Abstract
SIRT2 is a member of NAD+-dependent sirtuins and its inhibition has been proposed as a promising therapeutic approach for treating human diseases, including neurodegenerative diseases, cancer, and infections. Expanding SIRT2 inhibitors based on the 3-aminobenzyloxy nicotinamide core structure, we have synthesized and evaluated constrained analogs and selected stereoisomers. Our structure-activity relationship (SAR) study has revealed that 2,3-constrained (S)-isomers possess enhanced in vitro enzymatic inhibitory activity against SIRT2 and retain excellent selectivity over SIRT1 and SIRT3, provided that a suitable ring A is used. This current study further explores SIRT2 inhibitors based on the 3-aminobenzyloxy nicotinamide scaffold and contributes to the discovery of potent, selective SIRT2 inhibitors that have been actively pursued for their potential therapeutic applications.
1. Introduction
Sirtuins are a group of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylase that use NAD+ as a cofactor to remove an acetyl or other acyl group from the lysine residue of various substrates [1,2]. Among the seven human sirtuins (SIRT1-SIRT7), SIRT2 is the only one that mainly resides in the cytoplasm; however, it can be localized to the nucleus [3] and can also be detected in the mitochondria [4,5]. SIRT2 is implicated in different human diseases, including neurodegenerative diseases, cancer, and infections [6]. Therefore, as the most abundant sirtuin homolog in the brain [7,8], selective inhibition of SIRT2 has been proposed as a promising therapeutic approach for Parkinson’s disease [9] and Alzheimer’s disease [10]. While the roles of SIRT2 are complex and might vary in different cancers, pharmaceutical inhibition of SIRT2 holds promise as a potential treatment for certain types of cancer [11]. More recently, SIRT2 as a proviral host factor has been investigated, suggesting that SIRT2 is a viable antiviral target [12,13,14,15].
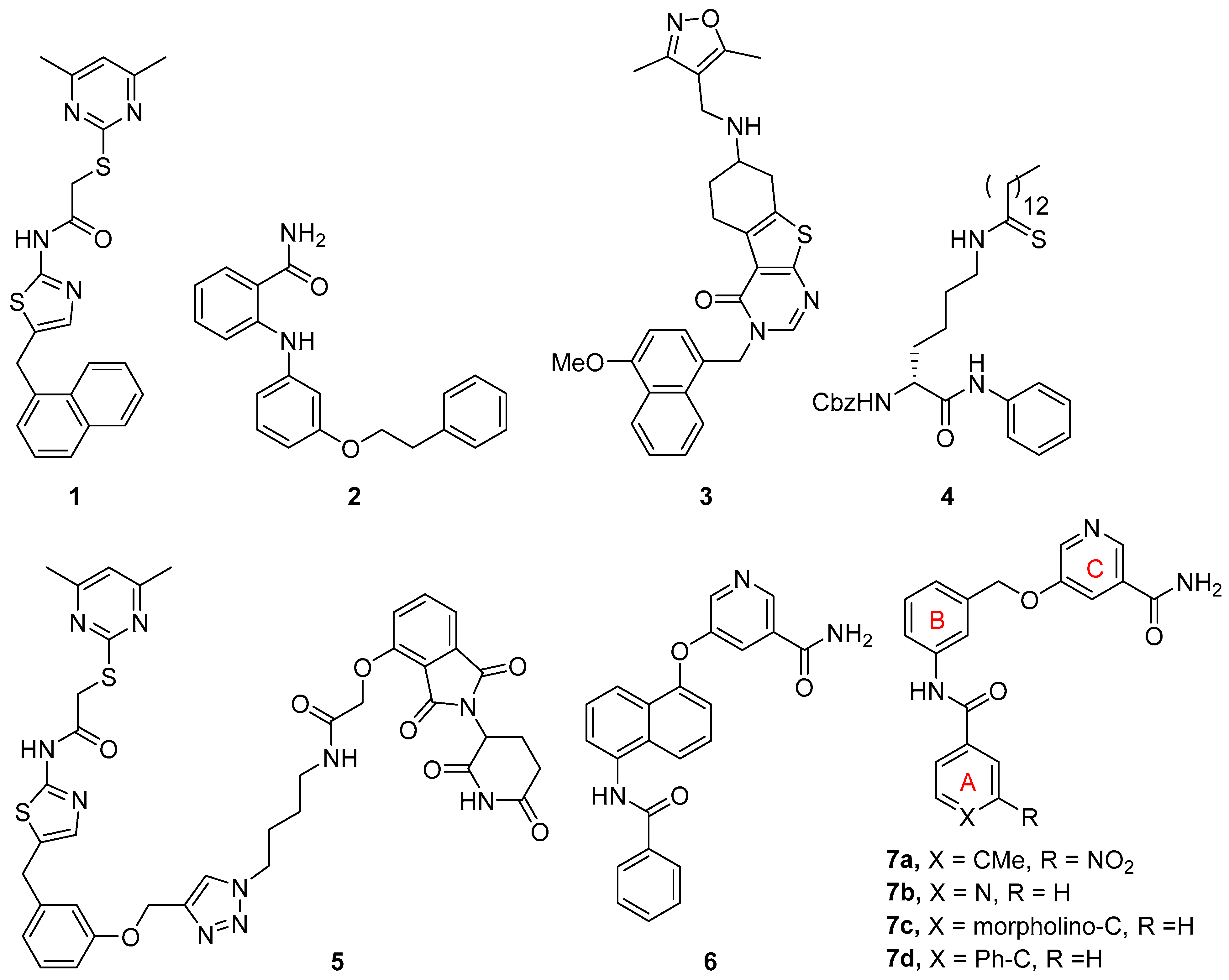
Interest in discovering potent and selective SIRT2 inhibitors and exploring their potential therapeutic applications in various diseases has resulted in a number of SIRT2 inhibitors, which have been updated and reviewed [11,16,17,18]. Remarkable among these SIRT2 inhibitors are those that bind into the ligand-induced, hydrophobic extended C-site and selectivity pocket, including SirReal2 (1, Figure 1) and its analogs [19,20], 2-anilinobenzamide 2 [21], and thienopyrimidinone 3 [22], which all possess excellent selectivity over SIRT1 and SIRT3. Mechanism-based SIRT2 inhibitors, such as thioamide 4, have also been developed [23,24,25]. Another significant development is the discovery of SIRT2 degraders based on the proteolysis-targeting chimera (PROTAC) strategy by introducing an E3 ligase-engaging moiety [26]. This new class of SIRT2 inhibitors is exemplified by compound 5 [27], which is based on the structure of SirReal inhibitors, and one derived from a mechanism-based inhibitor [28].
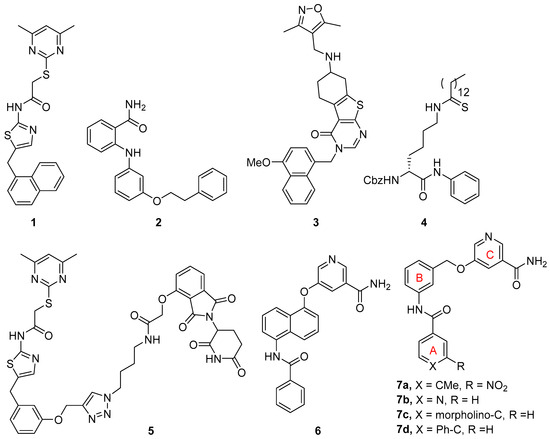
Figure 1.
Selected SIRT2 inhibitors.
Through a fragment-based approach, we have discovered potent and selective SIRT2 inhibitors based on a 5-aminonaphthalen-1-yloxy nicotinamide core structure as exemplified by compound 6 (Figure 1) [29]. Further simplification of the central naphthalene ring has resulted in SIRT2 inhibitors that feature a 3-aminobenzyloxy nicotinamide scaffold as represented by compound 7 [30]. An extensive structure-activity relationship (SAR) study on ring A has led to excellent SIRT2 inhibitors including compounds 7a–d. In our continued efforts to discover novel SIRT2 inhibitors, here we report constrained analogs based on the 3-aminobenzyloxy nicotinamide core structure. In these constrained compounds, we chose to rigidify the benzyloxy linker between rings B and C (Figure 1) by incorporating a five- or six-membered ring as part of the linker. We aimed to discover constrained inhibitors that allow for optimal projection of the terminal nicotinamide moiety into the C-pocket, which is occupied by the nicotinamide portion of NAD during the deacylation reaction.
