
Compact Combustion Mechanisms of Typical n-Alkanes Developed by the Minimized Reaction Network Method

Abstract
:
1. Introduction
2. Results and Discussion of the Developed Mechanisms for n-Alkanes
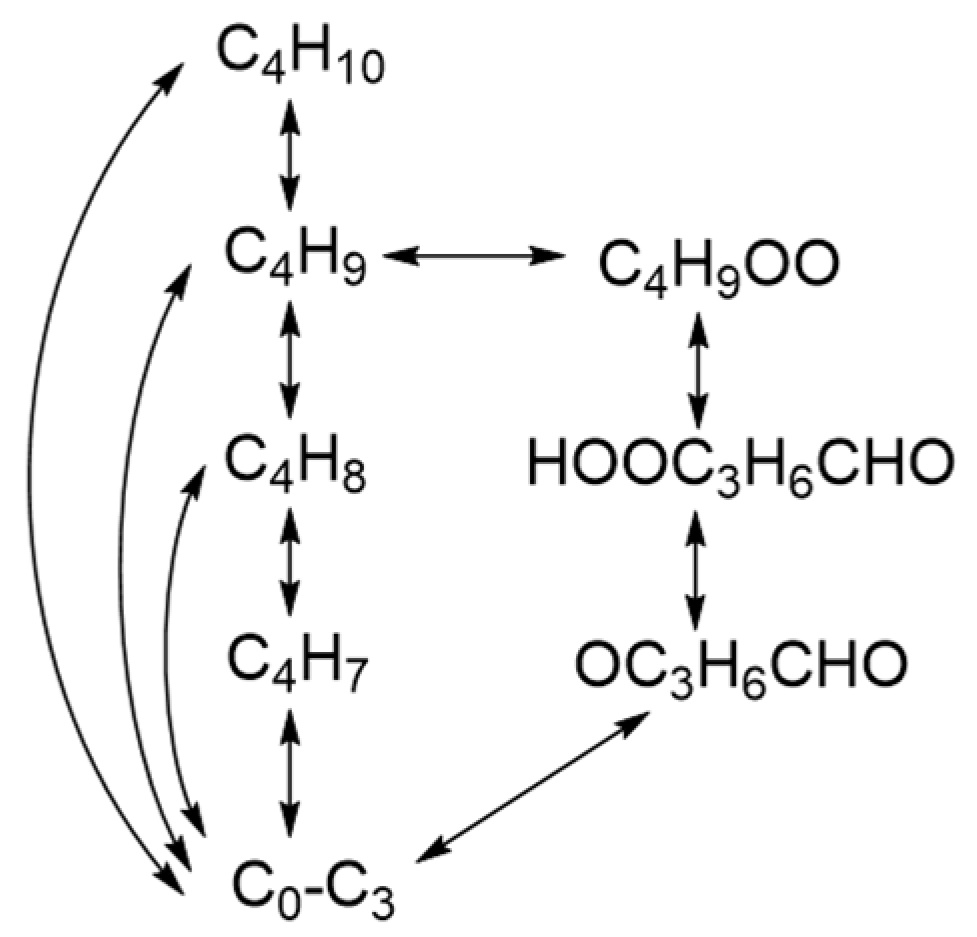
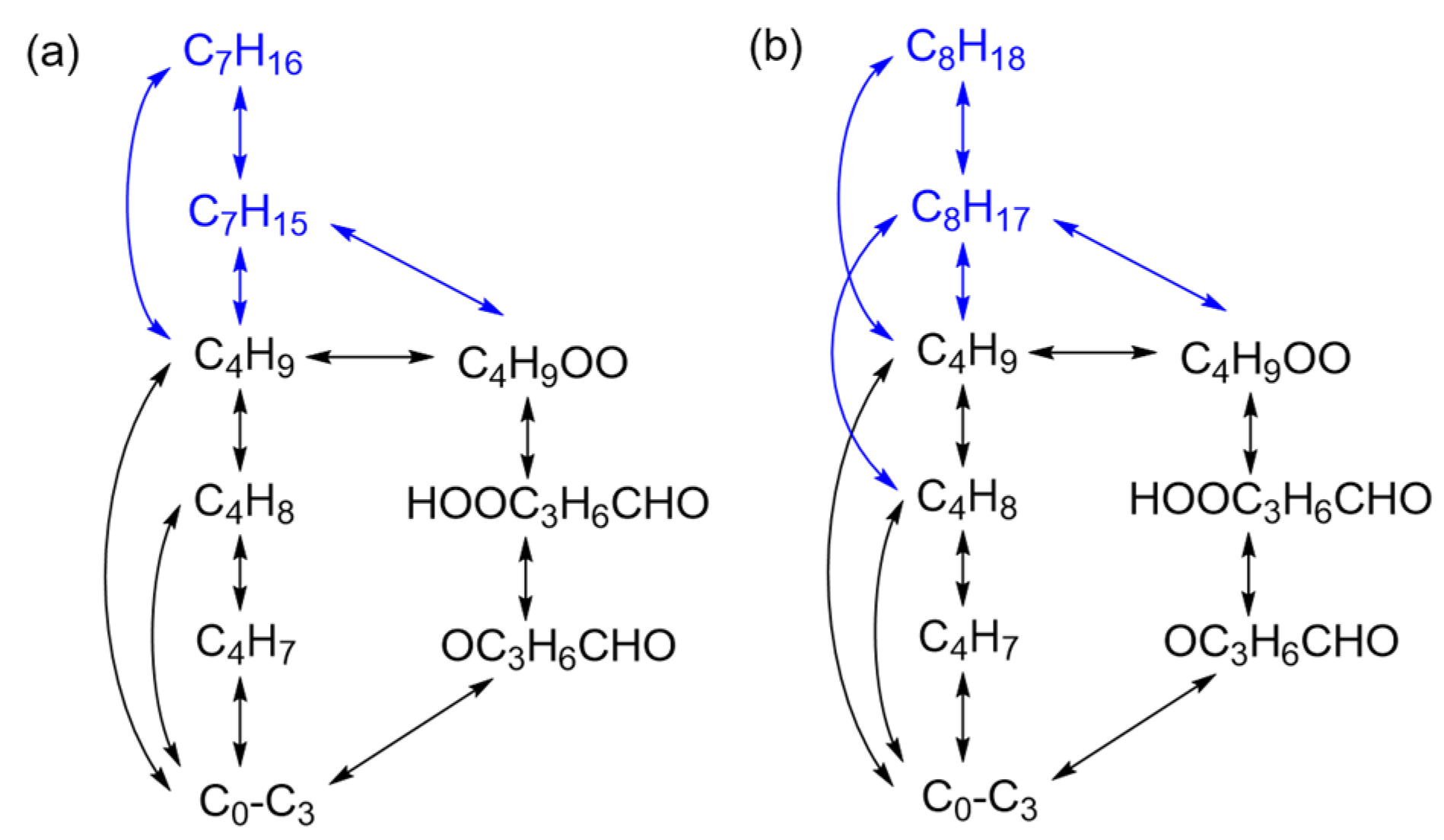
2.1. Reaction Mechanisms for Typical n-Alkanes
2.2. Mechanism Validations
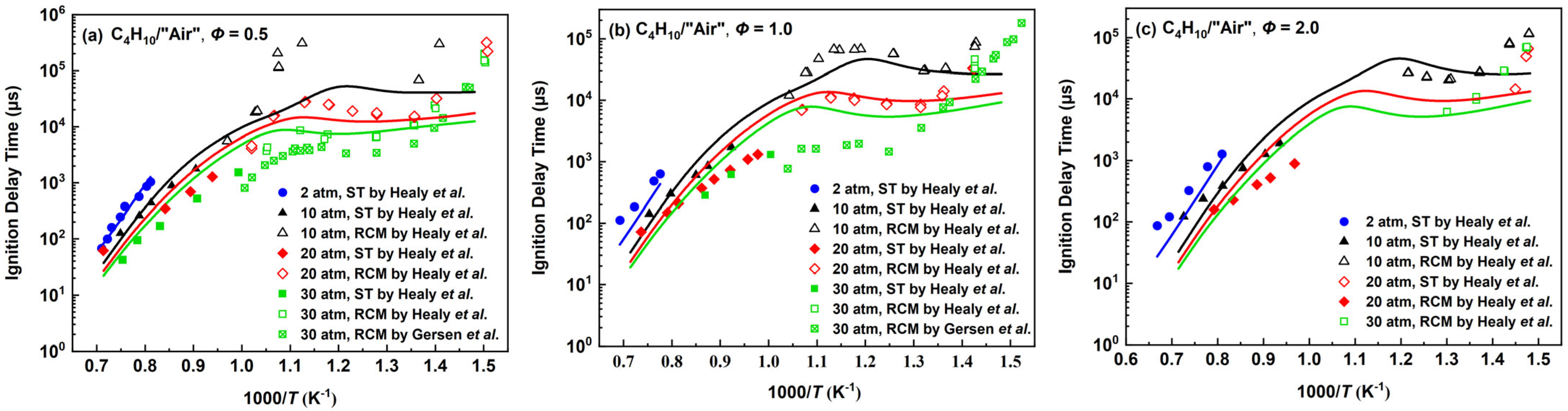
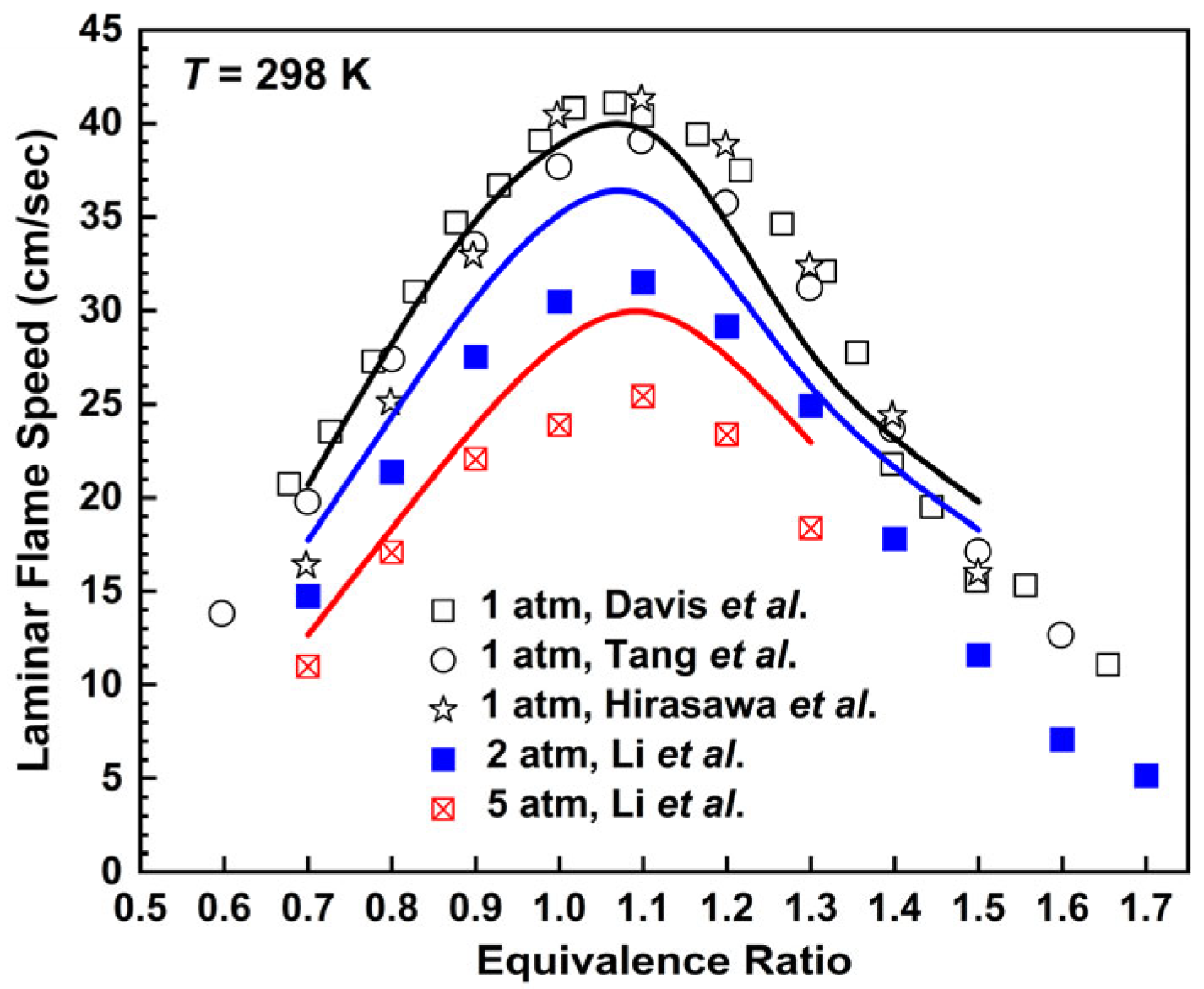
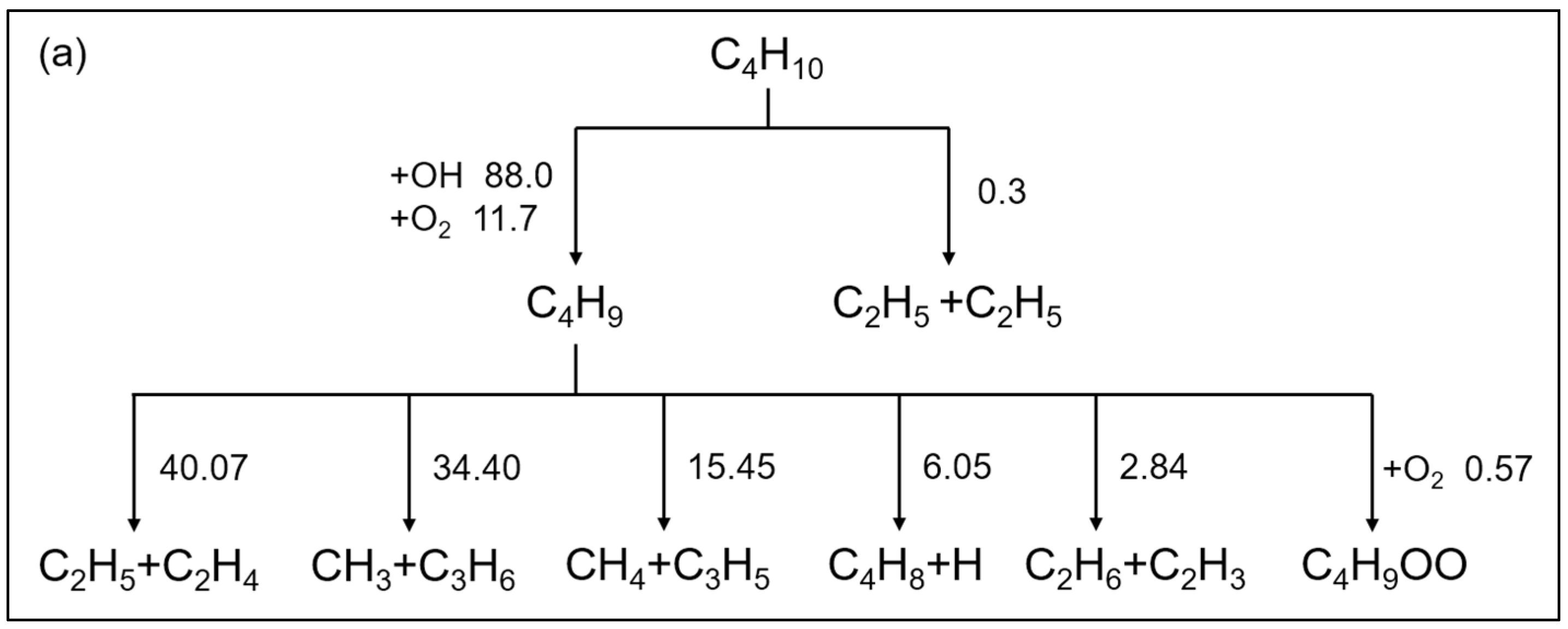
2.2.1. n-Butane
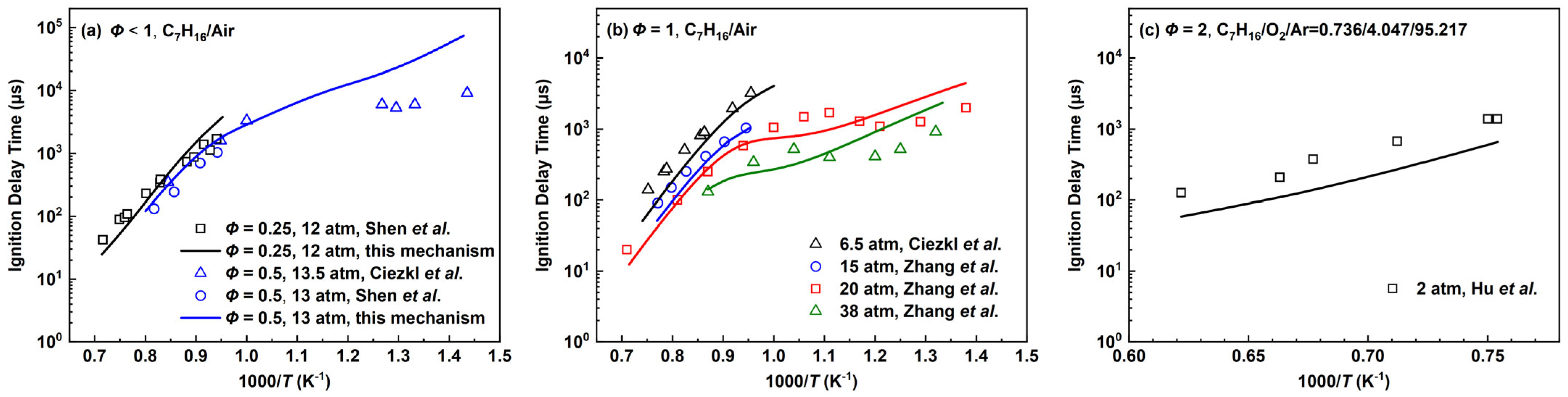
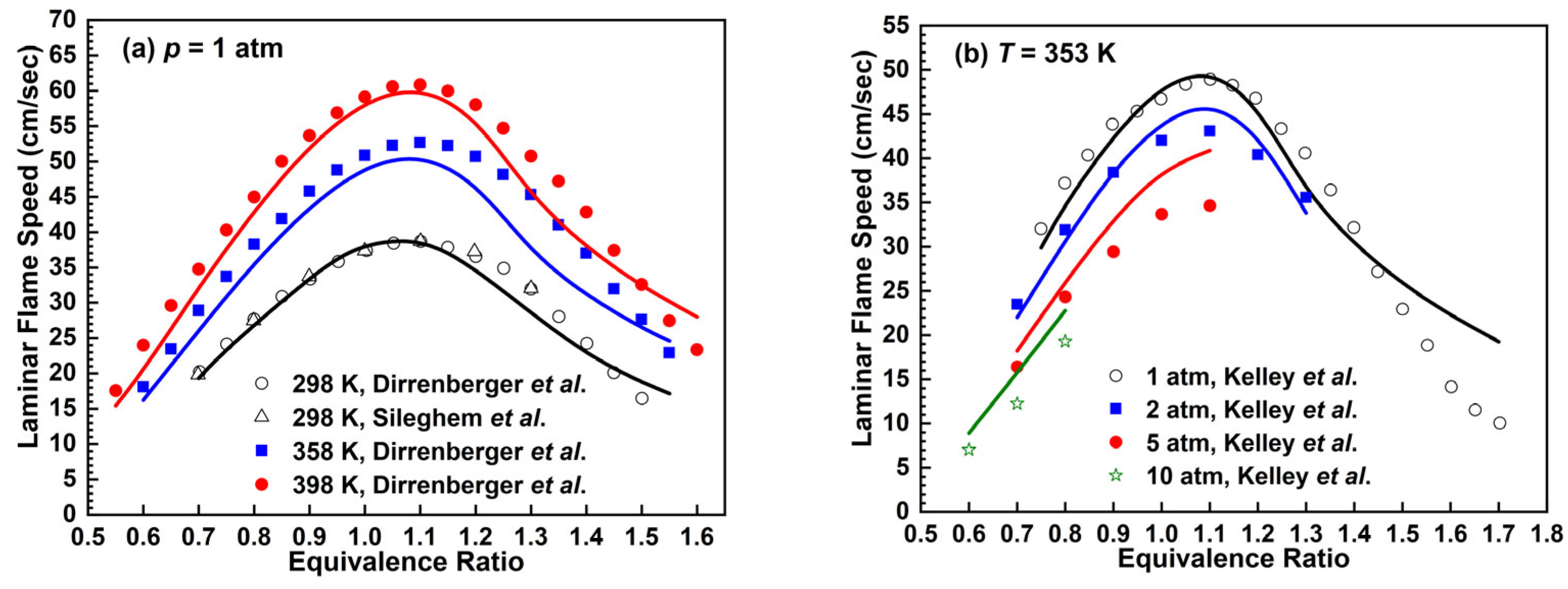
2.2.2. n-Heptane
