1. Introduction
Diabetes, a non-communicable disease (NCD), contributes significantly to the rising global death rate [
1]. By 2030, it is projected to be the leading cause of death in Africa [
2]. This condition is characterized by persistently high blood glucose levels due to insufficient insulin secretion, cellular insensitivity to insulin, or both, and is often linked to pancreatic β-cell failure [
3]. With alarming mortality rates, diabetes has emerged as a major underlying cause of death worldwide [
4,
5]. The International Diabetes Federation (IDF) reported a surge in global diabetes prevalence from 151 million adults in 2000 to 451 million in 2017, with a predicted increase to 693 million by 2045, especially in lower to middle-income countries, if no action is taken [
6].
While several conventional medications exist for diabetes management, their affordability for low-income individuals and issues related to prolonged use, such as adverse effects and poor patient compliance, remain be to challenges [
7]. These therapies often fail to halt disease progression, leading to eventual insulin dependence [
8]. The primary goal of treatment is to achieve normal glycemia levels to prevent complications. However, global efforts to find a successful treatment have not yet been realized. Consequently, the exploration of new antidiabetic therapeutics continues, with medicinal plants being considered a viable alternative. Medicinal plants are rich sources of phytochemicals, possessing the ability to alleviate various ailments and diseases. Their pharmacological properties stem from secondary metabolites, which are natural sources of bioactive compounds. Since diabetes affects multiple pathways in different tissues, these pathways serve as targets for drug development in monotherapy [
9]. Extracts containing bioactive secondary metabolites can be utilized in polytherapy to target multiple pathways, enhancing treatment outcomes [
10]. While drug discovery typically focuses on isolating single lead compounds, identifying and characterizing bioactive phytochemical compounds within extracts that collectively modulate multiple pathways to ameliorate disease progression is crucial.
Traditionally, plants belonging to the
Ficus genus, including
Ficus lutea, have been used for their medicinal properties, such as antidiabetic, anthelmintic, hypotensive, mild laxative, antirheumatic, digestive, and anti-dysentery remedies [
11,
12]. These properties are attributed to chemical constituents like triterpenes, sterols, polyphenols, flavonoids, coumarins, alkaloids, and other metabolites [
13].
F. lutea, in particular, has been identified for its potential in managing diabetes [
13].
Ficus lutea Vahl, a member of the Moraceae family, is commonly referred to as the African wild fig or yellow leaf rock fig. This distinctive fig tree species is native to various regions of Africa and is characterized by a spreading canopy that can reach heights of 20 m or more, with an extensive root system enabling its growth on rocky surfaces, both solitary and in clusters. The bark of the tree starts grayish-brown, appearing smooth but gradually developing slight fissures with age, eventually becoming rougher and more textured as the tree matures. The leaves are large, simple, and arranged alternately along the branches, featuring prominent veins, an oval to elliptical shape, and a pointed tip. While they are typically dark green, they can also exhibit a yellow tint. The fruit of
F. lutea, known as syconium, begins as green and ripens to a yellow shade. Inside the syconium, small flowers and seeds are enclosed [
14].
Studies have highlighted the antidiabetic potential of
F. lutea leaf crude acetone extract. This potential was demonstrated through the inhibition of digestive enzymes (α-amylase and α-glucosidase), stimulation of glucose utilization in muscle and adipocytes, and the insulin-releasing action of insulinoma cells [
13,
15]. In particular, the antidiabetic potential of the
F. lutea leaf crude acetone extract was found to be stronger in the ethyl acetate fraction, which exhibited potent in vitro antidiabetic activity [
16]. Furthermore, in vivo studies demonstrated the potential of the ethyl acetate fraction of
F. lutea extract to ameliorate hyperglycemia and obesity in an obese mouse model to some extent [
16]. The possible additive or synergistic therapeutic effects of the bioactive phytochemicals within the ethyl acetate fraction may likely be responsible for the perceived effects and may be beneficial for diabetes management [
10].