2. Results and Discussion
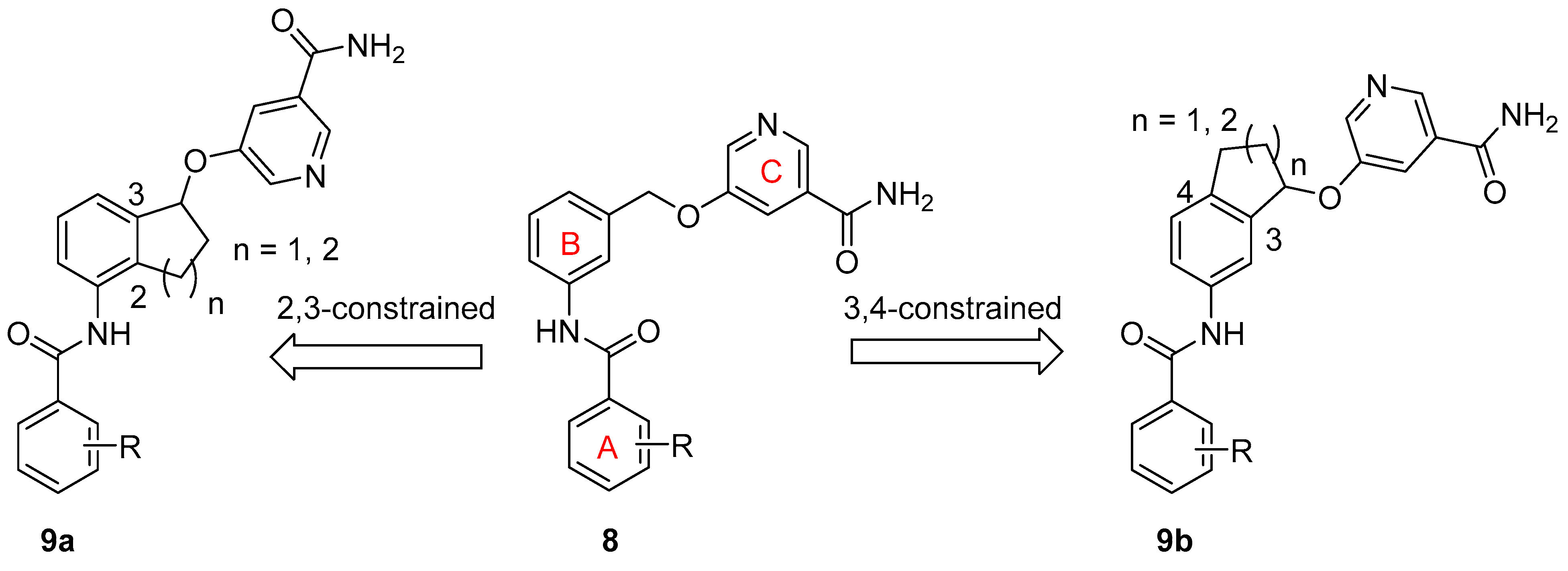
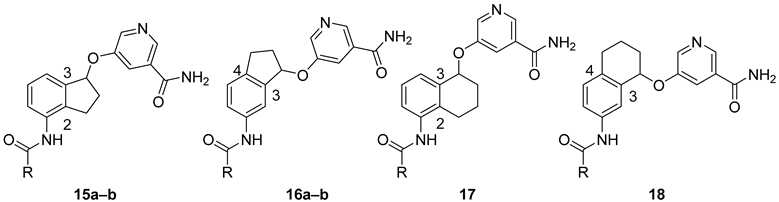
SAR Study. There are two approaches to designing constrained analogs of compound 8 by introducing a five-member ring through linking either positions 2 and 3 (9a, Figure 2) or positions 3 and 4 (9b). Besides a five-member ring, a six-member ring can also be explored (Figure 2). Accordingly, we first prepared compounds 15a–b and 16a–b, which featured a 2,3-five-member ring and a 3,4-five-member ring, respectively. Furthermore, six-member ring analogs 17 and 18 were also obtained. For these initial compounds, 4-methyl-3-nitrophenyl or 4-pyridyl was used as ring A because they were identified as good moieties in our previous SAR study (Table 1) [30].
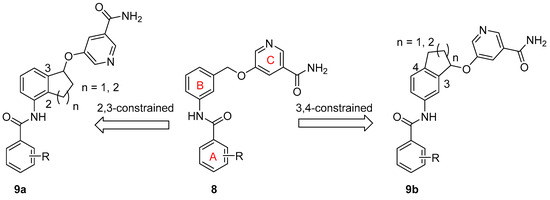
Figure 2.
Design of constrained SIRT2 inhibitors.

Table 1.
Inhibitory activity of the constrained analogs a.
These racemic compounds were subsequently tested against human SIRT1-3 (Table 1) with their non-constrained counterparts 7a–b as reference compounds. The enzymatic results showed that constrained analogs retained selectivity against SIRT2 over SIRT1 and SIRT3. For 2,3-constrained five-member ring inhibitors, compound 15a was about 3-fold less active than its non-constrained counterpart 7a. However, when 4-pyridyl was used as ring A, the resulting 15b exhibited anti-SIRT2 activity comparable to that of 7b, suggesting that the nature of ring A could be an important factor. For 3,4-constrained isomers, a 5- and 3-fold reduction in SIRT2 inhibition was observed in 16a and 16b when compared with non-constrained 7a and 7b, respectively. On the other hand, incorporation of a six-member ring in 17 or 18 led to an approximately 30-fold reduction in SIRT2 inhibition. These results indicated that a five-member ring was tolerated while a six-member one was clearly detrimental to anti-SIRT2 activity.
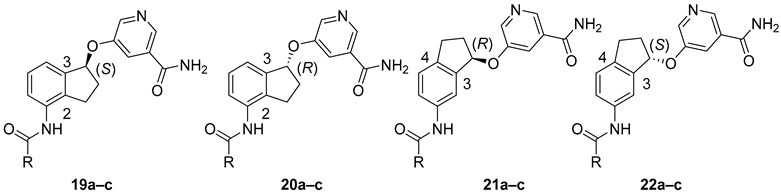
Having established the viability of five-member ring constrained analogs, we proceeded to assess the effect of stereochemistry. To that end, we prepared compounds 19–22 (Table 2). For ring A, we chose to evaluate three variations (4-pyridyl, 4-morpholinophenyl, and 1,1′-biphenyl), which all showed high inhibitory activity against SIRT2 and good selectivity in the previous study [30]. These new compounds and their non-constrained counterparts 7b–d were then evaluated in our in-house SIRT1-3 assays. In these assays, NAD and the peptide substrate were used at the KM values determined for each enzyme, allowing for accurate assessment of an inhibitor’s selectivity against SIRT1-3. We first focused on 2,3-constrained stereoisomers. Among the (S)-isomers, 19a (with 4-pyridyl as its ring A) possessed anti-SIRT2 activity nearly identical to that of non-constrained 7b, a trend that was also observed in its racemic counterpart 15b (Table 1); however, 19a displayed improved selectivity over SIRT1 and SIRT3 when compared with 7b. In comparison, (S)-isomer 19b’s anti-SIRT2 capacity was slightly weaker than its non-constrained counterpart 7c but retained a similar selectivity profile. When 1,1′-biphenyl was used as ring A, the resulting non-constrained 7d exhibited excellent selectivity over SIRT1 and SIRT3. Remarkably, 7d-derived (S)-isomer 19c showed more than 3-fold improvement in anti-SIRT2 activity (64 nM), leading to >1500-fold selectivity against SIRT2 over SIRT1 and SIRT3. With its low nanomolar inhibitory activity and excellent selectivity profile, compound 19c is among the most potent and selective SIRT2 inhibitors reported [11,16,17,18]. Also noticeably, ring A appeared to have different effects on constrained (S)-isomers’ ability to inhibit SIRT2 in comparison with their corresponding non-constrained ones. While 4-pyridyl had essentially no effect, 4-morpholinophenyl and 1,1′-biphenyl reduced and enhanced anti-SIRT2 inhibition, respectively. There is no definitive explanation for this phenomenon, but it might be because ring A binds into the relatively hydrophobic lysine substrate channel (see Computational Modeling below). Therefore, a lipophilic substitution (at position 4 of ring A) like a phenyl ring in 1,1′-biphenyl is preferred while a relatively polar morpholine ring in 4-morpholinophenyl is not well tolerated and 4-pyridyl has a minimal effect. We next examined (R)-isomers 20a–c, which exhibited approximately a 5-, 12-, and 2-fold reduction in anti-SIRT2 activity, respectively; however, these compounds remained relatively selective against SIRT2. These results showed that a lipophilic ring A was still preferred in (R)-isomers. It was also clear that the anti-SIRT2 activity and selectivity of (S)-isomers were significantly higher than the corresponding (R)-isomers (Table 2). Taken together, for 2,3-constrained inhibitors, (S)-isomers were superior to (R)-isomers especially when a lipophilic ring A like 1,1′-biphenyl was used.
Table 2.
Inhibitory activity of the constrained analogs: The effect of stereochemistry a.
We continued to examine 3,4-constrained stereoisomers 21 and 22. While (R)-isomers 21a–c and (S)-isomers 22a–c exhibited very similar anti-SIRT2 activity and selectivity profiles, their inhibitory capacity against SIRT2 was significantly diminished when compared with their non-constrained counterparts 7b–d as well as 2,3-constrained (S)-isomers 19a–c, respectively. These results clearly demonstrated that a 3,4-five-member ring was not a favorable structural feature. Taken together, 2,3-constrained (S)-isomers were the preferred isomers among those studied. Equipped with a suitable ring A like 1,1′-biphenyl, these stereoisomers could possess superior inhibitory activity and selectivity against SIRT2.
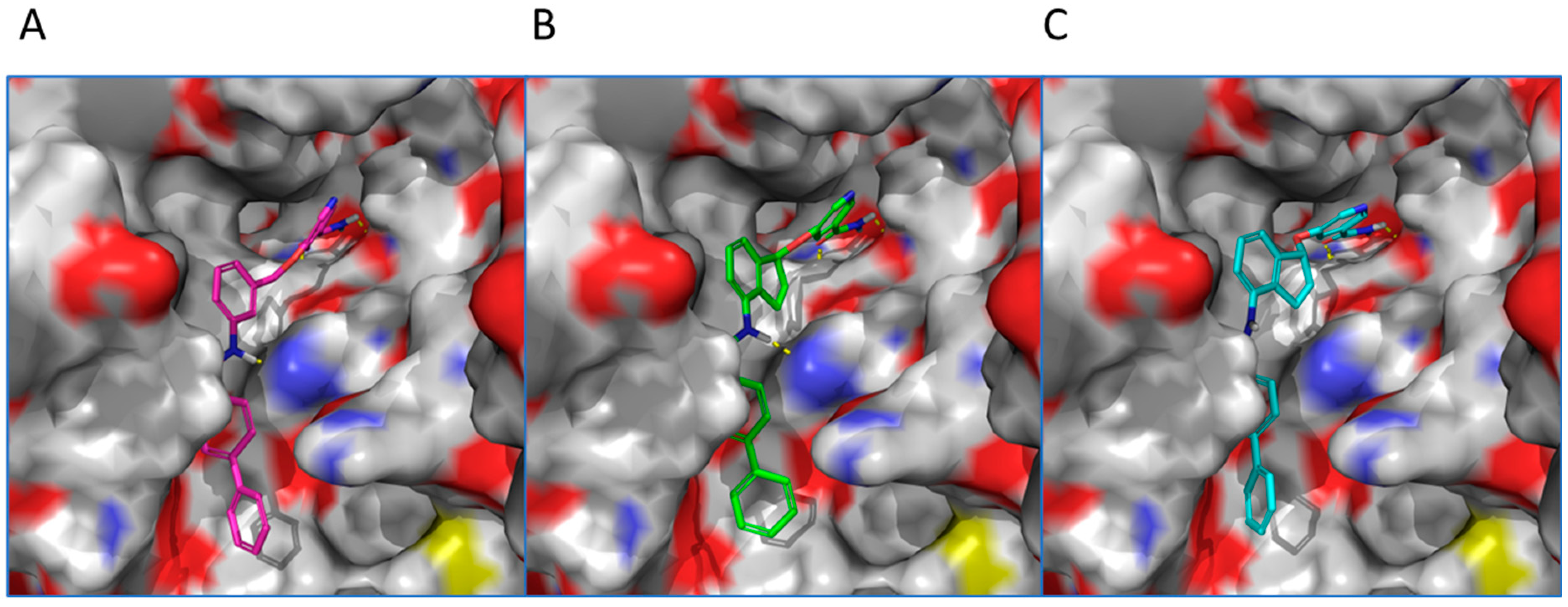
Computational Modeling. To understand the binding modes of constrained analogs versus non-constrained ones, compounds 19c and 20c were selected for computational docking. Reference compound 7d was first docked into human SIRT2 (PDB: 1j8f) [31]. A top pose showed that compound 7d’s nicotinamide moiety (ring C) was projected into the C-site of the NAD+ binding pocket and the remaining of the molecule fitted in SIRT2′s substrate binding channel (Figure 3). The primary amide of nicotinamide formed hydrogen bonds with the backbone NH of Ile 169 and the side chain carboxylate of Asp170. The aromatic rings were engaged in multiple π-π interactions: nicotinamide with Phe 96, the central ring B with Phe 119 and His 187, and ring A with Phe 235. The docking of constrained (S)-isomer 19c revealed a binding mode very similar to that of 7d (Figure 3). The primary amide participated in hydrogen bonds with Ile 169 and Asp170. Rings A, B, and C formed π-π interactions with Phe 96, Phe 119, and Phe 235, respectively. Furthermore, like 7d, the NH of compound 19c’s secondary amide is positioned close to His 187; therefore, a hydrogen bond is possible. When (R)-isomer 20c was docked into human SIRT2, its top pose generally mirrored the binding orientation of compounds 7d and 19c. However, 20c’s binding mode lacked an π-π interaction with Phe 96. In addition, the NH of compound secondary amide is oriented away from His 187 to preclude a potential hydrogen bond interaction. These observations could explain compound 20c’s inferior anti-SIRT2 activity in comparison with 19c. Taken together, while incorporation of a 2,3-five-member ring did not drastically alter the binding modes, an (S)-isomer was desired due to its augmented protein-ligand interactions, which could account for (S)-isomer’s enhanced inhibitory activity.
