molecular Dynamics (MD) Simulations
3.3. ADMET Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Solvent accessible surface area |
References
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Harari, P.M. Epidermal growth factor receptor inhibition strategies in oncology. Endocr.-Relat. Cancer Endocr Relat Cancer 2004, 11, 689–708. [Google Scholar] [CrossRef]
- Yewale, C.; Baradia, D.; Vhora, I.; Patil, S.; Misra, A. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 2013, 34, 8690–8707. [Google Scholar] [CrossRef]
- Wu, W.W.; Ye, Z.J.; Xu, Y.M.; Zhang, J.; Tang, J.S. Ergosta-7, 22-diene-2β, 3α, 9α-triol (EGDT) from Ganoderma lucidum inhibits nasopharyngeal carcinoma cells by blocking EGFR signaling pathway. Chin. Herb. Med. 2018, 10, 27–33. [Google Scholar] [CrossRef]
- Yang, T.; Guo, Q.; Shi, X.; Wu, S.; Li, Y.; Sun, Y.; Zhao, Y.; Chai, L.; Gao, Y.; Lou, L.; et al. Panax notoginseng saponins promotes cerebral recovery from ischemic injury by downregulating LINGO-1 and activating the EGFR/PI3K/AKT signaling pathways in vivo. J. Tradit. Chin. Med. Sci. 2018, 5, 151–160. [Google Scholar] [CrossRef]
- Westover, D.; Zugazagoitia, J.; Cho, B.; Lovly, C.; Paz-Ares, L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef]
- Passaro, A.; Mok, T.; Peters, S.; Popat, S.; Ahn, M.J.; De Marinis, F. Recent advances on the role of EGFR tyrosine kinase inhibitors in the management of NSCLC with uncommon, non exon 20 insertions, EGFR mutations. J. Thorac. Oncol. 2021, 16, 764–773. [Google Scholar] [CrossRef]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.H.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, J.; Lu, S.; Zhang, Y.; Zhao, J.; Kim, D.W.; Soo, R.A.; Kim, S.W.; Pan, H.; et al. Tepotinib plus gefitinib in patients with EGFR-mutant non-small-cell lung cancer with MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitor (INSIGHT study): An open-label, phase 1b/2, multicentre, randomised trial. Lancet Respir. Med. 2020, 8, 1132–1143. [Google Scholar] [CrossRef]
- El Guerrab, A.; Bamdad, M.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Co-targeting EGFR and mTOR with gefitinib and everolimus in triple-negative breast cancer cells. Sci. Rep. 2020, 10, 6367. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, Y.M.; Pyo, H. Gefitinib radiosensitizes non-small cell lung cancer cells through inhibition of ataxia telangiectasia mutated. Mol. Cancer 2010, 9, 222. [Google Scholar] [CrossRef]
- Anderson, N.G.; Ahmad, T.; Chan, K.; Dobson, R.; Bundred, N.J. ZD1839 (Iressa), a novel epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, potently inhibits the growth of EGFR-positive cancer cell lines with or without erbB2 overexpression. Int. J. Cancer 2001, 94, 774–782. [Google Scholar] [CrossRef]
- Bencardino, K.; Manzoni, M.; Delfanti, S.; Riccardi, A.; Danova, M.; Corazza, G. Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small-cell lung cancer: Results and open issues. Intern. Emerg. Med. 2007, 2, 3–12. [Google Scholar] [CrossRef]