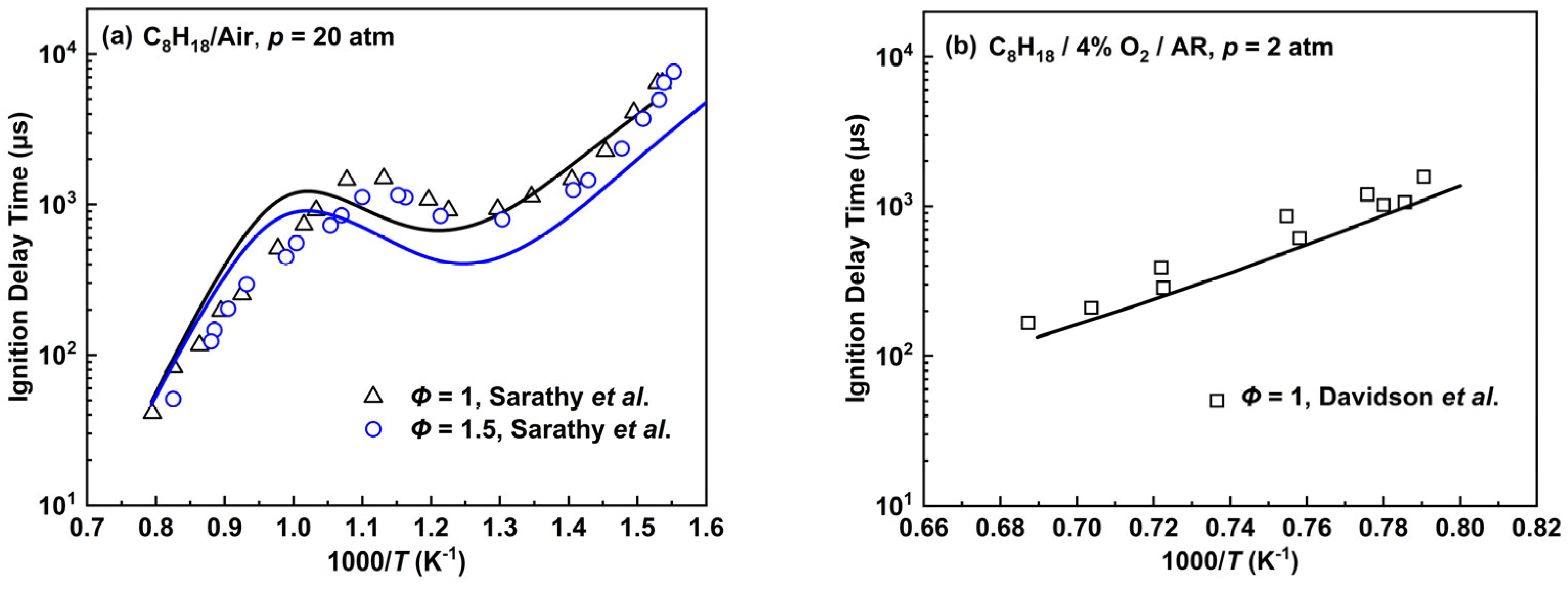
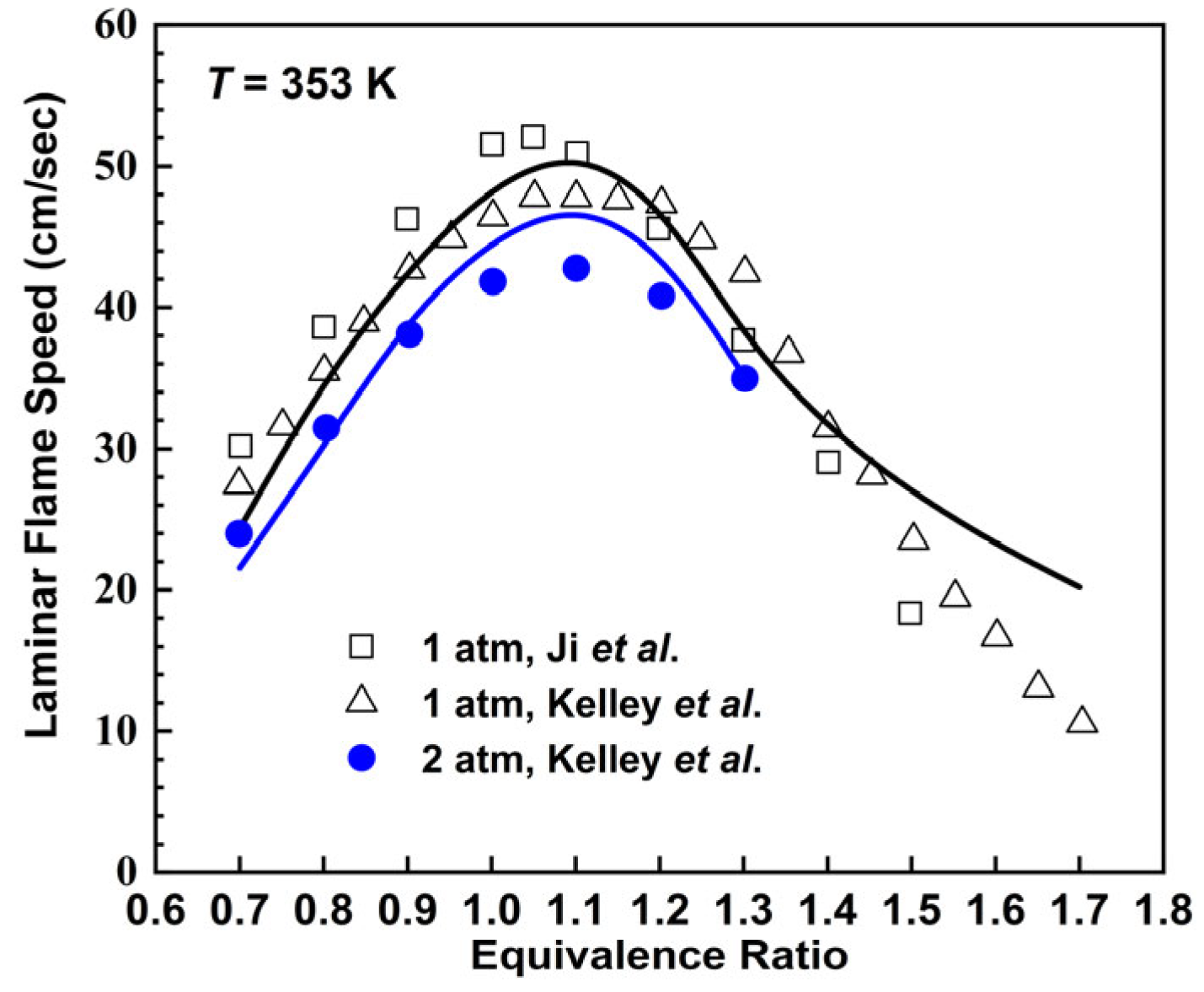
2.2.3. n-Octane
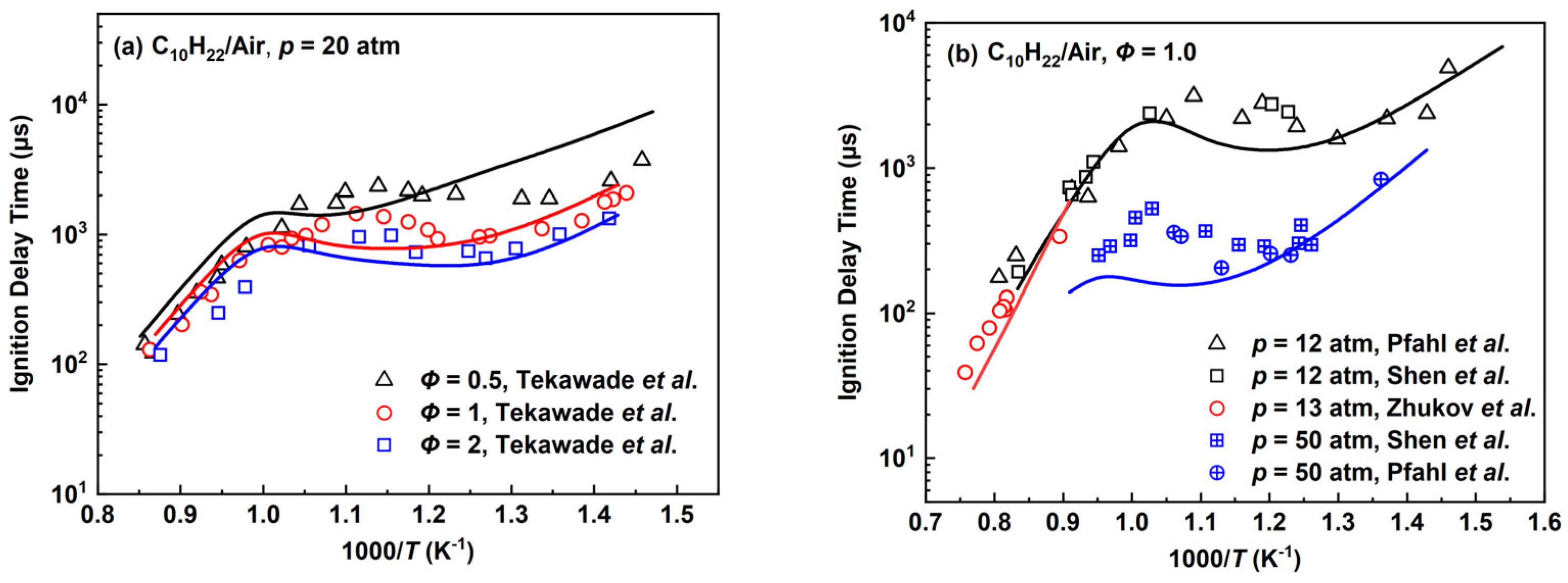
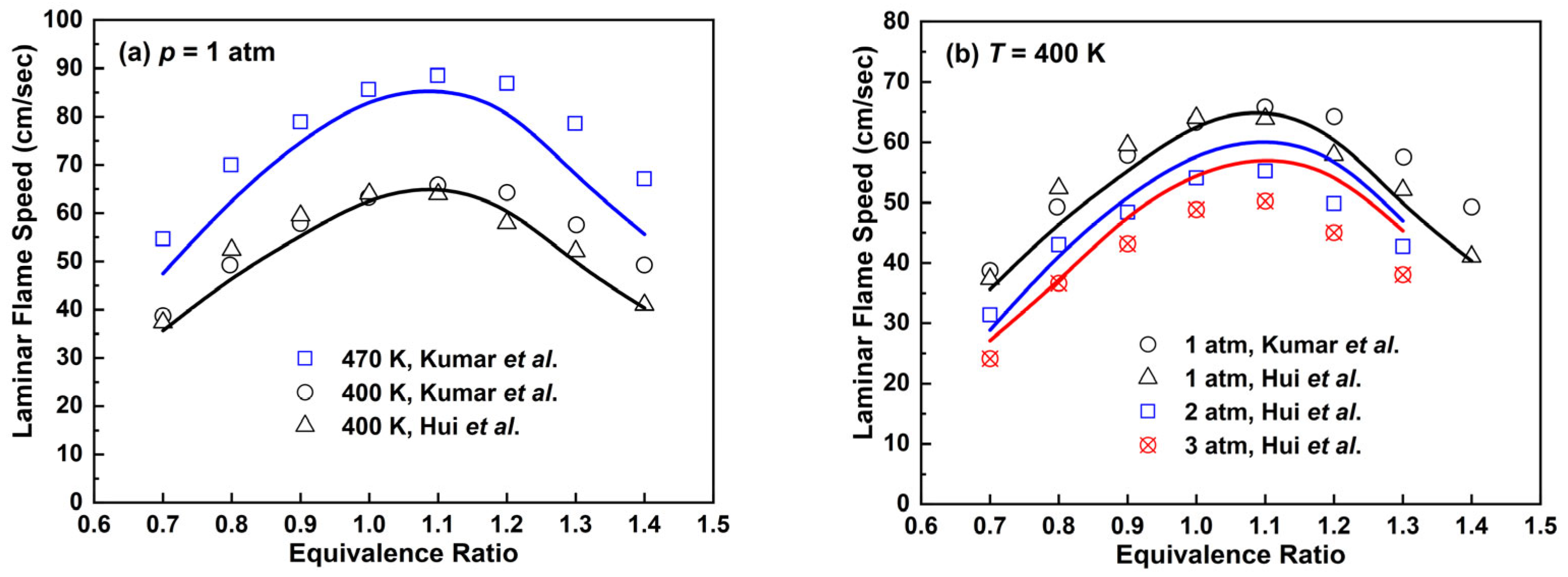
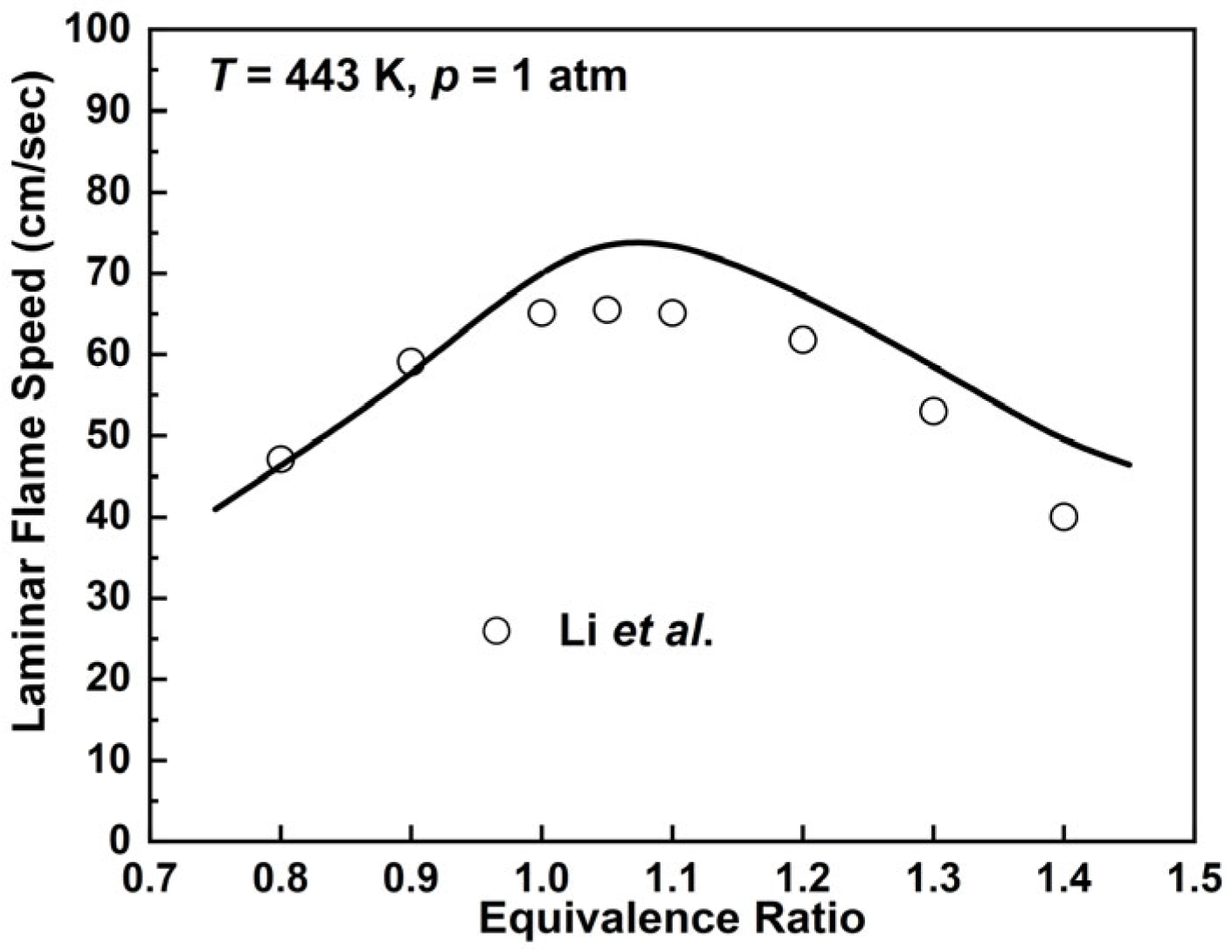
2.2.4. n-Decane
2.2.5. n-Dodecane
2.2.6. n-Hexadecane
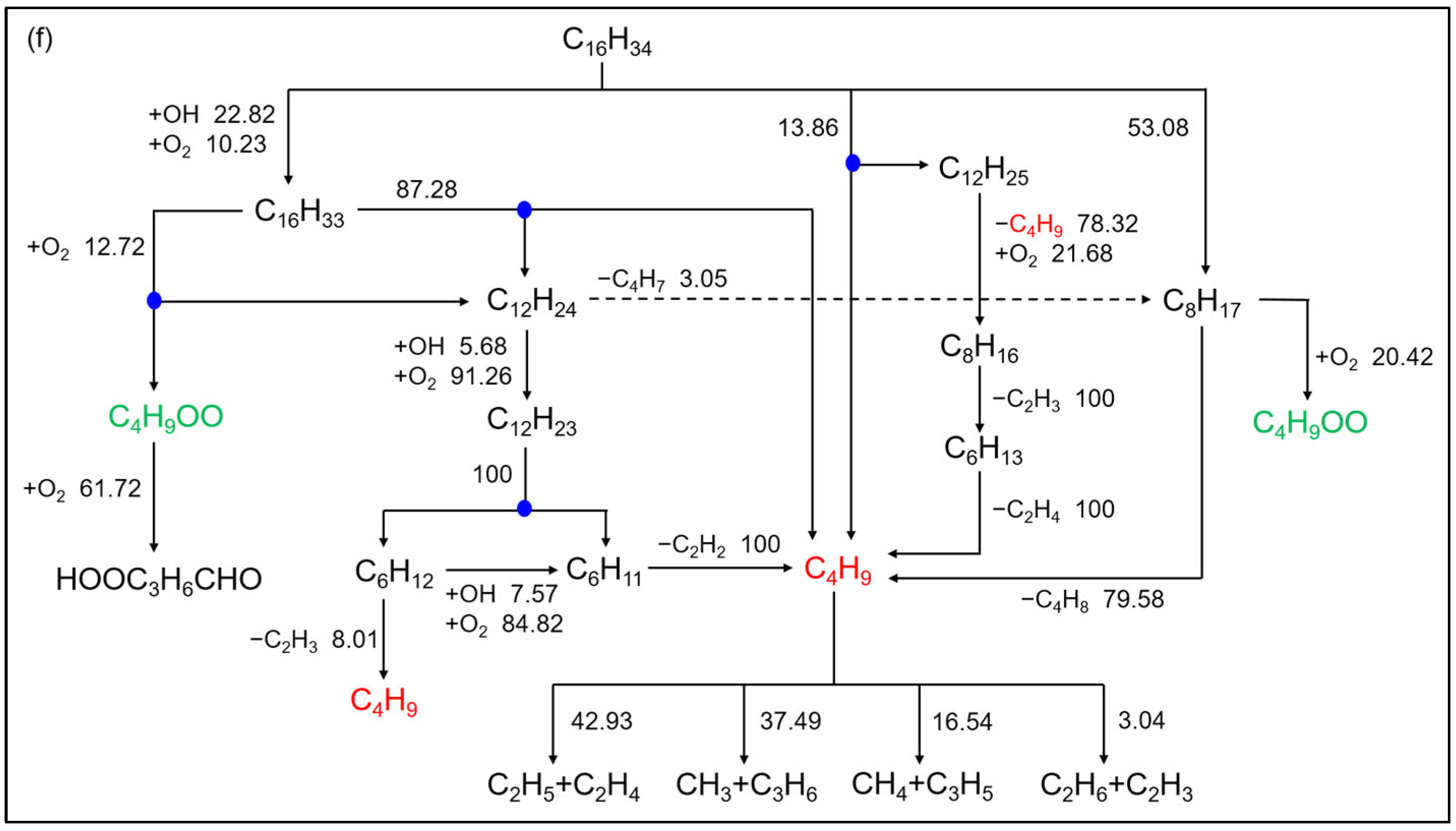
2.3. Reaction Path Analysis
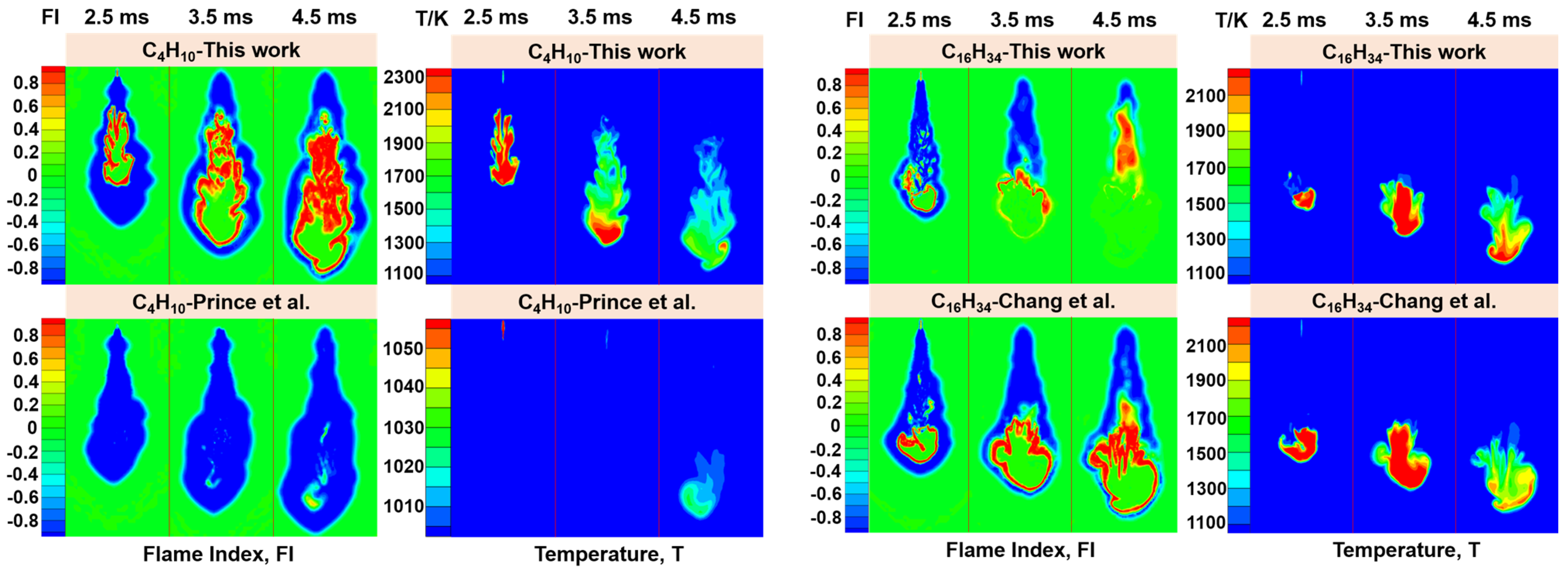
2.4. CFD Calculation
3. Methods
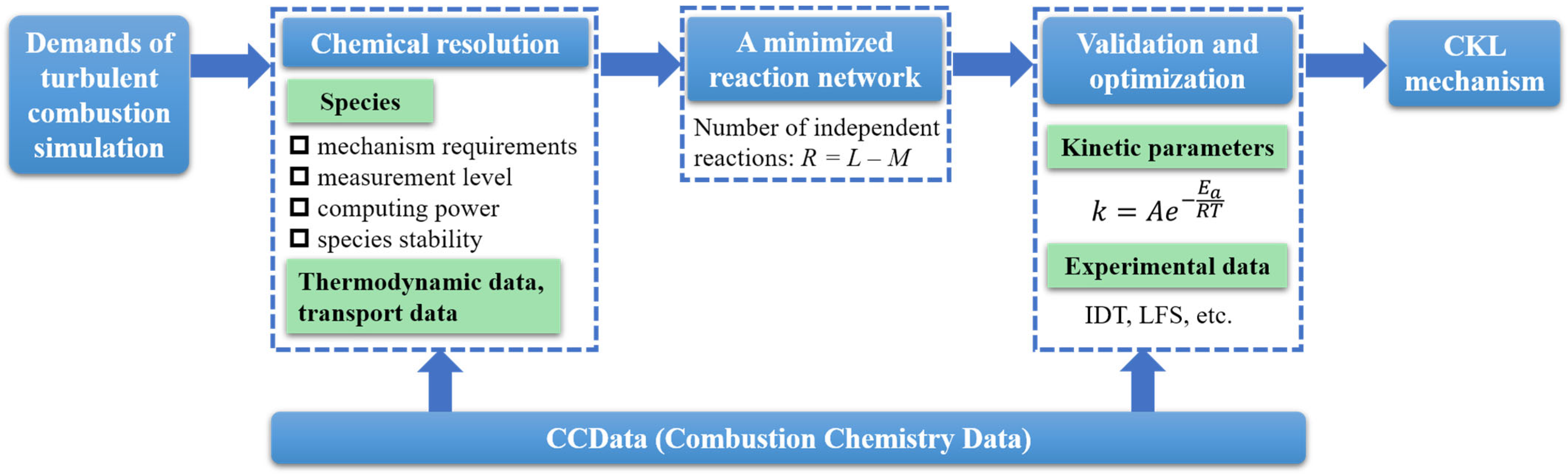
3.1. Chemical Resolution
3.2. The Determination of Minimized Reaction Network