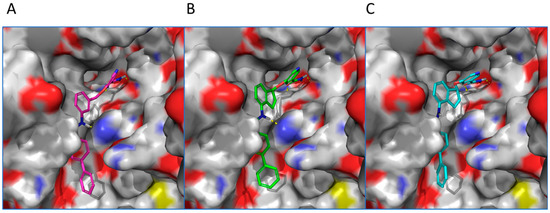
Figure 3.
Proposed binding modes of selected SIRT2 inhibitors docked into the active site of human SIRT2 (PDB: 1j8f) (surface representation). (A) Compound 7d (magenta). (B) Compound 19c (green). (C) Compound 20c (cyan).
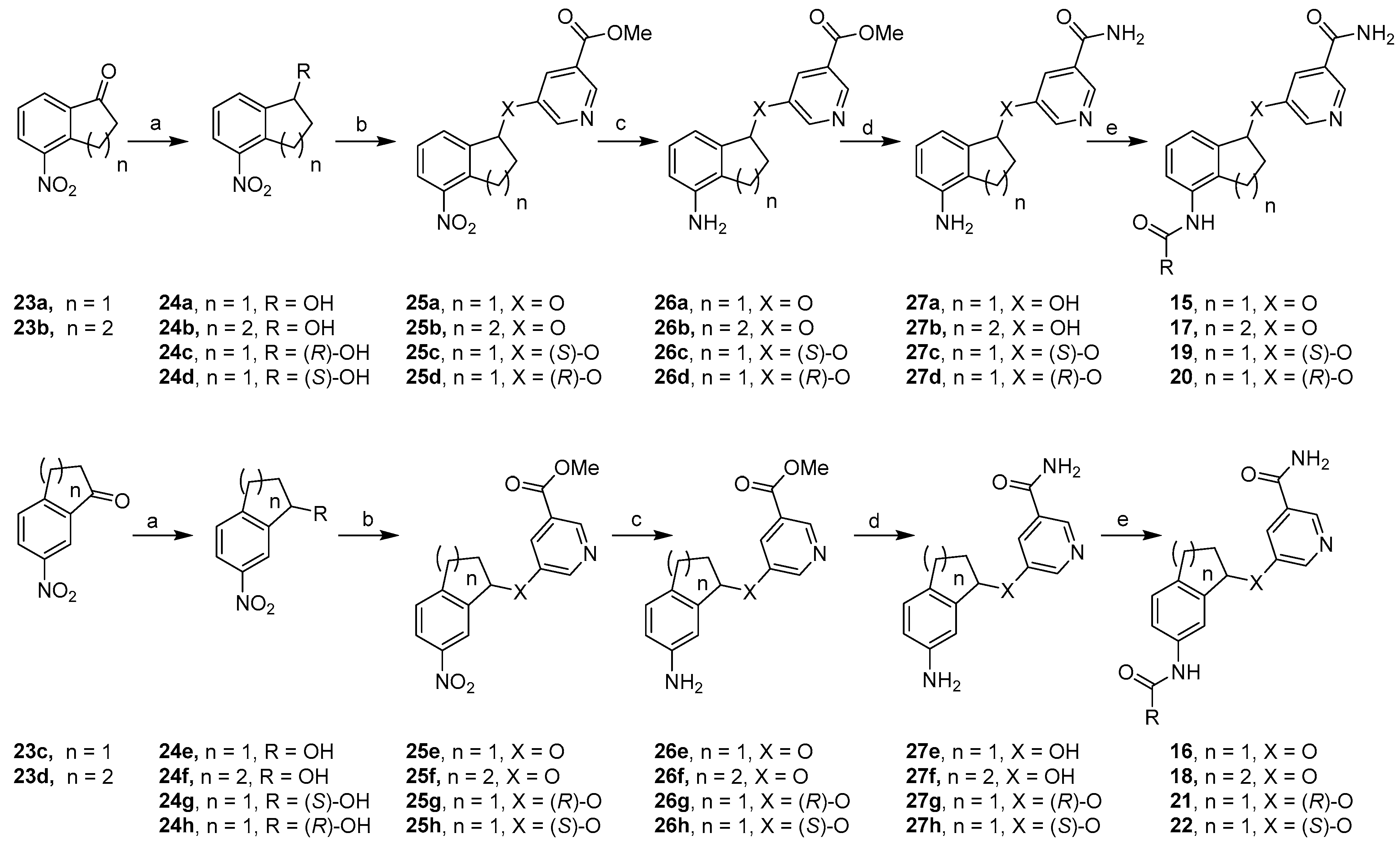
Chemical Synthesis. Scheme 1 depicted a general synthetic sequence that was used to prepare constrained analogs. The starting ketones 23a and 23c were prepared through nitration of 1-indanone according to reported procedures [32]. Nitration of tetralone under similar conditions gave ketones 23b and 23d in good yields. The synthesis of 2,3-constrained five-member ring analogs 15a–b started with ketone 23a, which was reduced to secondary alcohol 24a in excellent yields. Through Mitsunobu reaction, a methyl nicotinate moiety was introduced in good yields to give methyl ester 25a, whose nitro group was further reduced in moderate yields to form aniline 26a. The methyl ester functionality was subsequently converted into the corresponding primary amide in excellent yields, resulting in aniline 27a. Upon amide formation, final compounds 15a–b were obtained in low yields. This general synthetic sequence was also adopted to prepare 3,4-constrained five-member ring compounds 16a–b as well as six-member ring analogs 17 and 18 in similar yields. To prepare (S)-isomers 19a–c, enantioselective Corey-Bakshi-Shibata reduction was used [33]. (S)-CBS catalyst provided an (R)-hydroxyl group as shown in intermediate 24c in excellent yields, which was then subjected to the same general synthetic sequence. Among the ensuing chemical transformations was Mitsunobu reaction, which inverted the stereochemistry of the chiral center, eventually leading to (S)-isomers 19a–c in low overall yields. Analogously, 24d was obtained by using (R)-CBS catalyst, and this intermediate was transformed into (R)-isomers 20a–c. Furthermore, 3,4-constrained five-member ring analogs 21a–c and 22a–c were prepared in similar yields via the same synthetic sequence (Scheme 1).
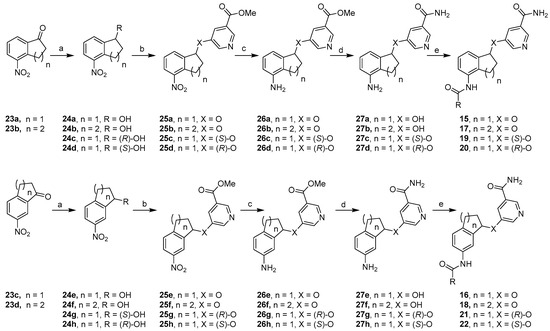
Scheme 1.
Reagents and conditions: (a) for 24a–b and 24 e–f, NaBH4, MeOH, 92–95%; for 24c and 24h, BH3·SMe2, (S)-CBS, CH2Cl2, −20 °C, 84% and 90%, respectively; for 24d and 24g, BH3·SMe2, (R)-CBS, CH2Cl2, −20 °C, both 89%; (b) methyl 5-hydroxynicotinate, DIAD, Ph3P, THF, 64–76%; (c) NaBH4, NiCl2·6H2O, MeOH, 59–94%; (d) 7 N methanolic NH3, 70 °C, 79–98%; (e) RCOCl, Et3N; or RCOOH, coupling agents, 4–42%.
3. Conclusions
Expanding our research on SIRT2 inhibitors based on the 3-aminobenzyloxy nicotinamide core structure, we have synthesized and evaluated constrained analogs by incorporating a five- or six-membered ring via different connection modes (either positions 2 and 3 or positions 3 and 4). While six-member rings are detrimental to anti-SIRT2 activity, introducing five-member rings is a viable structural modification. Examination of the stereoisomers derived from five-member rings reveals that 2,3-constrained isomers are preferred over 3,4-constrained ones. Furthermore, 2,3-constrained (S)-isomers are desired. When combined with a suitable ring A such as 1,1′-biphenyl, the resulting stereoisomer 19c possesses enhanced inhibitory activity against SIRT2 and retains excellent selectivity over SIRT1 and SIRT3. These results suggest that it is beneficial to increase the rigidity of the linker between rings B and C. Besides 2,3-constrained five-member rings examined in this study, linkers containing a rigid double or triple bond could also be envisioned. Our study has demonstrated that ring A is a key determinant of anti-SIRT2 activity and selectivity; therefore, it is important to explore the structural diversity of ring A to properly match modifications performed on other elements of SIRT2 inhibitors. Taken together, we have discovered potent and selective constrained SIRT2 inhibitors based on 3-aminobenzyloxy nicotinamide, representing a significant addition to the growing list of novel SIRT2 inhibitors. The important SAR information gathered in the current study has also shed light on key structural factors for SIRT2 inhibition and will certainly facilitate the discovery of new chemotypes as SIRT2 inhibitors.