To further explore the antidiabetic potential of the ethyl acetate fraction, a procedure was undertaken that fractionated the bioactive ethyl acetate fraction using silica gel column chromatography. This process led to the isolation of five compounds, which were elucidated using nuclear magnetic resonance (NMR) [
8]. While a previous study had delved into the antidiabetic potential of
F. lutea extract and its fractions [
16], there remained a gap in understanding the molecular-level interactions, including ligand-protein binding and potential metabolism. To address this, experimentation with isolated compounds was initiated. Consequently, this study had two main objectives: first, to investigate the in vitro antidiabetic potential of the five compounds from the bioactive ethyl acetate fraction of
F. lutea; and second, to predict, through in silico analysis, the binding interactions via molecular docking as well as the ADMET pharmacokinetic characteristics.
3. Discussion
The exploration of bioactive compounds in F. lutea has uncovered promising candidates for potential antidiabetic agents. Among the isolated compounds, epicatechin, epiafzelechin, and stigmasterol demonstrated inhibitory effects on α-glucosidase activity. α-Glucosidase is an enzyme in the small intestine crucial for glucose metabolism. It is involved in the breakdown of complex carbohydrates into simpler sugars like glucose, facilitating absorption and raising blood sugar levels. As α-Glucosidase inhibitors, epicatechin, epiafzelechin, and stigmasterol can slow down the enzyme’s action, reducing carbohydrate conversion into glucose, lowering post-prandial blood sugar levels, and aiding diabetes management. Furthermore, epicatechin and epiafzelechin exhibited enhanced glucose utilization in both C2C12 muscle cells and H-4-II-E liver cells. The skeletal muscle plays a crucial role in maintaining blood glucose homeostasis by serving as the primary site for glucose uptake, accounting for about 75% of glucose disposal after a meal. Additionally, the liver, a key organ in glycemic regulation, stores energy as glycogen and triglycerides. The ability of these two compounds to enhance glucose utilization in cells suggests their potential as antidiabetic agents. These findings implied that the antidiabetic potential of the ethyl acetate fraction may probably be due to the synergistic action of these bioactive compounds and/or other terpenoids, steroids, and flavonoids contributing to the plant’s antidiabetic properties. Following this, molecular docking studies were employed to elucidate the binding mechanisms of these compounds with receptors involved in antidiabetic activity, utilizing an in-silico approach.