3.3. Validation and Optimization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, W.; Li, Y.; Qi, F. Challenges and perspectives of combustion chemistry research. Sci. China Chem. 2017, 60, 1391–1401. [Google Scholar] [CrossRef]
- Kohse-Höinghaus, K. Combustion in the future: The importance of chemistry. Proc. Combust. Inst. 2021, 38, 1–56. [Google Scholar] [CrossRef]
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. Reaxff: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Curran, H.J. Developing detailed chemical kinetic mechanisms for fuel combustion. Proc. Combust. Inst. 2019, 37, 57–81. [Google Scholar] [CrossRef]
- Ranzi, E.; Frassoldati, A.; Grana, R.; Cuoci, A.; Faravelli, T.; Kelley, A.P.; Law, C.K. Hierarchical and comparative kinetic modeling of laminar flame speeds of hydrocarbon and oxygenated fuels. Prog. Energy Combust. Sci. 2012, 38, 468–501. [Google Scholar] [CrossRef]
- Zhou, C.; Li, Y.; Burke, U.; Banyon, C.; Somers, K.P.; Ding, S.; Khan, S.; Hargis, J.W.; Sikes, T.; Mathieu, O.; et al. An experimental and chemical kinetic modeling study of 1,3-butadiene combustion: Ignition delay time and laminar flame speed measurements. Combust. Flame 2018, 197, 423–438. [Google Scholar] [CrossRef]
- Martinez, S.; Baigmohammadi, M.; Patel, V.; Panigrahy, S.; Sahu, A.B.; Nagaraja, S.; Ramalingam, A.; Heufer, K.A.; Pekalski, A.; Curran, H.J. A comprehensive experimental and modeling study of the ignition delay time characteristics of ternary and quaternary blends of methane, ethane, ethylene, and propane over a wide range of temperature, pressure, equivalence ratio, and dilution. Combust. Flame 2021, 234, 111626. [Google Scholar] [CrossRef]
- Ren, H.S.; Wang, J.B.; Li, X.Y. Combustion Dynamics, Sichuan, CDS, Center for Combustion Dynamics, Sichuan University, September 2021. Available online: https://cds.scu.edu.cn (accessed on 1 December 2022).