4. Experimental
Chemical Synthesis. All commercial reagents were used as provided unless otherwise indicated. An anhydrous solvent dispensing system (J. C. Meyer) using 2 packed columns of neutral alumina was used for drying THF, Et2O, and CH2Cl2, whereas 2 packed columns of molecular sieves were used to dry DMF. Solvents were dispensed under argon. Flash chromatography was performed with Ultra Pure silica gel (Silicycle, Quebec City, QC, Canada) or with RediSep Rf silica gel columns on a Teledyne ISCO CombiFlash® Rf system using the solvents as indicated. Nuclear magnetic resonance spectra were recorded on a Varian 600 MHz with Me4Si or signals from residual solvent as the internal standard for 1H. Chemical shifts are reported in ppm, and signals are described as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), bs (broad singlet), and dd (double doublet). Values given for coupling constants are first order. High resolution mass spectra were recorded on an Agilent TOF II TOF/MS instrument (Santa Clara, CA, USA) equipped with either an ESI or APCI interface. Analysis of sample purity was performed on an Agilent 1200 Infinity series HPLC system with a Phenomenex Gemini C18 column (5 μ, 4.6 × 250 mm). HPLC conditions were as follows: solvent A = water, solvent B = MeCN or MeOH, and flow rate = 2.0 mL/min. Compounds were eluted with a gradient of from 30% to 100% MeCN/water or from 40% to 100% MeOH/water in 15 min. Purity was determined by the absorbance at 254 nm. All tested compounds have a purity of ≥95% except for compound 20a with a purity of 94% using the MeOH/water method and compound 22b with a purity of 92% using the MeCN/water method.
- 5-((4-(4-Methyl-3-nitrobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (15a). To a solution of 27a (57 mg, 0.21 mmol) and Et3N (33 mg, 0.32 mmol) in anhydrous DMF (2 mL) at rt was added 4-methyl-3-nitrobenzoyl chloride (84 mg, 0.42 mmol) dropwise. After the mixture was allowed to stir at rt for 12 h, the reaction was quenched with saturated NH4Cl (10 mL). The resulting mixture was extracted with EtOAc, and the organic phase was washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (MeOH/CH2Cl2) to afford compound 15a as a light yellow solid (37 mg, 42%). 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.67 (s, 1H), 8.57 (s, 1H), 8.49 (d, J = 2.4 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.16 (s, 1H), 7.92 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.63 (s, 1H), 7.47–7.43 (m, 1H), 7.34–7.29 (m, 2H), 6.06 (dd, J = 3.9, 5.7 Hz, 1H), 3.07–3.01 (m, 1H), 2.92–2.85 (m, 1H), 2.64–2.57 (m, 4H), 2.09–2.03 (m, 1H). HRMS (ESI+) calcd for C23H21N4O5 (M + H)+ 433.1506 found 433.1509.
The following compounds were prepared through an amide formation reaction in a manner similar to that described for the preparation of compound 15a.
- 5-((4-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (15b). White solid, 8%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.80 (d, J = 5.4 Hz, 2H), 8.67 (s, 1H), 8.49 (d, J = 3.0 Hz, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.88 (d, J = 4.8 Hz, 2H), 7.62 (s, 1H), 7.50–7.47 (m, 1H), 7.34–7.30 (m, 2H), 6.08–6.04 (m, 1H), 3.08–3.02 (m, 1H), 2.92–2.86 (m, 1H), 2.64–2.57 (m, 1H), 2.10–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1463.
- 5-((6-(4-Methyl-3-nitrobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (16a). Light yellow solid, 19%. 1H NMR (DMSO-d6, 600 MHz) δ 10.48 (s, 1H), 8.67 (s, 1H), 8.56 (d, J = 1.2 Hz, 1H), 8.50 (d, J = 2.4 Hz, 1H), 8.20 (d, J = 6.6 Hz, 1H), 8.16 (s, 1H), 7.90 (s, 1H), 7.85 (s, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.35 (d, J = 9.0 Hz, 1H), 6.05 (dd, J = 3.6, 3.6 Hz, 1H), 3.07–3.01 (m, 1H), 2.92–2.86 (m, 1H), 2.69–2.63 (m, 1H), 2.59 (s, 3H), 2.09–2.04 (m, 1H). HRMS (ESI+) calcd for C23H21N4O5 (M + H)+ 433.1506 found 433.1509.
- 5-((6-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (16b). White solid, 17%. 1H NMR (DMSO-d6, 600 MHz) δ 10.51 (s, 1H), 8.77 (d, J = 6.0 Hz, 2H), 8.67 (s, 1H), 8.49 (d, J = 2.4 Hz, 1H), 8.15 (s, 1H), 7.90 (s, 1H), 7.88–7.83 (m, 3H), 7.73 (d, J = 7.8 Hz, 1H), 7.62 (s, 1H), 7.35 (d, J = 7.8 Hz, 1H), 6.06–6.02 (m, 1H), 3.08–3.02 (m, 1H), 2.94–2.86 (m, 1H), 2.68–2.62 (m, 1H), 2.10–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1458.
- 5-((5-(4-Methyl-3-nitrobenzamido)-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (17). White solid, 16%. 1H NMR (DMSO-d6, 600 MHz) δ 10.14 (s, 1H), 8.66 (s, 1H), 8.58 (s, 1H), 8.50 (s, 1H), 8.22 (d, J = 7.8 Hz, 1H), 8.16 (s, 1H), 7.95 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.63 (s, 1H), 7.36–7.30 (m, 2H), 7.29–7.25 (m, 1H), 5.73–5.69 (m, 1H), 2.84–2.76 (m, 1H), 2.67–2.59 (m, 4H), 2.06–1.93 (m, 2H), 1.90–1.81 (m, 1H), 1.80–1.73 (m, 1H). HRMS (ESI+) calcd for C24H23N4O5 (M + H)+ 447.1663 found 447.1667.
- 5-((7-(4-Methyl-3-nitrobenzamido)-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (18). White solid, 15%. 1H NMR (DMSO-d6, 600 MHz) δ 10.41 (s, 1H), 8.66 (s, 1H), 8.55 (s, 1H), 8.51 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.15 (s, 1H), 7.94 (s, 1H), 7.75–7.00 (m, 2H), 7.66 (d, J = 7.2 Hz, 1H), 7.63 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 5.68–5.64 (m, 1H), 2.86–2.79 (m, 1H), 2.76–2.68 (m, 1H), 2.58 (s, 3H), 2.07–1.95 (m, 2H), 1.92–1.83 (m, 1H), 1.82–1.74 (m, 1H). HRMS (ESI+) calcd for C23H21ClN3O3 (M + H)+ 447.1663 found 447.1671.
- (S)-5-((4-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (19a). White solid, 13%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.80 (d, J = 5.4 Hz, 2H), 8.67 (s, 1H), 8.49 (d, J = 2.4 Hz, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.88 (d, J = 4.8 Hz, 2H), 7.63 (s, 1H), 7.51–7.47 (m, 1H), 7.35–7.30 (m, 2H), 6.06 (dd, J = 3.6, 6.0 Hz, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.65–2.56 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1460.
- (S)-5-((4-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (19b). White solid, 5%. 1H NMR (CD3OD, 600 MHz) δ 8.65 (s, 1H), 8.45 (s, 1H), 7.97 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 7.2 Hz,1H), 7.34–7.26 (m, 2H), 7.03 (d, J = 8.4 Hz, 2H), 6.04–5.99 (m, 1H), 3.88–3.78 (m, 4H), 3.35–3.25 (m, 4H), 3.16–3.08 (m, 1H), 3.00–2.92 (m, 1H), 2.72–2.62 (m, 1H), 2.24–2.16 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027 found 459.2031.
- (S)-5-((4-([1,1′-Biphenyl]-4-ylcarboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (19c). White solid, 28%. 1H NMR (DMSO-d6, 600 MHz) δ 10.10 (s, 1H), 8.67 (s, 1H), 8.50 (d, J = 1.8 Hz, 1H), 8.17 (s, 1H), 8.08 (d, J = 8.4 Hz, 2H), 7.92 (s, 1H), 7.84 (d, J = 7.8 Hz, 2H), 7.76 (d, J = 7.8 Hz, 2H), 7.63 (s, 1H), 7.55–7.47 (m, 3H), 7.43 (dd, J = 7.2, 7.2 Hz, 1H), 7.34–7.28 (m, 2H), 6.08–6.04 (m, 1H), 3.10–3.02 (m, 1H), 2.95–2.88 (m, 1H), 2.65–2.56 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1812.
- (R)-5-((4-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (20a). White solid, 10%. 1H NMR (DMSO-d6, 600 MHz) δ 10.33 (s, 1H), 8.80 (d, J = 4.2 Hz, 2H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.91 (s, 1H), 7.88 (d, J = 4.2 Hz, 2H), 7.63 (s, 1H), 7.49 (d, J = 4.8 Hz, 1H), 7.35–7.30 (m, 2H), 6.08–6.04 (m, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.65–2.56 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1465.
- (R)-5-((4-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (20b). White solid, 9%. 1H NMR (DMSO-d6, 600 MHz) δ 9.75 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.93–7.85 (m, 3H), 7.63 (s, 1H), 7.46 (d, J = 7.2 Hz, 1H), 7.29–7.23 (m, 2H), 7.03 (d, J = 9.0 Hz, 2H), 6.06–6.02 (m, 1H), 3.79–3.72 (m, 4H), 3.28–3.22 (m, 4H), 3.08–3.00 (m, 1H), 2.91–2.84 (m, 1H), 2.63–2.55 (m, 1H), 2.09–2.02 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027e found 459.2039.
- (R)-5-((4-([1,1′-Biphenyl]-4-carboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (20c). White solid, 5%. 1H NMR (CD3OD, 600 MHz) δ 8.65 (s, 1H), 8.46 (s, 1H), 8.06 (d, J = 7.8 Hz, 2H), 7.99 (s, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.52–7.46 (m, 3H), 7.42–7.39 (m, 1H), 7.38–7.32 (m, 2H), 6.06–6.02 (m, 1H), 3.20–3.13 (m, 1H), 3.03–2.96 (m, 1H), 2.74–2.66 (m, 1H), 2.26–2.19 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1819.