- Hochmair, M.J.; Buder, A.; Schwab, S.; Burghuber, O.C.; Prosch, H.; Hilbe, W.; Cseh, A.; Fritz, R.; Filipits, M. Liquid-biopsy-based identification of EGFR T790M mutation-mediated resistance to afatinib treatment in patients with advanced EGFR mutation-positive NSCLC, and subsequent response to osimertinib. Target. Oncol. 2019, 14, 75–83. [Google Scholar] [CrossRef]
- Lin, N.U.; Winer, E.P.; Wheatley, D.; Carey, L.A.; Houston, S.; Mendelson, D.; Munster, P.; Frakes, L.; Kelly, S.; Garcia, A.A.; et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res. Treat. 2012, 133, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Gregorc, V.; Karachaliou, N.; Rosell, R.; Santarpia, M. Mechanisms of resistance to osimertinib. J. Thorac. Dis. 2020, 12, 2851. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Yin, B.; Fang, D.M.; Zhou, X.L.; Gao, F. Natural products as important tyrosine kinase inhibitors. Eur. J. Med. Chem. 2019, 182, 111664. [Google Scholar] [CrossRef]
- Abou-Zied, H.A.; Youssif, B.; Mohamed, M.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, M.A.; Musa, A.; Almalki, A.H.; Alzarea, S.I.; Mostafa, E.M.; Hegazy, M.M.; Mostafa-Hedeab, G.; Ghoneim, M.M.; Parambi, D.G.; Bakr, R.B.; et al. Novel phenolic compounds as potential dual EGFR and COX-2 inhibitors: Design, semisynthesis, in vitro biological evaluation and in silico Insights. Drug Des. Dev. Ther. 2021, 15, 2325–2337. [Google Scholar] [CrossRef]
- Acevedo, C.H.; Scotti, L.; Scotti, M.T. In Silico Studies Designed to Select Sesquiterpene Lactones with Potential Antichagasic Activity from an In-House Asteraceae Database. ChemMedChem 2018, 13, 634–645. [Google Scholar] [CrossRef]
- Wheeler, D.; Dunn, E.; Harari, P. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/Protein Kinase B Is Constitutively Active in Non-Small Cell Lung Cancer Cells and Promotes Cellular Survival and Resistance to Chemotherapy and Radiation. Cancer Res. 2001, 61, 3986. [Google Scholar]
- Pan, Q.; Lu, Y.; Xie, L.; Wu, D.; Liu, R.; Gao, W.; Luo, K.; He, B.; Pu, Y. Recent Advances in Boosting EGFR Tyrosine Kinase Inhibitors-Based Cancer Therapy. Mol. Pharm. 2023, 20, 829–852. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Fan, M.; Shi, S.; Song, X.; Wang, F.; He, H.; Qi, B. Dual target inhibitors based on EGFR: Promising anticancer agents for the treatment of cancers (2017-). Eur. J. Med. Chem. 2022, 227, 113963. [Google Scholar] [CrossRef] [PubMed]
- Choowongkomon, K.; Sawatdichaikul, O.; Songtawee, N.; Limtrakul, J. Receptor-Based Virtual Screening of EGFR Kinase Inhibitors from the NCI Diversity Database. Molecules 2010, 15, 4041–4054. [Google Scholar] [CrossRef]
- Chuang, C.H.; Cheng, T.C.; Leu, Y.L.; Chuang, K.H.; Tzou, S.C.; Chen, C.S. Discovery of Akt Kinase Inhibitors through Structure-Based Virtual Screening and Their Evaluation as Potential Anticancer Agents. Int. J. Mol. Sci. 2015, 16, 3202–3212. [Google Scholar] [CrossRef]
- Lopez-Vallejo, F.; Caulfield, T.; Martinez-Mayorga, K.; A. Giulianotti, M.; Nefzi, A.; A. Houghten, R.; L. Medina-Franco, J. Integrating Virtual Screening and Combinatorial Chemistry for Accelerated Drug Discovery. Comb. Chem. High Throughput Screen. 2011, 14, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Bono, A.; Lauria, A.; La Monica, G.; Alamia, F.; Mingoia, F.; Martorana, A. In Silico Design of New Dual Inhibitors of SARS-CoV-2 MPRO through Ligand- and Structure-Based Methods. Int. J. Mol. Sci. 2023, 24, 8377. [Google Scholar] [CrossRef]