- Green, W.H.; West, R.H. RMG—Reaction Mechanism Generator. Available online: https://rmg.mit.edu/ (accessed on 15 March 2023).
- Ning, H.B.; Li, Z.R.; Li, X.Y. Progress in combustion kinetics. Acta Phys.-Chim. Sin. 2016, 32, 131–153. [Google Scholar] [CrossRef]
- Liu, L.; Xu, H.; Zhu, Q.; Ren, H.; Li, X. Soot formation of n-decane pyrolysis: A mechanistic view from reaxff molecular dynamics simulation. Chem. Phys. Lett. 2020, 760, 137983. [Google Scholar] [CrossRef]
- Saggese, C.; Ferrario, S.; Camacho, J.; Cuoci, A.; Frassoldati, A.; Ranzi, E.; Wang, H.; Faravelli, T. Kinetic modeling of particle size distribution of soot in a premixed burner-stabilized stagnation ethylene flame. Combust. Flame 2015, 162, 3356–3369. [Google Scholar] [CrossRef]
- Saggese, C.; Wan, K.; Xu, R.; Tao, Y.; Bowman, C.T.; Park, J.; Lu, T.; Wang, H. A physics-based approach to modeling real-fuel combustion chemistry—V. NOx formation from a typical Jet A. Combust. Flame 2020, 212, 270–278. [Google Scholar] [CrossRef]
- Sun, W.; Zhao, Q.; Curran, H.J.; Deng, F.; Zhao, N.; Zheng, H.; Kang, S.; Zhou, X.; Kang, Y.; Deng, Y.; et al. Further insights into the core mechanism of H2/CO/NOx reaction system. Combust. Flame 2022, 245, 112308. [Google Scholar] [CrossRef]
- Westbrook, C.K.; Pitz, W.J.; Herbinet, O.; Curran, H.J.; Silke, E.J. A comprehensive detailed chemical kinetic reaction mechanism for combustion of n-alkane hydrocarbons from n-octane to n-hexadecane. Combust. Flame 2009, 156, 181–199. [Google Scholar] [CrossRef]
- Ranzi, E.; Cavallotti, C.; Cuoci, A.; Frassoldati, A.; Pelucchi, M.; Faravelli, T. New reaction classes in the kinetic modeling of low temperature oxidation of n-alkanes. Combust. Flame 2015, 162, 1679–1691. [Google Scholar] [CrossRef]
- Yao, M.; Zheng, Z.; Liu, H. Progress and recent trends in homogeneous charge compression ignition (HCCI) engines. Prog. Energy Combust. Sci. 2009, 35, 398–437. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Bi, G.; Zhang, L.; Song, J. A phenomenological model of diesel combustion characteristics under CO2/O2 atmosphere. Fuel Process. Technol. 2022, 229, 107167. [Google Scholar] [CrossRef]
- Ji, L.; Li, Y.; Wang, J.; Ning, A.; Zhang, N.; Liang, S.; He, J.; Zhang, T.; Qu, Z.; Gao, J. Community reaction network reduction for constructing a coarse-grained representation of combustion reaction mechanisms. J. Chem. Inf. Model. 2022, 62, 2352–2364. [Google Scholar] [CrossRef]
- Chang, Y.; Jia, M.; Liu, Y.; Li, Y.; Xie, M.; Yin, H. Application of a decoupling methodology for development of skeletal oxidation mechanisms for heavy n-alkanes from n-octane to n-hexadecane. Energy Fuels 2013, 27, 3467–3479. [Google Scholar] [CrossRef]
- Xue, J.; Xi, S.; Wang, F. An extensive study on skeletal mechanism reduction for the oxidation of C0-C4 fuels. Combust. Flame 2020, 214, 184–198. [Google Scholar] [CrossRef]
- Wang, H.; Xu, R.; Wang, K.; Bowman, C.T.; Hanson, R.K.; Davidson, D.F.; Brezinsky, K.; Egolfopoulos, F.N. A physics-based approach to modeling real-fuel combustion chemistry—I. Evidence from experiments, and thermodynamic, chemical kinetic and statistical considerations. Combust. Flame 2018, 193, 502–519. [Google Scholar] [CrossRef]
- Frassoldati, A.; Cuoci, A.; Faravelli, T.; Ranzi, E. Kinetic modeling of the oxidation of ethanol and gasoline surrogate mixtures. Combust. Sci. Technol. 2010, 182, 653–667. [Google Scholar] [CrossRef]
- Li, X.Y.; Yao, X.X.; Shentu, J.T.; Sun, X.H.; Li, J.Q.; Liu, M.X.; Xu, S.M. Combustion reaction mechanism construction by two-parameter rate constant method. Chem. J. Chin. Univ.-Chin. 2020, 41, 512–520. [Google Scholar] [CrossRef]
- Wang, H.; Sheen, D.A. Combustion kinetic model uncertainty quantification, propagation and minimization. Prog. Energy Combust. Sci. 2015, 47, 1–31. [Google Scholar] [CrossRef]
- Li, X.Y.; Shentu, J.T.; Li, Y.W.; Li, J.Q.; Wang, J.B. Combustion mechanism construction based on minimized reaction network: Hydrogen-oxygen combustion. Chem. J. Chin. Univ.-Chin. 2020, 41, 772–779. [Google Scholar] [CrossRef]
- Li, Y.W.; Shentu, J.T.; Wang, J.B.; Li, X.Y. Combustion mechanism construction based on minimized reaction network: C1-oxygen combustion. Chem. J. Chin. Univ.-Chin. 2021, 42, 1871–1880. [Google Scholar] [CrossRef]
- Chemkin-Pro 15112; Reaction Design: San Diego, CA, USA, 2011.