- (R)-5-((6-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (21a). White solid, 4%. 1H NMR (DMSO-d6, 600 MHz) δ 10.51 (s, 1H), 8.77 (d, J = 6.0 Hz, 2H), 8.67 (s, 1H), 8.50 (s, 1H), 8.16 (s, 1H), 7.90 (s, 1H), 7.88–7.82 (m, 3H), 7.73 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.35 (d, J = 8.4 Hz, 1H), 6.06–6.01 (m, 1H), 3.08–3.01 (m, 1H), 2.94–2.86 (m, 1H), 2.68–2.62 (m, 1H), 2.10–2.03 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1461.
- (R)-5-((6-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (21b). White solid, 27%. 1H NMR (DMSO-d6, 600 MHz) δ 9.98 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.93–7.83 (m, 4H), 7.72 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 7.8 Hz, 2H), 6.03–5.98 (m, 1H), 3.78–3.71 (m, 4H), 3.27–3.20 (m, 4H), 3.06–2.98 (m, 1H), 2.92–2.83 (m, 1H), 2.68–2.60 (m, 1H), 2.10–2.02 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027 found 459.2034.
- (R)-5-((6-([1,1′-Biphenyl]-4-ylcarboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (21c). White solid, 30%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.68 (s, 1H), 8.51 (s, 1H), 8.16 (s, 1H), 8.05 (d, J = 7.8 Hz, 2H), 7.91 (s, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.78–7.73 (m, 3H), 7.63 (s, 1H), 7.51 (dd, J = 7.2, 7.2 Hz, 2H), 7.45–7.40 (m, 1H), 7.34 (d, J = 7.8 Hz, 1H), 6.06–6.02 (m, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.70–2.62 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1820.
- (S)-5-((6-(Isonicotinamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (22a). White solid, 9%. 1H NMR (CD3OD, 600 MHz) δ 8.72 (d, J = 5.4 Hz, 2H), 8.65 (s, 1H), 8.45 (s, 1H), 7.99–7.96 (m, 1H), 7.87 (d, J = 5.4 Hz, 2H), 7.83 (s, 1H), 7.68–7.64 (m, 1H), 7.35 (d, J = 8.4 Hz, 1H), 6.01–5.97 (m, 1H), 3.17–3.10 (m, 1H), 3.00–2.93 (m, 1H), 2.74–2.67 (m, 1H), 2.26–2.19 (m, 1H). HRMS (ESI+) calcd for C21H19N4O3 (M + H)+ 375.1452 found 375.1465.
- (S)-5-((6-(4-Morpholinobenzamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (22b). White solid, 7%. 1H NMR (DMSO-d6, 600 MHz) δ 9.98 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.16 (s, 1H), 7.93–7.83 (m, 4H), 7.72 (d, J = 8.4 Hz, 1H), 7.63 (s, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 6.03–5.98 (m, 1H), 3.78–3.70 (m, 4H), 3.28–3.21 (m, 4H), 3.06–2.98 (m, 1H), 2.92–2.83 (m, 1H), 2.68–2.60 (m, 1H), 2.10–2.02 (m, 1H). HRMS (ESI+) calcd for C26H27N4O4 (M + H)+ 459.2027 found 459.2037.
- (S)-5-((6-([1,1′-Biphenyl]-4-ylcarboxamido)-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (22c). White solid, 25%. 1H NMR (DMSO-d6, 600 MHz) δ 10.32 (s, 1H), 8.67 (s, 1H), 8.51 (s, 1H), 8.17 (s, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.91 (s, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.78–7.73 (m, 3H), 7.63 (s, 1H), 7.51 (dd, J = 7.5, 7.5 Hz, 2H), 7.45–7.41 (m, 1H), 7.34 (d, J = 7.2 Hz, 1H), 6.06–6.02 (m, 1H), 3.08–3.01 (m, 1H), 2.93–2.85 (m, 1H), 2.70–2.62 (m, 1H), 2.11–2.03 (m, 1H). HRMS (ESI+) calcd for C28H24N3O3 (M + H)+ 450.1812 found 450.1820.
- 4-Nitro-2,3-dihydro-1H-inden-1-one (23a) and 6-Nitro-2,3-dihydro-1H-inden-1-one (23c) [32]. To a solution of 1-indanone (20 g, 151 mmol) in concentrated H2SO4 (210 mL) at 0 °C was added KNO3 (15.3 g, 151 mmol) in several portions. The reaction mixture was allowed to stir for 1 h and poured over ice (1 L). The mixture was extracted with EtOAc, and the organic phase was washed with brine and dried over Na2SO4. After filtration and removal of the solvent, the residue was purified by flash column chromatography (EtOAc/hexanes) to afford 23a as a yellow solid (3.00 g, 23%) and 23c as a yellow solid (4.00 g, 30%). 1H NMR data were consistent with those reported [32].
- 5-Nitro-3,4-dihydronaphthalen-1(2H)-one (23b) and 7-Nitro-3,4-dihydronaphthalen-1(2H)-one (23d). In a manner similar to that described for the preparation of compounds 23a and 23c, tetralone (5.53g, 37.8 mmol) was subjected to a nitration reaction to afford 23b as a yellow solid (1.48 g, 20%). 1H NMR (DMSO-d6, 600 MHz) δ 8.22 (d, J = 7.8 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 7.61 (dd, J = 7.5, 7.5 Hz, 1H), 3.09 (t, J = 6.3 Hz, 2H), 2.68 (t, J = 6.9 Hz, 2H), 2.06 (p, J = 6.3 Hz, 2H). HRMS (ESI−) calcd for C10H8NO3 (M − H)- 190.0510, found 190.0505 and 23d as a yellow solid (4.00 g, 55%). 1H NMR (DMSO-d6, 600 MHz) δ 8.54 (s, 1H), 8.36 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 3.08 (t, J = 5.7 Hz, 2H), 2.70 (t, J = 6.6 Hz, 2H), 2.09 (p, J = 6.3 Hz, 2H). HRMS (ESI−) calcd for C10H8NO3 (M − H)− 190.0510 found 190.0511.
- 4-Nitro-2,3-dihydro-1H-inden-1-ol (24a). To a solution of 23a (1.50 g, 8.47 mmol) in MeOH was added NaBH4 (641 mg, 16.9 mmol) in several portions. The reaction mixture was allowed to stir at rt for 2 h. After the reaction was quenched with 1 N HCl, the aqueous solution was extracted with EtOAc and the organic phase washed with brine and dried over Na2SO4. After filtration and removal of the solvent, the residue was purified by flash column chromatography (EtOAc/hexanes) to afford 24a as a light yellow solid (1.40 g, 92%). 1H NMR (CDCl3, 600 MHz) δ 8.13 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 7.2 Hz, 1H), 7.44 (dd, J = 7.5, 7.5 Hz, 1H), 5.33 (d, J = 6.6 Hz, 1H), 3.60–3.54 (m, 1H), 3.32–3.26 (m, 1H), 2.63–2.56 (m, 1H), 2.06–2.00 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0513.
- 5-Nitro-1,2,3,4-tetrahydronaphthalen-1-ol (24b). In a manner similar to that described for the preparation of compound 24a, 23b (1.35 g, 7.06 mmol) was reduced with NaBH4 to afford 24b as a light yellow solid (1.25 g, 92%). 1H NMR (DMSO-d6, 600 MHz) δ 8.26 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 5.55 (d, J = 6.0 Hz, 1H), 4.66–4.61 (m, 1H), 2.88–2.75 (m, 2H), 2.01–1.94 (m, 1H), 1.93–1.85 (m, 1H), 1.76–1.62 (m, 2H). HRMS (ESI−) calcd for C10H10NO3 (M − H)− 192.0666 found 192.0659.
- (R)-4-Nitro-2,3-dihydro-1H-inden-1-ol (24c). To a solution of compound 23a (1.77 g, 10.0 mmol) and (S)-CBS (139 mg, 0.50 mmol) in CH2Cl2 (30 mL) at –20 °C was slowly added BH3.SMe2 (1.00 mL, 10.0 mmol), and the mixture was stirred at this temperature for 1 h. After the reaction was quenched with saturated NH4Cl (20 mL) and stirred at rt for 3 h, the mixture was extracted with EtOAc. The organic phase was washed with water and brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/hexanes) to afford compound 24c as a white solid (1.61 g, 90%). [α]D20 = +51.4 (c = 0.43, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.12 (d, J = 8.4 Hz, 1H), 7.72 (d, J = 7.2 Hz, 1H), 7.43 (dd, J = 7.5, 7.5 Hz, 1H), 5.32 (dd, J = 6.6, 6.6 Hz, 1H), 3.60–3.52 (m, 1H), 3.32–3.24 (m, 1H), 2.63–2.56 (m, 1H), 2.06–2.00 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0512.
- (S)-4-Nitro-2,3-dihydro-1H-inden-1-ol (24d). In a manner similar to that described for the preparation of compound 24c, 23a (1.00 g, 5.65 mmol) was reduced with BH3.SMe2 (0.57 mL, 5.65 mmol) in the presence of (R)-CBS (78 mg, 0.28 mmol) to give 24d as a light yellow solid (900 mg, 89%). [α]D20 = –47.3 (c = 0.75, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.13 (d, J = 8.4 Hz, 1H), 7.73(d, J = 7.2 Hz, 1H), 7.44 (dd, J = 7.5, 7.5 Hz, 1H), 5.33 (dd, J = 6.6, 6.6 Hz, 1H), 3.60–3.52 (m, 1H), 3.32–3.24 (m, 1H), 2.63–2.56 (m, 1H), 2.06–2.00 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0513.
- 6-Nitro-2,3-dihydro-1H-inden-1-ol (24e). In a manner similar to that described for the preparation of compound 24a, 23c (2.50 g, 14.1 mmol) was reduced with NaBH4 (1.07 g, 28.2 mmol) in MeOH to give 24e as a light yellow solid (2.40 g, 95%). 1H NMR (CDCl3, 600 MHz) δ 8.26–8.22 (m, 1H), 8.14–8.10 (m, 1H), 7.39–7.35 (m, 1H), 5.34–5.28 (m, 1H), 3.16–3.09 (m, 1H), 2.94–2.86 (m, 1H), 2.63–2.56 (m, 1H), 2.09–2.01 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0512.