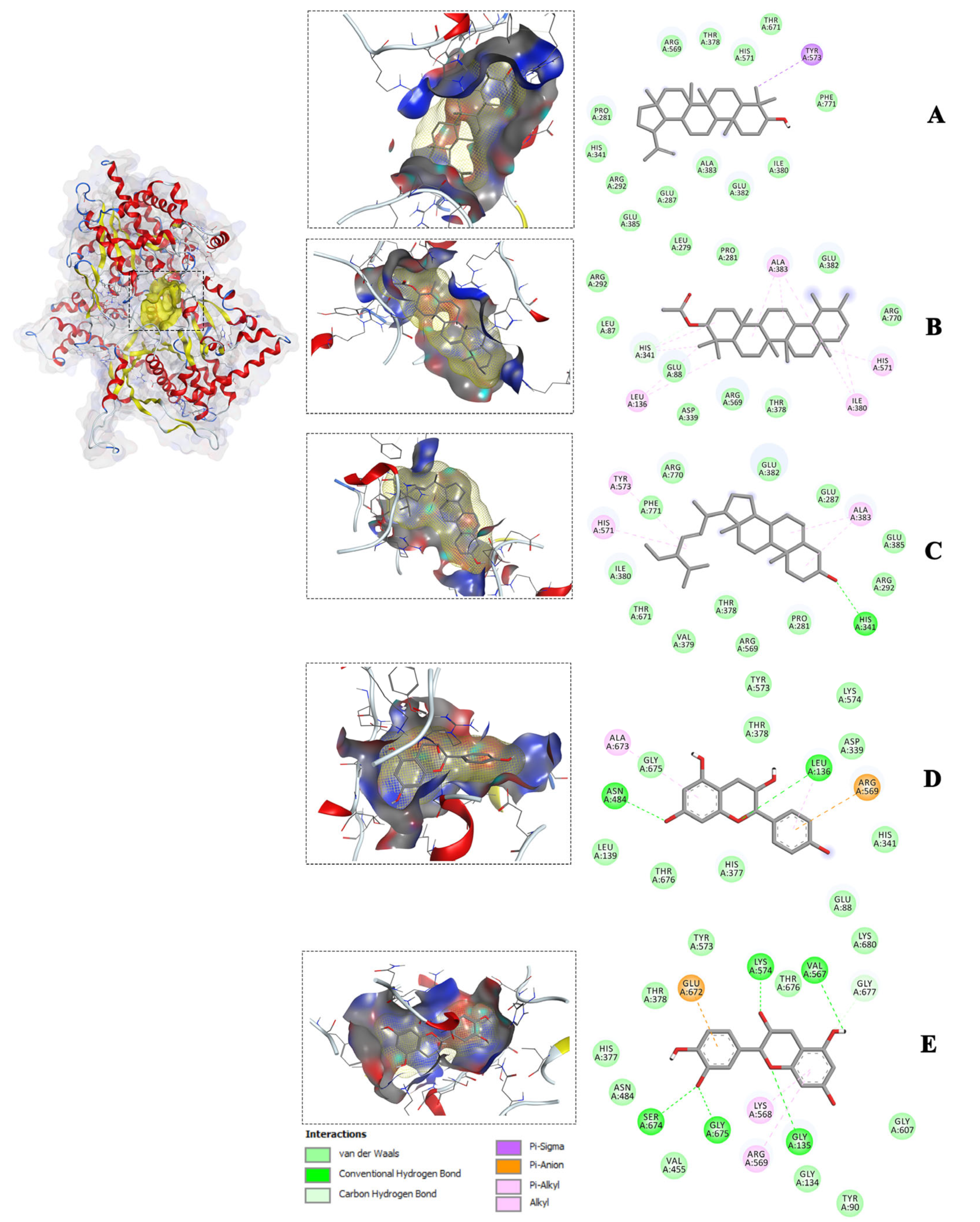
Molecular docking studies are frequently employed in drug design to predict interactions between ligands and proteins. This is achieved by calculating the binding affinity and visualizing the amino acid interactions contributing to it. Docking enables the prediction of antidiabetic activity by assessing the binding affinity of isolated compounds for proteins involved in glucose metabolism. Antidiabetic therapies are typically developed to target various mechanisms of glucose metabolism, involving multiple pathways [
23]. In this study, molecular docking was performed against twelve receptors (α-amylase, α-glucosidase, PPAR-γ, IGF1R, DPP-IV, GLUT1, SUR, GP, IR, GK, PTP1B, and SGT2) identified in the literature as playing important roles in glucose metabolism to determine their efficacy [
23]. A total of 125 docking analyses were conducted for the five isolated compounds against these twenty-five receptors. The binding conformation of the compounds within the active site of the receptors was assessed based on the scoring function and predicting the strength of the compound-receptor interaction. Forty molecular docking interactions were selected because they had the best (lower scores) free energy of binding (ΔG kcal/mol). All the compounds interacted with the receptors to varying degrees. The binding affinities, evaluated through scoring functions, identified stigmasterol as the most promising compound, demonstrating a strong affinity for a broad spectrum of receptors, followed closely by lupeol. These compounds exhibited superior binding to multiple receptors, suggesting their potential as candidates for antidiabetic drug development. However, these in silico results contrasted with the findings from the in vitro α-glucosidase inhibitory and glucose utilization assays in cells, where epicatechin and epiafzelechin demonstrated favorable activity. Notably, epicatechin, epiafzelechin, and the conventional antidiabetic drugs (i.e., the positive controls) did not exhibit superior binding affinities for the protein receptors. This observation might indicate that epicatechin, epiafzelechin, and the positive controls interacted with the protein receptors in a similar manner, suggesting a limitation in the results of the molecular docking analysis.