- Healy, D.; Donato, N.S.; Aul, C.J.; Petersen, E.L.; Zinner, C.M.; Bourque, G.; Curran, H.J. N-butane: Ignition delay measurements at high pressure and detailed chemical kinetic simulations. Combust. Flame 2010, 157, 1526–1539. [Google Scholar] [CrossRef]
- Gersen, S.; Mokhov, A.V.; Darmeveil, J.H.; Levinsky, H.B. Ignition properties of n-butane and iso-butane in a rapid compression machine. Combust. Flame 2010, 157, 240–245. [Google Scholar] [CrossRef]
- Davis, S.G.; Law, C.K. Determination of and fuel structure effects on laminar flame speeds of C1 to C8 hydrocarbons. Combust. Sci. Technol. 1998, 140, 427–449. [Google Scholar] [CrossRef]
- Tang, C.L.; Huang, Z.H.; Law, C.K. Determination, correlation, and mechanistic interpretation of effects of hydrogen addition on laminar flame speeds of hydrocarbon–air mixtures. Proc. Combust. Inst. 2011, 33, 921–928. [Google Scholar] [CrossRef]
- Hirasawa, T.; Sung, C.J.; Joshi, A.; Yang, Z.; Wang, H.; Law, C.K. Determination of laminar flame speeds using digital particle image velocimetry: Binary fuel blends of ethylene, n-butane, and toluene. Proc. Combust. Inst. 2002, 29, 1427–1434. [Google Scholar] [CrossRef]
- Li, W.; Wang, G.; Li, Y.; Li, T.; Zhang, Y.; Cao, C.; Zou, J.; Law, C.K. Experimental and kinetic modeling investigation on pyrolysis and combustion of n-butane and i-butane at various pressures. Combust. Flame 2018, 191, 126–141. [Google Scholar] [CrossRef]
- Zhang, K.; Banyon, C.; Bugler, J.; Curran, H.J.; Rodriguez, A.; Herbinet, O.; Battin-Leclerc, F.; B’Chir, C.; Heufer, K.A. An updated experimental and kinetic modeling study of n-heptane oxidation. Combust. Flame 2016, 172, 116–135. [Google Scholar] [CrossRef]
- Ciezki, H.K.; Adomeit, G. Shock-tube investigation of self-ignition of n-heptane-air mixtures under engine relevant conditions. Combust. Flame 1993, 93, 421–433. [Google Scholar] [CrossRef]
- Shen, H.S.; Steinberg, J.; Vanderover, J.; Oehlschlaeger, M.A. A shock tube study of the ignition of n-heptane, n-decane, n-dodecane, and n-tetradecane at elevated pressures. Energy Fuels 2009, 23, 2482–2489. [Google Scholar] [CrossRef]
- Hu, E.; Gao, Z.; Liu, Y.; Yin, G.; Huang, Z. Experimental and modeling study on ignition delay times of dimethoxy methane/n-heptane blends. Fuel 2017, 189, 350–357. [Google Scholar] [CrossRef]
- Dirrenberger, P.; Glaude, P.A.; Bounaceur, R.; Le Gall, H.; Da Cruz, A.P.; Konnov, A.A.; Battin-Leclerc, F. Laminar burning velocity of gasolines with addition of ethanol. Fuel 2014, 115, 162–169. [Google Scholar] [CrossRef]
- Sileghem, L.; Alekseev, V.A.; Vancoillie, J.; Van Geem, K.M.; Nilsson, E.J.K.; Verhelst, S.; Konnov, A.A. Laminar burning velocity of gasoline and the gasoline surrogate components iso-octane, n-heptane and toluene. Fuel 2013, 112, 355–365. [Google Scholar] [CrossRef]
- Kelley, A.P.; Smallbone, A.J.; Zhu, D.L.; Law, C.K. Laminar flame speeds of C5 to C8 n-alkanes at elevated pressures: Experimental determination, fuel similarity, and stretch sensitivity. Proc. Combust. Inst. 2011, 33, 963–970. [Google Scholar] [CrossRef]
- Sarathy, S.M.; Westbrook, C.K.; Mehl, M.; Pitz, W.J.; Togbe, C.; Dagaut, P.; Wang, H.; Oehlschlaeger, M.A.; Niemann, U.; Seshadri, K.; et al. Comprehensive chemical kinetic modeling of the oxidation of 2-methylalkanes from C7 to C20. Combust. Flame 2011, 158, 2338–2357. [Google Scholar] [CrossRef]
- Davidson, D.F.; Ranganath, S.C.; Lam, K.Y.; Liaw, M.; Hong, Z.; Hanson, R.K. Ignition delay time measurements of normal alkanes and simple oxygenates. J. Propul. Power 2010, 26, 280–287. [Google Scholar] [CrossRef]
- Ji, C.; Sarathy, S.M.; Veloo, P.S.; Westbrook, C.K.; Egolfopoulos, F.N. Effects of fuel branching on the propagation of octane isomers flames. Combust. Flame 2012, 159, 1426–1436. [Google Scholar] [CrossRef]
- Tekawade, A.; Xie, T.; Oehlschlaeger, M.A. Comparative study of the ignition of 1-decene, trans-5-decene, and n-decane: Constant-volume spray and shock-tube experiments. Energy Fuels 2017, 31, 6493–6500. [Google Scholar] [CrossRef]
- Pfahl, U.; Fieweger, K.; Adomeit, G. Self-ignition of diesel-relevant hydrocarbon-air mixtures under engine conditions. Symp. Int. Combust. 1996, 26, 781–789. [Google Scholar] [CrossRef]
- Zhukov, V.P.; Sechenov, V.A.; Starikovskii, A.Y. Autoignition of n-decane at high pressure. Combust. Flame 2008, 153, 130–136. [Google Scholar] [CrossRef]
- Munzar, J.D.; Akih-Kumgeh, B.; Denman, B.M.; Zia, A.; Bergthorson, J.M. An experimental and reduced modeling study of the laminar flame speed of jet fuel surrogate components. Fuel 2013, 113, 586–597. [Google Scholar] [CrossRef]
- Kim, H.H.; Won, S.H.; Santner, J.; Chen, Z.; Ju, Y. Measurements of the critical initiation radius and unsteady propagation of n-decane/air premixed flames. Proc. Combust. Inst. 2013, 34, 929–936. [Google Scholar] [CrossRef]
- Hui, X.; Sung, C. Laminar flame speeds of transportation-relevant hydrocarbons and jet fuels at elevated temperatures and pressures. Fuel 2013, 109, 191–200. [Google Scholar] [CrossRef]
- Singh, D.; Nishiie, T.; Qiao, L. Experimental and kinetic modeling study of the combustion of n-Decane, Jet-A, and S-8 in laminar premixed flames. Combust. Sci. Technol. 2011, 183, 1002–1026. [Google Scholar] [CrossRef]
- Kumar, K.; Sung, C. Laminar flame speeds and extinction limits of preheated n-decane/O2/N2 and n-dodecane/O2/N2 mixtures. Combust. Flame 2007, 151, 209–224. [Google Scholar] [CrossRef]
- Shao, J.; Choudhary, R.; Peng, Y.; Davidson, D.F.; Hanson, R.K. A shock tube study of n-heptane, iso-octane, n-dodecane and iso-octane/n-dodecane blends oxidation at elevated pressures and intermediate temperatures. Fuel 2019, 243, 541–553. [Google Scholar] [CrossRef]
- Vasu, S.S.; Davidson, D.F.; Hong, Z.; Vasudevan, V.; Hanson, R.K. N-dodecane oxidation at high-pressures: Measurements of ignition delay times and oh concentration time-histories. Proc. Combust. Inst. 2009, 32, 173–180. [Google Scholar] [CrossRef]
- Mao, Y.; Raza, M.; Wu, Z.; Zhu, J.; Yu, L.; Wang, S.; Zhu, L.; Lu, X. An experimental study of n-dodecane and the development of an improved kinetic model. Combust. Flame 2020, 212, 388–402. [Google Scholar] [CrossRef]
- Haylett, D.R.; Davidson, D.F.; Hanson, R.K. Ignition delay times of low-vapor-pressure fuels measured using an aerosol shock tube. Combust. Flame 2012, 159, 552–561. [Google Scholar] [CrossRef]
- Li, B.; Liu, N.; Zhao, R.; Zhang, H.; Egolfopoulos, F.N. Flame propagation of mixtures of air with high molecular weight neat hydrocarbons and practical jet and diesel fuels. Proc. Combust. Inst. 2013, 34, 727–733. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Wang, J.; Liu, H. Effect of co2 and its concentration variation on the ignition and combustion of diesel surrogate fuel: Optical experiments and numerical simulations. J. Energy Inst. 2023, 110, 101350. [Google Scholar] [CrossRef]