- 7-Nitro-1,2,3,4-tetrahydronaphthalen-1-ol (24f). In a manner similar to that described for the preparation of compound 24a, 23d (1.50 g, 7.85 mmol) was reduced with NaBH4 to give 24f as a white solid (1.40 g, 92%). 1H NMR (DMSO-d6, 600 MHz) δ 7.76 (d, J = 7.8 Hz, 2H), 7.42 (dd, J = 7.8, 7.8 Hz, 1H), 5.43 (d, J = 5.4 Hz, 1H), 4.65–4.60 (m, 1H), 2.89–2.82 (m, 1H), 2.80–2.73 (m, 1H), 1.96–1.84 (m, 2H), 1.74–1.64 (m, 2H). HRMS (ESI−) calcd for C10H10NO3 (M − H)− 192.0666 found 192.0669.
- (S)-6-Nitro-2,3-dihydro-1H-inden-1-ol (24g). In a manner similar to that described for the preparation of compound 24c, 23c (1.00 g, 5.65 mmol) was reduced with BH3.SMe2 (0.57 mL, 5.65 mmol) in the presence of (R)-CBS (78 mg, 0.28 mmol) to give 24g as a light yellow solid (900 mg, 89%). [α]D20 = +46.1 (c = 0.42, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.26–8.22 (m, 1H), 8.14 (dd, J = 8.4, 2.4 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 5.32 (dd, J = 6.6, 6.6 Hz, 1H), 3.16–3.09 (m, 1H), 2.94–2.86 (m, 1H), 2.64–2.58 (m, 1H), 2.09–2.01 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0513.
- (R)-6-Nitro-2,3-dihydro-1H-inden-1-ol (24h): In a manner similar to that described for the preparation of compound 24c, 23c (1.00 g, 5.65 mmol) was reduced with BH3.SMe2 (0.57 mL, 5.65 mmol) in the presence of (S)-CBS (78 mg, 0.28 mmol) in CH2Cl2 to give 24h (850 mg, 84%) as a light yellow solid. [α]D20 = –46.6 (c = 0.70, MeOH). 1H NMR (CDCl3, 600 MHz) δ 8.26–8.22 (m, 1H), 8.14 (dd, J = 8.4, 2.4 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 5.32 (dd, J = 6.6, 6.6 Hz, 1H), 3.16–3.09 (m, 1H), 2.94–2.86 (m, 1H), 2.64–2.58 (m, 1H), 2.09–2.01 (m, 1H). HRMS (ESI−) calcd for C9H8NO3 (M − H)− 178.0510 found 178.0514.
- Methyl 5-((4-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25a). To a solution of 24a (1.28 g, 7.16 mmol), methyl 5-hydroxynicotinate (1.32 g, 8.59 mmol), and Ph3P (2.82 g, 10.7 mmol) in anhydrous THF (45 mL) at rt was added DIAD (2.17 g, 10.7 mmol) dropwise. After the mixture was stirred for 12 h and the solvent removed, the residue was purified by flash column chromatography (EtOAc/hexanes) to afford 25a as a yellow solid (1.70 g, 76%). 1H NMR (DMSO-d6, 600 MHz) δ 8.74 (s, 1H), 8.61 (d, J = 3.0 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 7.93 (s, 1H), 7.88 (d, J = 6.6 Hz, 1H), 7.59 (dd, J = 7.8, 7.8 Hz, 1H), 6.17 (dd, J = 6.6, 4.2 Hz, 1H), 3.91 (s, 3H), 3.52–3.45 (m, 1H), 3.40–3.34 (m, 1H), 2.71–2.64 (m, 1H), 2.17–2.10 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0983.
- Methyl 5-((5-Nitro-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (25b). In a manner similar to that described for the preparation of compound 25a, 24b (468 mg, 2.42 mmol) and methyl 5-hydroxynicotinate (371 mg, 2.42 mmol) were treated with DIAD and Ph3P to afford 25b as a yellow solid (600 mg, 75%). 1H NMR (CDCl3, 600 MHz) δ 8.88 (s, 1H), 8.53 (d, J = 3.0 Hz, 1H), 7.91 (s, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.38 (dd, J = 8.1, 8.1 Hz, 1H), 5.50 (dd, J = 4.8, 4.8 Hz, 1H), 3.98 (s, 3H), 3.17–3.11 (m, 1H), 3.04–2.96 (m, 1H), 2.22–2.15 (m, 1H), 2.12–1.98 (m, 2H), 1.93–1.86 (m, 1H). HRMS (ESI+) calcd for C17H17N2O5 (M + H)+ 329.1132 found 329.1139.
- (S)-Methyl 5-((4-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25c). In a manner similar to that described for the preparation of compound 25a, 24c (500 mg, 2.79 mmol) and methyl 5-hydroxynicotinate (513 g, 3.35 mmol) were treated with DIAD and Ph3P to afford 25c as a yellow solid (580 g, 66%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.90 (s, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.82 (dd, J = 6.6, 3.6 Hz, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.66–2.60 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0984.
- (R)-Methyl 5-((4-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25d). In a manner similar to that described for the preparation of compound 25a, 24d (500 mg, 2.79 mmol) and methyl 5-hydroxynicotinate (513 g, 3.35 mmol) were treated with DIAD and Ph3P to afford 25d as a yellow solid (560 g, 64%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (s, 1H), 7.90 (s, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.82 (dd, J = 6.6, 3.6 Hz, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.66–2.60 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0985.
- Methyl 5-((6-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25e). In a manner similar to that described for the preparation of compound 25a, 25e was prepared from 24e (1.14 g, 6.34 mmol) as a yellow solid (1.30 g, 65%). 1H NMR (CDCl3, 600 MHz) δ 8.89 (s, 1H), 8.54 (d, J = 3.0 Hz, 1H), 8.28 (s, 1H), 8.23 (dd, J = 7.8, 1.8 Hz, 1H), 7.91 (s, 1H), 7.48 (s, 1H), 5.91–5.88 (m, 1H), 3.98 (s, 3H), 3.29–3.23 (m, 1H), 3.11–3.05 (m, 1H), 2.79–2.73 (m, 1H), 2.34–2.28 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0988.
- Methyl 5-((7-Nitro-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (25f). In a manner similar to that described for the preparation of compound 25a, 24f (505 mg, 3.30 mmol) and methyl 5-hydroxynicotinate (638 mg, 3.30 mmol) were treated with DIAD and Ph3P to afford 25f as a yellow solid (810 mg, 75%). 1H NMR (CDCl3, 600 MHz) δ 8.89 (s, 1H), 8.55 (d, J = 2.4 Hz, 1H), 8.24 (s, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.92 (s, 1H), 7.35 (d, J = 8.4, 1H), 5.51 (dd, J = 4.8, 4.8 Hz, 1H), 3.98 (s, 3H), 3.05–2.99 (m, 1H), 2.91–2.85 (m, 1H), 2.17–2.10 (m, 2H), 2.08–2.02 (m, 1H), 1.93–1.87 (m, 1H). HRMS (ESI+) calcd for C17H17N2O5 (M + H)+ 329.1132 found 329.1134.
- (R)-Methyl 5-((6-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25g). In a manner similar to that described for the preparation of compound 25a, 24g (550 mg, 3.07 mmol) and methyl 5-hydroxynicotinate (554 g, 3.68 mmol) were treated with DIAD and Ph3P to afford 25g as a yellow solid (629 g, 64%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 3.0 Hz, 1H), 7.89 (dd, J = 2.4, 2.4 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (dd, J = 8.1, 1.8 Hz, 1H), 5.76 (dd, J = 6.0, 4.2 Hz, 1H), 3.97 (s, 3H), 3.08–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.22–2.14 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0981.
- (S)-Methyl 5-((6-Nitro-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (25h). In a manner similar to that described for the preparation of compound 25a, 24h (700 mg, 3.91 mmol) and methyl 5-hydroxynicotinate (718 g, 4.69 mmol) were treated with DIAD and Ph3P to afford 25h as a yellow solid (800 g, 65%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.89 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.88–5.74 (m, 1H), 3.97 (s, 3H), 3.08–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.22–2.14 (m, 1H). HRMS (ESI+) calcd for C16H15N2O5 (M + H)+ 315.0975 found 315.0986.
- Methyl 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26a). To a solution of compound 25a (500 mg, 1.60 mmol) and NiCl2·6H2O (760 mg, 4.48 mmol) in MeOH (800 mL) was slowly added NaBH4 (250 mg, 6.40 mmol), and the mixture was stirred at rt for 2 h. The reaction was quenched with saturated NH4Cl (50 mL) and extracted with EtOAc. The organic phase was washed with water and brine, dried over anhydrous K2CO3, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/hexanes) to afford compound 26a as a white solid (290 mg, 64%). 1H NMR (DMSO-d6, 600 MHz) δ 8.70 (s, 1H), 8.56 (d, J = 3.0 Hz, 1H), 7.88–7.86 (m, 1H), 6.93 (dd, J = 7.8, 7.8 Hz, 1H), 6.58 (d, J = 7.2 Hz, 1H), 6.56 (d, J = 8.4, 1H), 5.95 (dd, J = 6.6, 3.3 Hz, 1H), 5.02 (s, 2H), 3.90 (s, 3H), 2.82–2.77 (m, 1H), 2.69–2.64 (m, 1H), 2.57–2.51 (m, 1H), 2.05–2.00 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1241.
- Methyl 5-((5-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (26b). In a manner similar to that described for the preparation of compound 26a, 25b (737 mg, 2.24 mmol) was reduced to afford 26b as a light yellow solid (620 mg, 94%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.53 (s, 1H), 7.91 (s, 1H), 7.06 (dd, J = 7.8, 7.8 Hz, 1H), 6.78 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 8.4 Hz, 1H), 5.45–5.40 (m, 1H), 3.96 (s, 3H), 3.68 (bs, 2H), 2.64–2.57 (m, 1H), 2.49–2.41 (m, 1H), 2.19–2.06 (m, 2H), 2.03–1.95 (m, 1H), 1.94–1.86 (m, 1H). HRMS (ESI+) calcd for C17H19N2O3 (M + H)+ 299.1390 found 299.1400.