In drug development, effective binding to the target is not only essential but also ensures oral bioavailability [
24] and drug-likeness properties [
25]. In this regard, examining the physicochemical properties of the compounds is crucial for drug development [
26]. The adherence to Lipinski’s rule [
22] by epiafzelechin and epicatechin positions them as active drug candidates. Lipinski’s rule of five is a set of criteria used to evaluate the drug-likeness of small molecules, which includes molecular weight ≤ 500 g/mol, Log P (octanol-water partition coefficient) ≤ 5, hydrogen bond acceptors ≤ 10, hydrogen donors ≤ 5, and topological polar surface area (≤140), with only one violation permitted (
Table 4). Compounds that violate more than one of Lipinski’s rules are unlikely to be active drug candidates [
27]. Compounds with high log
p-values like lupeol, stigmasterol, and α-amyrin-acetate may pose challenges in reaching therapeutic targets due to their lipophilicity, potentially limiting their efficacy [
28]. Favorable bioavailability scores (0.55) predict good suitability for oral drug applications [
29,
30], implying that a smaller quantity of the compound is required to achieve the expected therapeutic outcome, reducing the risk of side effects and toxicity. Also, compounds with molecular weights > 250 g/mol ≤ 350 g/mol and lower hydrophobicity than those specified by Lipinski’s rule are positioned as lead-like candidates [
31]. In this regard, epiafzelechin and epicatechin exhibit lead-like characteristics [
30,
32], indicating their potential as candidates in the drug discovery process. Consequently, lead-likeness, adherence to Lipinski’s rule, and favorable oral bioavailability are suggested as the characteristics of compounds pivotal in drug discovery.
To develop an effective therapeutic agent, it is crucial a drug candidate reaches its target location in sufficient concentration, inducing the desired pharmacologic effect while minimizing side effects. Predictive ADMET analyses offer valuable insights into how compounds behave in the human body, revealing their interactions with proteins, distribution patterns, and metabolism pathways [
33]. Epicatechin and epiafzelechin exhibited excellent gastrointestinal absorption, indicating their potential as orally administered agents crucial for antidiabetic therapy [
24,
34]. The intestine, with its large surface area, serves as the primary site for drug absorption when administered orally [
35,
36]. P-glycoprotein (P-gp), an ATP-binding cassette efflux transporter, acts as a biological barrier [
37], limiting the absorption of drugs and natural compounds from the gut by pumping xenobiotics out of cells to protect against toxic substances [
37]. While substrates of P-gp are easily pumped out of cells, inhibitors of P-gp I/II can enhance the absorption and distribution of chemicals, leading to therapeutic or adverse effects. Epicatechin and epiafzelechin, functioning as P-glycoprotein substrates, might have their absorption affected. In contrast, lupeol, stigmasterol, and α-amyrin acetate, acting as P-glycoprotein inhibitors, exhibited low water solubility and poor intestinal absorption. These findings offer valuable insights for designing effective therapeutic regimens [
38]. Depending on the mode and type of P-gpI/II inhibition, lupeol, stigmasterol, and α-amyrin acetate could potentially facilitate the absorption of flavonoid compounds into cells when the ethyl acetate fraction is administered. Among the compounds, only epicatechin, with a recorded negative value, is predicted to have moderate Caco-2 absorption. Caco-2, a human colon epithelial cancer cell line, is modeled to simulate the properties of the human small intestine. It expresses enterocytes, transporters, cytochrome P450 enzymes, and efflux proteins [
37,
39], making it a gold standard for predicting in vitro human intestinal chemical permeability. All the isolated compounds show poor absorption through the skin, a transdermal route irrelevant in the administration of antidiabetic therapeutics.
The compounds’ potential for distribution within the body was evaluated using parameters such as steady-state volume of distribution (VDss) and compound-protein binding. Compounds with higher VDss values (VDss > 0.45), such as epicatechin and epiafzelechin, tend to distribute more in tissues than in plasma [
36], potentially enhancing therapeutic effects. The ability of lupeol, stigmasterol, and α-amyrin-acetate to bind to proteins in the blood can impact efficacy because the unbound fraction of a compound is the portion that can exert therapeutic effects [
40], as it is available to permeate through the cell membrane [
36,
37]. Strong protein binding may also lead to prolonged therapeutic effects. Furthermore, compounds like lupeol, stigmasterol, and α-amyrin-acetate, which can cross the blood-brain barrier [
33], raise concerns about potential neurological effects that may induce positive, negative, or toxic effects.