- López-López, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef]
- Tarozzi, A.; Marchetti, C.; Nicolini, B.; D’Amico, M.; Ticchi, N.; Pruccoli, L.; Tumiatti, V.; Simoni, E.; Lodola, A.; Mor, M.; et al. Combined inhibition of the EGFR/AKT pathways by a novel conjugate of quinazoline with isothiocyanate. Eur. J. Med. Chem. 2016, 117, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cai, C.; Du, J.; Liu, B.; Cui, L.; Fan, X.; Wu, Q.; Fang, J.; Xie, L. TCMIO: A comprehensive database of traditional Chinese medicine on immuno-oncology. Front. Pharmacol. 2020, 11, 439. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef] [PubMed]
- Wissner, A.; Fraser, H.L.; Ingalls, C.L.; Dushin, R.G.; Floyd, M.B.; Cheung, K.; Nittoli, T.; Ravi, M.R.; Tan, X.; Loganzo, F. Dual irreversible kinase inhibitors: Quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorg. Med. Chem. 2007, 15, 3635–3648. [Google Scholar] [CrossRef]
- Yang, Y.A.; Tang, W.J.; Zhang, X.; Yuan, J.W.; Liu, X.H.; Zhu, H.L. Synthesis, molecular docking and biological evaluation of glycyrrhizin analogs as anticancer agents targeting EGFR. Molecules 2014, 19, 6368–6381. [Google Scholar] [CrossRef]
- Peng, Y.H.; Shiao, H.Y.; Tu, C.H.; Liu, P.M.; Hsu, J.T.A.; Amancha, P.K.; Wu, J.S.; Coumar, M.S.; Chen, C.H.; Wang, S.Y.; et al. Protein Kinase Inhibitor Design by Targeting the Asp-Phe-Gly (DFG) Motif: The Role of the DFG Motif in the Design of Epidermal Growth Factor Receptor Inhibitors. J. Med. Chem. 2013, 56, 3889–3903. [Google Scholar] [CrossRef]
- Zarezade, V.; Abolghasemi, M.; Rahim, F.; Veisi, A.; Behbahani, M. In silico assessment of new progesterone receptor inhibitors using molecular dynamics: A new insight into breast cancer treatment. J. Mol. Model. 2018, 24, 337. [Google Scholar] [CrossRef]
- Zarezade, V.; Rezaei, H.; Shakerinezhad, G.; Safavi, A.; Nazeri, Z.; Veisi, A.; Azadbakht, O.; Hatami, M.; Sabaghan, M.; Shajirat, Z. The identification of novel inhibitors of human angiotensin-converting enzyme 2 and main protease of Sars-Cov-2: A combination of in silico methods for treatment of COVID-19. J. Mol. Struct. 2021, 1237, 130409. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Méndez-Luna, D.; Martínez-Archundia, M.; Maroun, R.C.; Ceballos-Reyes, G.; Fragoso-Vázquez, M.; González-Juárez, D.; Correa-Basurto, J. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J. Biomol. Struct. Dyn. 2015, 33, 2161–2172. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Sneha, P.; Doss, C.G.P.; Siva, R.; Zayed, H. A profound computational study to prioritize the disease-causing mutations inPRPS1gene. Metab. Brain Dis. 2017, 33, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.; Sumitha, R.; Doss, C.; Rajasekaran, C.; Siva, R. Molecular Docking and Molecular Dynamics to Identify a Novel Human Immunodeficiency Virus Inhibitor from Alkaloids of Toddalia asiatica. Pharmacogn. Mag. 2015, 11, S414. [Google Scholar] [PubMed]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA methods in virtual screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J.-Comput.-Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef]