- (S)-Methyl 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26c). In a manner similar to that described for the preparation of compound 26a, 25c (250 mg, 0.80 mmol) was reduced to afford 26c as a light yellow solid (180 mg, 80%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.90 (dd, J = 1.8, 1.8 Hz, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.8 Hz, 1H), 5.83 (dd, J = 6.0, 3.6 Hz, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.67–2.60 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1237.
- (R)-Methyl 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26d). In a manner similar to that described for the preparation of compound 26a, 25d (250 mg, 0.80 mmol) was reduced to afford 26d as a light yellow solid (160 mg, 70%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (s, 1H), 7.90 (s, 1H), 7.11 (dd, J = 7.8, 7.8 Hz, 1H), 6.85 (d, J = 7.2 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 5.84–5.80 (m, 1H), 3.96 (s, 3H), 3.00–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.67–2.59 (m, 1H), 2.29–2.22 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1241.
- Methyl 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26e). In a manner similar to that described for the preparation of compound 26a, 25e (500 mg, 1.60 mmol) was reduced to afford 26e as a light yellow solid (270 mg, 59%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (d, J = 1.8 Hz, 1H), 8.53 (d, J = 3.0 Hz, 1H), 7.89 (dd, J = 3.0, 1.8 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (d, J = 2.4 Hz, 1H), 6.69 (dd, J = 8.4, 2.4 Hz, 1H), 5.76 (dd, J = 6.6, 4.2 Hz, 1H), 3.97 (s, 3H), 3.65 (bs, 2H), 3.08–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.02–1.95 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1243.
- Methyl 5-((7-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinate (26f). In a manner similar to that described for the preparation of compound 26a, 25f (682 mg, 2.08 mmol) was reduced to afford 26f as a white solid (580 mg, 94%). 1H NMR (CDCl3, 600 MHz) δ 8.83 (s, 1H), 8.53 (s, 1H), 7.90 (s, 1H), 6.96 (d, J = 8.4 Hz, 1H), 6.66–6.61 (m, 2H), 5.38–5.34 (m, 1H), 3.96 (s, 3H), 3.60 (bs, 2H), 2.81–2.75 (m, 1H), 2.71–2.63 (m, 1H), 2.12–2.05 (m, 1H), 2.04–1.92 (m, 2H), 1.81–1.74 (m, 1H). HRMS (ESI+) calcd for C17H19N2O3 (M + H)+ 299.1390 found 299.1390.
- (R)-Methyl 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26g). In a manner similar to that described for the preparation of compound 26a, 25g (314 mg, 1.00 mmol) was reduced to afford 26g as a yellow solid (200 mg, 70%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (s, 1H), 8.52 (d, J = 2.4 Hz, 1H), 7.89 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.77–5.74 (m, 1H), 3.96 (s, 3H), 3.08–3.01 (m, 1H), 2.88–2.81 (m, 1H), 2.64–2.56 (m, 1H), 2.22–2.14 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found285.1239.
- (S)-Methyl 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinate (26h). In a manner similar to that described for the preparation of compound 25a, 25h (314 mg, 1.00 mmol) was reduced to afford 26h as a yellow solid (220 mg, 77%). 1H NMR (CDCl3, 600 MHz) δ 8.84 (d, J = 1.2 Hz, 1H), 8.52 (d, J = 3.0 Hz, 1H), 7.89 (dd, J = 2.4, 2.4 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (d, J = 1.8 Hz, 1H), 6.69 (dd, J = 7.8, 1.8 Hz, 1H), 5.76 (dd, J = 6.0, 3.6 Hz, 1H), 3.97 (s, 3H), 3.07–3.02 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.21–2.14 (m, 1H). HRMS (ESI+) calcd for C16H17N2O3 (M + H)+ 285.1234 found 285.1239.
- 5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27a). A solution of methyl ester 26a (280 mg, 0.98 mmol) in NH3/MeOH (~7 N, 10 mL) in a seal tube was heated at 70 °C for 24 h. After the solvent was evaporated in vacuo, the residue was dissolved in EtOAc (150 mL) and the organic layer was washed with H2O (150 mL) and then with brine (100 mL). After the organic layer was dried over Na2SO4 and filtered, the filtrate was concentrated and the residue was purified by flash column chromatography (5% MeOH/CH2Cl2) to afford compound 27a as a light yellow solid (260 mg, 98%). 1H NMR (DMSO-d6, 600 MHz) δ 8.64 (s, 1H), 8.43 (d, J = 1.8 Hz, 1H), 8.14 (s, 1H), 7.86 (s, 1H), 7.61 (s, 1H), 6.93 (dd, J = 7.5, 7.5 Hz, 1H), 6.58 (d, J = 7.8, 1H), 6.50 (d, J = 7.2 Hz, 1H), 5.90 (dd, J = 6.6, 3.6 Hz, 1H), 5.02 (s, 2H), 2.82–2.77 (m, 1H), 2.68–2.63 (m, 1H), 2.58–2.52 (m, 1H), 2.04–2.00 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1247.
- 5-((5-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (27b). In a manner similar to that described for the preparation of compound 27a, aminolysis of methyl ester 26b (400 mg, 1.34 mmol) in the presence of CaCl2 (149 mg, 1.34 mmol) afforded 27b as a white solid (320 mg, 84%). 1H NMR (CD3OD, 600 MHz) δ 8.62 (s, 1H), 8.40 (s, 1H), 7.95 (s, 1H), 6.95 (dd, J = 7.5, 7.5 Hz, 1H), 6.71 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.54–5.48 (m, 1H), 2.67–2.60 (m, 1H), 2.50–2.42 (m, 1H), 2.16–2.01 (m, 2H), 2.00–1.95 (m, 1H), 1.92–1.80 (m, 1H). HRMS (ESI+) calcd for C16H18N3O2 (M + H)+ 284.1394 found 284.1397.
- (S)-5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27c). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26c (180 mg, 0.63 mmol) afforded 27c as a white solid (160 mg, 94%). 1H NMR (CDCl3, 600 MHz) δ 8.57 (d, J = 1.8 Hz, 1H), 8.50 (d, J = 3.0 Hz, 1H), 7.81 (dd, J = 2.4, 2.4 Hz, 1H), 7.11 (dd, J = 7.8 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.68 (d, J = 7.2 Hz, 1H), 5.84 (dd, J = 6.6, 3.6 Hz, 1H), 2.99–2.92 (m, 1H), 2.80–2.73 (m, 1H), 2.68–2.61 (m, 1H), 2.28–2.22 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1249.
- (R)-5-((4-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27d). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26d (160 mg, 0.56 mmol) afforded 27d as a white solid (140 mg, 93%). 1H NMR (CDCl3, 600 MHz) δ 8.59 (d, J = 1.8 Hz, 1H), 8.48 (d, J = 3.0 Hz, 1H), 7.81 (dd, J = 2.4, 2.4 Hz, 1H), 7.11 (dd, J = 7.8 Hz, 1H), 6.85 (d, J = 7.8 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 5.83 (dd, J = 7.2, 3.6 Hz, 1H), 2.98–2.91 (m, 1H), 2.78–2.72 (m, 1H), 2.66–2.60 (m, 1H), 2.27–2.21 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1235.
- 5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27e). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26e (250 mg, 0.88 mmol) afforded 27e as a light yellow solid (230 mg, 97%). 1H NMR (DMSO-d6, 600 MHz) δ 8.65 (s, 1H), 8.45 (d, J = 2.4 Hz, 1H), 8.14 (s, 1H), 7.87 (s, 1H), 7.61 (s, 1H), 6.97 (d, J = 7.8 Hz, 1H), 6.59 (s, 1H), 6.56 (d, J = 7.2, 1H), 5.86 (dd, J = 5.1, 5.1 Hz, 1H), 4.96 (s, 2H), 2.90–2.85 (m, 1H), 2.75–2.69 (m, 1H), 2.57–2.50 (m, 1H), 2.00–1.95 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1237.
- 5-((7-Amino-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)nicotinamide (27f). In a manner similar to that described for the preparation of compound 27a, aminolysis of methyl ester 26f (500 mg, 1.73 mmol) in the presence of CaCl2 (192 mg, 1.73 mmol) afforded 27f as a white solid (410 mg, 85%). 1H NMR (DMSO-d6, 600 MHz) δ 8.64 (d, J = 1.8 Hz, 1H), 8.45 (d, J = 3.0 Hz, 1H), 8.14 (s, 1H), 7.89 (dd, J = 1.8, 1.8 Hz, 1H), 7.61 (s, 1H), 6.82 (d, J = 8.4 Hz, 1H), 6.52–6.47 (m, 2H), 5.49 (t, J = 4.8 Hz, 1H), 4.86 (s, 2H), 2.68–2.62 (m, 1H), 2.60–2.53 (m, 1H), 1.96–1.91 (m, 2H), 1.86–1.79 (m, 1H), 1.73–1.66 (m, 1H). HRMS (ESI+) calcd for C16H18N3O2 (M + H)+ 284.1394 found 284.1399.
- (R)-5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27g). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26g (220 mg, 0.77 mmol) afforded 27g as a light yellow solid (180 mg, 86%). 1H NMR (CDCl3, 600 MHz) δ 8.57 (s, 1H), 8.50 (d, J = 3.0 Hz, 1H), 7.81 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.77 (dd, J = 4.8, 4.8 Hz, 1H), 3.08–3.01 (m, 1H), 2.88–2.82 (m, 1H), 2.63–2.56 (m, 1H), 2.20–2.14 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1240.
- (S)-5-((6-Amino-2,3-dihydro-1H-inden-1-yl)oxy)nicotinamide (27h). In a manner similar to that described for the preparation of compound 27a, aminolysis of 26h (200 mg, 0.70 mmol) afforded 27h as a light yellow solid (150 mg, 79%). 1H NMR (CDCl3, 600 MHz) δ 8.58 (d, J = 1.2 Hz, 1H), 8.49 (d, J = 3.0 Hz, 1H), 7.80 (dd, J = 2.4, 2.4 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 6.69 (dd, J = 8.4, 2.4 Hz, 1H), 5.77 (dd, J = 6.0, 4.2 Hz, 1H), 3.07–3.00 (m, 1H), 2.87–2.81 (m, 1H), 2.62–2.55 (m, 1H), 2.20–2.13 (m, 1H). HRMS (ESI+) calcd for C15H16N3O2 (M + H)+ 270.1237 found 270.1249.