- Senecal, P.K.; Pomraning, E.; Richards, K.J.; Briggs, T.E.; Choi, C.Y.; Mcdavid, R.M.; Patterson, M.A. Multi-dimensional modeling of direct-injection diesel spray liquid length and flame lift-off length using cfd and parallel detailed chemistry. SAE Trans. 2003, 112, 1331–1351. [Google Scholar] [CrossRef]
- Smagorinsky, J. General circulation experiments with the primitive equations: I. The basic experiment. Mon. Weather Rev. 1963, 91, 99–164. [Google Scholar] [CrossRef]
- Prince, J.C.; Treviño, C.; Williams, F.A. A reduced reaction mechanism for the combustion of n-butane. Combust. Flame 2017, 175, 27–33. [Google Scholar] [CrossRef]
- Yamashita, H.; Shimada, M.; Takeno, T. A numerical study on flame stability at the transition point of jet diffusion flames. Symp. Int. Combust. 1996, 26, 27–34. [Google Scholar] [CrossRef]
- Sharma, D.; Mahapatra, S.; Garnayak, S.; Arghode, V.K.; Bandopadhyay, A.; Dash, S.K.; Reddy, V.M. Development of the reduced chemical kinetic mechanism for combustion of H2/CO/C1-C4 hydrocarbons. Energy Fuels 2021, 35, 718–742. [Google Scholar] [CrossRef]
- Weltin, E. Let a computer balance your chemical equations and determine the number of independent reactions. J. Chem. Educ. 1994, 71, 295. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | # | Reaction | A 1 | Ea 1 |
|---|---|---|---|---|
| C4H10 | R1 | C4H10(+M)<=>2C2H5(+M) | 1.52 × 1016 | 82,898 |
| Low pressure limit | 4.72 × 1018 | 49,580 | ||
| R2 | C4H10<=>C4H9+H | 6.32 × 1017 | 90,000 | |
| R3 | C4H10+O2<=>C4H9+HO2 | 6.00 × 1014 | 45,340 | |
| R4 | C4H10+OH<=>C4H9+H2O | 3.11 × 1014 | 3250 | |
| C4H9 | R5 | C4H9<=>CH4+C3H5 | 1.72 × 1013 | 28,900 |
| R6 | C4H9<=>C2H5+C2H4 | 4.47 × 1013 | 28,900 | |
| R7 | C4H9<=>C2H6+C2H3 | 3.16 × 1012 | 28,900 | |
| R8 | C4H9<=>CH3+C3H6 | 4.19 × 1013 | 28,900 | |
| R9 | C4H9<=>H+C4H8 | 1.29 × 1013 | 28,900 | |
| R10 | C4H9+CH4<=>C3H8+C2H5 | 7.23 × 1013 | 28,900 | |
| R11 | C4H9+H<=>C4H8+H2 | 5.42 × 1012 | 0 | |
| R12 | C4H9+O2<=>C4H9OO | 6.60 × 1011 | 1840 | |
| C4H9OO | R13 | C4H9OO+O2<=>HOOC3H6CHO+OH | 3.03 × 1011 | 1000 |
| HOOC3H6CHO | R14 | HOOC3H6CHO<=>OH+OC3H6CHO | 1.05 × 1013 | 40,000 |
| OC3H6CHO | R15 | OC3H6CHO<=>CH3CHO+CH3CO | 7.01 × 1013 | 1750 |
| C4H8 | R16 | C4H8<=>C3H5+CH3 | 1.00 × 1016 | 72,900 |
| R17 | C4H8<=>C4H7+H | 6.32 × 1016 | 90,000 | |
| R18 | C4H8+O2<=>C4H7+HO2 | 2.00 × 1015 | 30,900 | |
| R19 | C4H8+OH<=>C4H7+H2O | 3.52 × 1014 | 3970 | |
| C4H7 | R20 | C4H7<=>C2H4+C2H3 | 1.83 × 1014 | 27,200 |
| Species | # | Reaction | A 1 | Ea 1 |
|---|---|---|---|---|
| C7H16 | R21 | C7H16<=>C3H7+C4H9 | 5.00 × 1017 | 77,000 |
| R22 | C7H16<=>C7H15+H | 6.32 × 1016 | 90,000 | |
| R23 | C7H16+O2<=>C7H15+HO2 | 4.20 × 1013 | 30,800 | |
| R24 | C7H16+OH<=>C7H15+H2O | 1.28 × 1014 | 23,140 | |
| R25 | C7H16+HO2<=>C7H15+H2O2 | 5.36 × 1015 | 3880 | |
| C7H15 | R26 | C7H15<=>C3H6+C4H9 | 1.42 × 1014 | 30,000 |
| R27 | C7H15+O2<=>C3H6+C4H9OO | 9.08 × 1012 | 1840 |
| Species | # | Reaction | A 1 | Ea 1 |
|---|---|---|---|---|
| C8H18 | R28 | C8H18<=>2C4H9 | 2.50 × 1018 | 76,200 |
| R29 | C8H18<=>C8H17+H | 6.32 × 1016 | 90,000 | |
| R30 | C8H18+O2<=>C8H17+HO2 | 4.20 × 1014 | 30,800 | |
| R31 | C8H18+OH<=>C8H17+H2O | 2.81 × 1015 | 2470 | |
| R32 | C8H18+HO2<=>C8H17+H2O2 | 1.28 × 1014 | 23,140 | |
| C8H17 | R33 | C8H17<=>C4H8+C4H9 | 1.42 × 1014 | 30,000 |
| R34 | C8H17+O2<=>C4H8+C4H9OO | 2.20 × 1012 | 1840 |
| Species | # | Reaction | A 1 | Ea 1 |
|---|---|---|---|---|
| C10H22 | R35 | C10H22<=>C6H13+C4H9 | 2.50 × 1018 | 76,200 |
| R36 | C10H22<=>C10H21+H | 6.32 × 1017 | 90,000 | |
| R37 | C10H22+O2<=>C10H21+HO2 | 4.20 × 1013 | 30,800 | |
| R38 | C10H22+OH<=>C10H21+H2O | 5.36 × 1014 | 3880 | |
| R39 | C10H22+HO2<=>C10H21+H2O2 | 1.28 × 1014 | 23,140 | |
| C10H21 | R40 | C10H21<=>C6H12+C4H9 | 5.68 × 1013 | 30,000 |
| R41 | C10H21+O2<=>C6H12+C4H9OO | 1.10 × 1012 | 1840 | |
| C6H13 | R42 | C6H13<=>C4H9+C2H4 | 5.68 × 1014 | 30,000 |
| C6H12 | R43 | C6H12+OH<=>C6H11+H2O | 1.20 × 1014 | 4950 |
| R44 | C6H12+O2<=>C6H11+HO2 | 2.00 × 1014 | 30,930 | |
| R45 | C6H12<=>C4H9+C2H3 | 1.89 × 1014 | 61,000 | |
| C6H11 | R46 | C6H11<=>C4H9+C2H2 | 7.12 × 1015 | 32,100 |
| Species | # | Reaction | A 1 | Ea 1 |
|---|---|---|---|---|
| C12H26 | R47 | C12H26<=>2C6H13 | 2.50 × 1018 | 76,200 |
| R48 | C12H26<=>C12H25+H | 6.32 × 1016 | 90,000 | |
| R49 | C12H26+O2<=>C12H25+HO2 | 4.20 × 1015 | 30,800 | |
| R50 | C12H26+OH<=>C12H25+H2O | 5.36 × 1016 | 3880 | |
| R51 | C12H26+HO2<=>C12H25+H2O2 | 8.28 × 1014 | 20,140 | |
| C12H25 | R52 | C12H25<=>C10H21+C2H4 | 5.68 × 1013 | 25,000 |
| R53 | C12H25+O2<=>C8H16+C4H9OO | 9.08 × 1012 | 1840 | |
| C8H16 | R54 | C8H16<=>C6H13+C2H3 | 1.89 × 1016 | 61,000 |
| R55 | C8H16+OH<=>C8H15+H2O | 1.20 × 1013 | 4950 | |
| C8H15 | R56 | C8H15<=>C5H9+C3H6 | 2.11 × 1015 | 50,310 |
| C5H9 | R57 | C5H9<=>C2H4+C3H5 | 2.11 × 1015 | 20,310 |
| Species | # | Reaction | A 1 | Ea 1 |
|---|---|---|---|---|
| C16H34 | R58 | C16H34<=>2C8H17 | 2.50 × 1019 | 76,200 |
| R59 | C16H34<=>C4H9+C12H25 | 2.50 × 1018 | 76,200 | |
| R60 | C16H34<=>C16H33+H | 1.58 × 1016 | 97,510 | |
| R61 | C16H34+O2<=>C16H33+HO2 | 4.20 × 1013 | 30,800 | |
| R62 | C16H34+OH<=>C16H33+H2O | 5.36 × 1014 | 3880 | |
| R63 | C16H34+HO2<=>H2O2+C16H33 | 1.28 × 1014 | 23,140 | |
| C16H33 | R64 | C16H33<=>C4H9+C12H24 | 8.42 × 1014 | 30,000 |
| R65 | C16H33+O2<=>C4H9OO+C12H24 | 7.08 × 1012 | 1840 | |
| C12H24 | R66 | C12H24<=>C4H7+C8H17 | 1.89 × 1014 | 61,000 |
| R67 | C12H24+O2<=>C12H23+HO2 | 2.00 × 1014 | 30,930 | |
| R68 | C12H24+OH<=>C12H23+H2O | 1.20 × 1014 | 4950 | |
| C12H23 | R69 | C12H23<=>C6H11+C6H12 | 7.12 × 1015 | 32,100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shentu, J.; Lu, Y.; Li, Y.; Li, J.; Mao, Y.; Li, X. Compact Combustion Mechanisms of Typical n-Alkanes Developed by the Minimized Reaction Network Method. Molecules 2023, 28, 7695. https://doi.org/10.3390/molecules28237695
Shentu J, Lu Y, Li Y, Li J, Mao Y, Li X. Compact Combustion Mechanisms of Typical n-Alkanes Developed by the Minimized Reaction Network Method. Molecules. 2023; 28(23):7695. https://doi.org/10.3390/molecules28237695
Chicago/Turabian StyleShentu, Jiangtao, Yanrong Lu, Yiwei Li, Juanqin Li, Yebing Mao, and Xiangyuan Li. 2023. "Compact Combustion Mechanisms of Typical n-Alkanes Developed by the Minimized Reaction Network Method" Molecules 28, no. 23: 7695. https://doi.org/10.3390/molecules28237695
APA StyleShentu, J., Lu, Y., Li, Y., Li, J., Mao, Y., & Li, X. (2023). Compact Combustion Mechanisms of Typical n-Alkanes Developed by the Minimized Reaction Network Method. Molecules, 28(23), 7695. https://doi.org/10.3390/molecules28237695