- Empereur-Mot, C.; Zagury, J.F.; Montes, M. Screening Explorer–An Interactive Tool for the Analysis of Screening Results. J. Chem. Inf. Model. 2016, 56, 2281–2286. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Yadav, A.V.; Shete, A.S.; Dabke, A.P.; Kulkarni, P.V.; Sakhare, S.S. Co-Crystals: A Novel Approach to Modify Physicochemical Properties of Active Pharmaceutical Ingredients. Indian J. Pharm. Sci. 2009, 71, 359–370. [Google Scholar] [CrossRef]
- Case, D.; Aktulga, H.; Belfon, K.; Ben-Shalom, I.; Berryman, J.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cisneros, G. In Proceedings of the Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Vassetti, D.; Pagliai, M.; Procacci, P. Assessment of GAFF2 and OPLS-AA General Force Fields in Combination with the Water Models TIP3P, SPCE, and OPC3 for the Solvation Free Energy of Druglike Organic Molecules. J. Chem. Theory Comput. 2019, 15, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Lu, W.C. AmberMDrun: A Scripting Tool for Running Amber MD in an Easy Way. Biomolecules 2023, 13, 635. [Google Scholar] [CrossRef]
- Roe, D.R.; Brooks, B.R. A protocol for preparing explicitly solvated systems for stable molecular dynamics simulations. J. Chem. Phys. 2020, 153, 054123. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 2008, 72, 2384–2393. [Google Scholar] [CrossRef]
- Miller, B.R.; Mcgee, T.D.; Swails, J.M.; Homeyer, N.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J Chem Theory Comput 8:3314-3321. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Cui, Z.; Zhang, Z.; Zhou, T.; Zhou, X.; Zhang, Y.; Meng, H.; Wang, W.; Liu, Y. A TastePeptides-Meta system including an umami/bitter classification model Umami_YYDS, a TastePeptidesDB database and an open-source package Auto_Taste_ML. Food Chem. 2023, 405, 134812. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | Docking Score (kcal/mol) | Interaction Type | Key Amino Acid | Ligand Efficiency (kcal/mol) |
|---|---|---|---|---|
| TCMIO5312 | −7.72 | H-donor | Glu831, Met742, | −0.276 |
| Asp831 | ||||
| H-acceptor | Asp831 | |||
| TCMIO89212 | −7.06 | H-acceptor | Met769, Gln767 | −0.252 |
| TCMIO90156 | −7.51 | H-acceptor | Met769, Lys721, | −0.256 |
| Gln767 | ||||
| H-acceptor | Met769 | |||
| TCMIO98874 | −7.96 | H-donor | Asp831 | −0.221 |
| H-acceptor | Met769, Gln767 | |||
| HMDB0012243 | −7.29 | H-donor | Asp831 | −0.270 |
| HMDB0014570 | −7.33 | H-acceptor | Met769 | −0.318 |
| HMDB0037450 | −7.85 | H-donor | Asp776, Gln767 | −0.231 |
| H-acceptor | Met769, Thr830, | |||
| Asp831, Lys721 | ||||
| Ionic | Lys721 | |||
| Erlotinib | −7.37 | H-acceptor | Met769 | −0.254 |
| Compound ID | Docking Score (kcal/mol) | Interaction Type | Key Amino Acid | Ligand Efficiency (kcal/mol) |
|---|---|---|---|---|
| TCMIO5312 | −8.08 | H-donor | Asn820, Glu236 | −0.289 |