Initial SIRT1-3 Biochemical Assays. Initial biochemical assays against human SIRT1-3 were performed at Reaction Biology Corporation (RBC) (Malvern, PA, USA, http://www.reactionbiology.com) as reported previously [29]. Briefly, the concentrations of SIRT1, SIRT2, and SIRT3 were 91, 233, and 917 nM, respectively. A fluorogenic 7-amino-4-methylcoumarin (AMC)-labeled peptide Ac-Arg-His-Lys-Lys(Ac)-AMC was used as the peptide substrate for SIRT1-3. The testing was performed in a two-step fashion. First, 50 μM AMC-labeled substrate was incubated for 2 h at 30 °C to produce the deacetylated substrate. Second, the deacetylated substrate was digested by a mixture of developer to release AMC that was detected at 360/460 Ex/Em. Inhibitors were tested in a 10-dose mode with a 3-fold serial dilution starting at 200 μM (200 μM–10.2 nM). The IC50 values were determined from the sigmoidal dose-response curves using GraphPad Prism.
In-house SIRT1-3 Biochemical Assays. These assays were performed as reported previously [30]. Briefly, SIRT1, SIRT2, and SIRT3 were used at 0.25, 0.10, and 1.5 µM, respectively. NAD and the peptide substrate ((Ac)RHKK(Ac)-AMC) were used at the KM values determined for each enzyme to accurately assess an inhibitor’s selectivity against SIRT1-3. The reaction was allowed to proceed at 37 °C for the desired time and then quenched by the addition of a developing buffer. After the mixture was developed for 20 min at 37 °C, the fluorescence intensity was measured using an excitation at 355 nm and an emission at 460 nm. The data were analyzed using either the Morrison equation (SIRT2) or the four-parameter dose response equation (SIRT1 and 3) in GraphPad Prism.
Molecular Modeling. The modeling study was carried out as described previously [29]. The X-ray crystal structure of human SIRT2 (PDB: 1j8f) [31] was used and the energy minimization was performed using OPLS 2005 forcefield. SIRT2 inhibitors were generated by LigPrep and then docked into the Glide grid (20 × 20 × 20 Å) encompassing the active site of SIRT2. Docking was carried out in both standard SP and XP modes. Structural visualization and representation were performed with PyMOL.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28227655/s1. 1H NMR spectra of the final compounds and synthetic intermediates; HPLC chromatograms (two solvent conditions) of the final compounds; and HRMS spectra of the final compounds.
Author Contributions
T.A. and D.J.W. performed the experiments and organized data. L.C. prepared the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Center for Drug Design at the University of Minnesota.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
The University of Minnesota Supercomputing Institute provided all the necessary computational resources.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
SIRT2, Sirtuin 2; NAD+, nicotinamide adenine dinucleotide; SAR, structure-activity relationship.
References
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Interphase nucleo-cytoplasmic shuttling and localization of SIRT2 during mitosis. PLoS ONE 2007, 2, e784. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, S.H.; Imbesi, M.; Nathan, W.J.; Zou, X.; Zhu, Y.; Jiang, H.; Parisiadou, L.; Gius, D. Loss of NAD-Dependent Protein Deacetylase Sirtuin-2 Alters Mitochondrial Protein Acetylation and Dysregulates Mitophagy. Antioxid. Redox Signal. 2017, 26, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduino, D.M.; Silva, D.F.; Viana, S.D.; Pereira, F.C.; Cardoso, S.M. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 1440–1462. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target Ther. 2022, 7, 402. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef] [PubMed]
- Pandithage, R.; Lilischkis, R.; Harting, K.; Wolf, A.; Jedamzik, B.; Luscher-Firzlaff, J.; Vervoorts, J.; Lasonder, E.; Kremmer, E.; Knoll, B.; et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 2008, 180, 915–929. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Sola-Sevilla, N.; Puerta, E. SIRT2 as a potential new therapeutic target for Alzheimer’s disease. Neural Regen. Res. 2024, 19, 124–131. [Google Scholar] [CrossRef]
- Roshdy, E.; Mustafa, M.; Shaltout, A.E.; Radwan, M.O.; Ibrahim, M.A.A.; Soliman, M.E.; Fujita, M.; Otsuka, M.; Ali, T.F.S. Selective SIRT2 inhibitors as promising anticancer therapeutics: An update from 2016 to 2020. Eur. J. Med. Chem. 2021, 224, 113709. [Google Scholar] [CrossRef] [PubMed]
- Piracha, Z.Z.; Kwon, H.; Saeed, U.; Kim, J.; Jung, J.; Chwae, Y.J.; Park, S.; Shin, H.J.; Kim, K. Sirtuin 2 Isoform 1 Enhances Hepatitis B Virus RNA Transcription and DNA Synthesis through the AKT/GSK-3beta/beta-Catenin Signaling Pathway. J. Virol. 2018, 92, e00955-18. [Google Scholar] [CrossRef] [PubMed]
- Duran-Castells, C.; Llano, A.; Kawana-Tachikawa, A.; Prats, A.; Martinez-Zalacain, I.; Kobayashi-Ishihara, M.; Oriol-Tordera, B.; Pena, R.; Galvez, C.; Silva-Arrieta, S.; et al. Sirtuin-2, NAD-Dependent Deacetylase, Is a New Potential Therapeutic Target for HIV-1 Infection and HIV-Related Neurological Dysfunction. J. Virol. 2023, 97, e0165522. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.L.; Remiszewski, S.; Todd, M.J.; Kulp, J.L., 3rd; Tang, L.; Welsh, A.V.; Barry, A.P.; De, C.; Reiley, W.W.; Wahl, A.; et al. An allosteric inhibitor of sirtuin 2 deacetylase activity exhibits broad-spectrum antiviral activity. J. Clin. Investig. 2023, 133, e158978. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, W.; Zhang, J.; Li, L.; Wan, Y.; Tang, X.; Chen, X.; Liu, S.; Yao, X. Tenovin-1 inhibited dengue virus replication through SIRT2. Eur. J. Pharmacol. 2021, 907, 174264. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, T.; Zhu, Q.; Xu, Y.; Zha, X. Recent advances in inhibitors of sirtuin1/2: An update and perspective. Future Med. Chem. 2018, 10, 907–934. [Google Scholar] [CrossRef]
- Yang, W.; Chen, W.; Su, H.; Li, R.; Song, C.; Wang, Z.; Yang, L. Recent advances in the development of histone deacylase SIRT2 inhibitors. RSC Adv. 2020, 10, 37382–37390. [Google Scholar] [CrossRef]
- Penteado, A.B.; Hassanie, H.; Gomes, R.A.; Silva Emery, F.D.; Goulart Trossini, G.H. Human sirtuin 2 inhibitors, their mechanisms and binding modes. Future Med. Chem. 2023, 15, 291–311. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Olah, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Olah, J.; Gerhardt, S.; Ovadi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure-Activity Relationship Study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef]
- Suzuki, T.; Khan, M.N.; Sawada, H.; Imai, E.; Itoh, Y.; Yamatsuta, K.; Tokuda, N.; Takeuchi, J.; Seko, T.; Nakagawa, H.; et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [Google Scholar] [CrossRef] [PubMed]
- Sundriyal, S.; Moniot, S.; Mahmud, Z.; Yao, S.; Di Fruscia, P.; Reynolds, C.R.; Dexter, D.T.; Sternberg, M.J.; Lam, E.W.; Steegborn, C.; et al. Thienopyrimidinone Based Sirtuin-2 (SIRT2)-Selective Inhibitors Bind in the Ligand Induced Selectivity Pocket. J. Med. Chem. 2017, 60, 1928–1945. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Price, I.R.; Bai, J.J.; Lin, H. A Glycoconjugated SIRT2 Inhibitor with Aqueous Solubility Allows Structure-Based Design of SIRT2 Inhibitors. ACS Chem. Biol. 2019, 14, 1802–1810. [Google Scholar] [CrossRef] [PubMed]
- Mellini, P.; Itoh, Y.; Tsumoto, H.; Li, Y.; Suzuki, M.; Tokuda, N.; Kakizawa, T.; Miura, Y.; Takeuchi, J.; Lahtela-Kakkonen, M.; et al. Potent mechanism-based sirtuin-2-selective inhibition by an in situ-generated occupant of the substrate-binding site, “selectivity pocket” and NAD(+)-binding site. Chem. Sci. 2017, 8, 6400–6408. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.S.; Hong, J.Y.; Cao, J.; Lu, X.; Price, I.R.; Zhao, Q.; Kosciuk, T.; Yang, M.; Bai, J.J.; Lin, H. Novel Lysine-Based Thioureas as Mechanism-Based Inhibitors of Sirtuin 2 (SIRT2) with Anticancer Activity in a Colorectal Cancer Murine Model. J. Med. Chem. 2019, 62, 4131–4141. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, Y.; Liang, B.; Yin, Y.; Yao, J.; Wang, Q.; Liu, Y.; Neamati, N. The application of PROTAC in HDAC. Eur. J. Med. Chem. 2023, 260, 115746. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Olah, J.; Ovadi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef]
- Hong, J.Y.; Jing, H.; Price, I.R.; Cao, J.; Bai, J.J.; Lin, H. Simultaneous Inhibition of SIRT2 Deacetylase and Defatty-Acylase Activities via a PROTAC Strategy. ACS Med. Chem. Lett. 2020, 11, 2305–2311. [Google Scholar] [CrossRef]
- Cui, H.; Kamal, Z.; Ai, T.; Xu, Y.; More, S.S.; Wilson, D.J.; Chen, L. Discovery of potent and selective sirtuin 2 (SIRT2) inhibitors using a fragment-based approach. J. Med. Chem. 2014, 57, 8340–8357. [Google Scholar] [CrossRef]
- Ai, T.; Wilson, D.J.; More, S.S.; Xie, J.; Chen, L. 5-((3-Amidobenzyl)oxy)nicotinamides as Sirtuin 2 Inhibitors. J. Med. Chem. 2016, 59, 2928–2941. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Pavletich, N.P. Structure of the histone deacetylase SIRT2. Nat. Struct. Biol. 2001, 8, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.L.; Cochran, F.R.; Kelley, J.L.; McLean, E.W.; Selph, J.L.; Rigdon, G.C.; Orr, G.F.; Davis, R.G.; Cooper, B.R.; Styles, V.L.; et al. Indanylidenes. 1. Design and synthesis of (E)-2-(4,6-difluoro-1-indanylidene)acetamide, a potent, centrally acting muscle relaxant with antiinflammatory and analgesic activity. J. Med. Chem. 2003, 46, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Helal, C.J. Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem. Int. Ed. Engl. 1998, 37, 1986–2012. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).