| Ionic | Glu279 | |||
| TCMIO89212 | −8.24 | H-donor | Met229, Met282 | −0.294 |
| H-acceptor | Ala232 | |||
| TCMIO90156 | −8.28 | H-donor | Glu236 | −0.285 |
| H-donor | Asp831 | |||
| TCMIO98874 | −8.00 | H-donor | Asp440 | −0.222 |
| Ionic | Asp440 | |||
| HMDB0012243 | −8.17 | H-donor | Met229 | |
| H-acceptor | Ala232, Asp293 | |||
| Lys181 | ||||
| HMDB0014570 | −8.15 | H-donor | Glu230 | −0.354 |
| H-acceptor | Asp293, Ala232 | |||
| HMDB0037450 | −9.32 | H-donor | Glu230, Gln236 | −0.274 |
| H-acceptor | Lys181, Glu236, | |||
| Asp440 | ||||
| Ionic | Lys181 | |||
| A-443654 | −8.42 | H-donor | Glu230, Asn280, | −0.280 |
| Asp290, Glu236 | ||||
| H-acceptor | Ala232, Lys181 | |||
| Ionic | Asp293 |
| Ligands | SASA | ||||
|---|---|---|---|---|---|
| TCMIO90156 | −47.01 | −131.48 | 147.32 | −4.57 | −35.74 |
| TCMIO98874 | −29.94 | −243.41 | 247.05 | −3.36 | −29.66 |
| TCMIO5312 | −36.35 | −146.94 | 160.09 | −4.45 | −27.65 |
| TCMIO89212 | −42.79 | −149.06 | 169.92 | −4.27 | −26.20 |
| HMDB0012243 | −36.89 | −49.90 | 70.77 | −4.12 | −20.14 |
| HMDB0014570 | −40.65 | −16.51 | 45.41 | −3.92 | −15.67 |
| HMDB0037450 | −15.32 | −49.39 | 54.12 | −2.45 | −13.05 |
| Erlotinib | −47.65 | −146.57 | 167.75 | −4.32 | −30.79 |
| EGFR-backbone | −33.10 | −14.21 | 29.35 | −3.60 | −21.57 |
| Ligands | SASA | ||||
|---|---|---|---|---|---|
| TCMIO98874 | −52.28 | −647.65 | 648.81 | −5.20 | −56.32 |
| TCMIO89212 | −52.31 | −262.09 | 285.63 | −4.41 | −33.18 |
| TCMIO90156 | −31.11 | −310.42 | 312.20 | −3.50 | −29.83 |
| TCMIO5312 | −29.42 | −261.69 | 275.08 | −3.88 | −19.91 |
| HMDB0012243 | −35.02 | −61.14 | 78.35 | −3.87 | −21.68 |
| HMDB0014570 | −42.16 | −29.37 | 54.73 | −3.88 | −20.68 |
| HMDB0037450 | −36.89 | −74.53 | 111.81 | −4.39 | −4.00 |
| A-443654 | −42.52 | −270.18 | 278.73 | −4.4 | −38.37 |
| AKT-backbone | −42.98 | −274.20 | 276.02 | −4.43 | −45.59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Zhang, Z.; Liu, Q.; Yu, J.; Liu, C.; Lu, W. Identification of Dual-Target Inhibitors for Epidermal Growth Factor Receptor and AKT: Virtual Screening Based on Structure and Molecular Dynamics Study. Molecules 2023, 28, 7607. https://doi.org/10.3390/molecules28227607
Yang H, Zhang Z, Liu Q, Yu J, Liu C, Lu W. Identification of Dual-Target Inhibitors for Epidermal Growth Factor Receptor and AKT: Virtual Screening Based on Structure and Molecular Dynamics Study. Molecules. 2023; 28(22):7607. https://doi.org/10.3390/molecules28227607
Chicago/Turabian StyleYang, Hanyu, Zhiwei Zhang, Qian Liu, Jie Yu, Chongjin Liu, and Wencai Lu. 2023. "Identification of Dual-Target Inhibitors for Epidermal Growth Factor Receptor and AKT: Virtual Screening Based on Structure and Molecular Dynamics Study" Molecules 28, no. 22: 7607. https://doi.org/10.3390/molecules28227607
APA StyleYang, H., Zhang, Z., Liu, Q., Yu, J., Liu, C., & Lu, W. (2023). Identification of Dual-Target Inhibitors for Epidermal Growth Factor Receptor and AKT: Virtual Screening Based on Structure and Molecular Dynamics Study. Molecules, 28(22), 7607. https://doi.org/10.3390/molecules28227607