After absorption, the chemical compounds undergo metabolism in the liver, where cytochrome P450 isoenzymes (CYP1A2, CYP3A4, CYP2C9, CYP2C19, and CYP2D6) play a crucial role in drug safety, persistence, and bioactivation [
38]. None of the isolated compounds are predicted to be inhibitors of the CYP450 enzymes. This is favorable because inhibitors can block the substrate’s binding site, modify enzymatic activity, slow down metabolism, and lead to the accumulation of the substrate in the body [
40]. Lupeol, stigmasterol, and α-amyrin-acetate, however, are substrates of the CYP3A4 isoenzyme. As substrates, these compounds may be transformed into metabolites that could either be inactive for clearance or activated to produce beneficial or undesirable effects.
The amount of chemical compound removed from plasma in the vascular compartment per unit time is known as clearance [
37]. The total clearance score encompasses all hepatic and renal clearances of the compound excreted via the kidneys [
37,
41]. Stigmasterol exhibited the highest clearance score, indicating rapid elimination from the body. This suggests a shorter half-life and necessitates more frequent dosing to maintain therapeutic levels in the bloodstream. On the other hand, α-amyrin-acetate recorded a low clearance score, suggesting the compound is eliminated from the body at a relatively slow rate. This slower clearance results in a longer half-life, potentially requiring less frequent dosing to maintain therapeutic levels. Additionally, the renal uptake transporter Organic Cation Transporter 2 (OCT2) plays a vital role in drug disposition and renal clearance. None of the five compounds are substrates for OCT2, which is essential for the excretion of cationic molecules.
The toxicity below the detectable limit is a crucial factor when selecting a compound as a therapeutic candidate [
42]. The study evaluated the toxicity of potential therapeutic compounds using various parameters. The Maximum Tolerable Dose (MTD), estimating the highest dose at which a potential drug exhibits pharmacological activity without toxicity [
33,
36], predicted that all the compounds exhibited low MTD values (≤0.477 log mg/kg/day). Lupeol, stigmasterol, and α-amyrin-acetate had particularly unfavorable MTD values, specifying tolerated doses. The possibility of compounds causing adverse effects from repeated exposure over a long period of time in an oral rat chronic toxicity test [
43] was estimated. In the rat tests, lupeol and stigmasterol were predicted to cause adverse effects at low doses, known as the Lowest Observed Adverse Effect Level (LOAEL) [
33,
36]. None of the compounds was predicted to exert organ toxicity or mutagenicity. However, lupeol, stigmasterol, and α-amyrin-acetate were found to inhibit hERG II, indicating potential cardiotoxicity. The hERG (human ether-à-go-go-related gene) inhibition could lead to cardiac arrhythmias [
33]. This is because hERG encodes for a potassium channel with a fundamental role in cardiac action, the potential repolarization inhibition of which may lead to QT interval prolongation, ventricular tachycardia, and even death [
33]. These compounds were also predicted to exhibit immunotoxicity. Environmental impact predictions indicated moderate toxicity for most compounds against
T. pyriformis, with lupeol being relatively more toxic. The concentration that inhibited 50% growth (pIG
50) of
T. pyriformis with pIG
50 = −0.5 log µg/L is considered a toxic concentration [
44]. For minnow larvae, lupeol, stigmasterol, and α-amyrin-acetate were highly toxic. The concentration that caused the death of 50% of the Flathead minnows is considered highly toxic if LC
50 values are below 0.5 mM (log LC50 < 0.3) [
39]. The flavonoid compounds seemed more environmentally favorable compared to their non-flavonoid counterparts. Epiafzelechin and epicatechin were considered safer for oral consumption, while lupeol and stigmasterol were categorized as harmful if swallowed [
45]. To the best of our knowledge, apart from one study [
46] where epiafzelechin was among the metabolites docked against the PPARɣ receptor (PDB ID 2Q5S), no other study is available on the molecular docking of epiafzelechin against antidiabetic receptors. This study explored epiafzelechin’s potential in molecular docking against diabetes-related protein receptors. Epiafzelechin is a type B oligomer propelargonidin [
47], and pelargonidin and its glycosides have been demonstrated to possess antidiabetic potential by reducing hyperglycemia and glycation levels as well as stimulating insulin secretion in rodent pancreatic β-cells in vitro [
48,
49,
50], inferring the possible bioactivity of epiafzelechin. The molecular docking study could not unravel the potential activity of epiafzelechin in the same manner it failed with conventional therapeutics, indicating its limitations. However, this study suggests that epiafzelechin alone or in synergy with other compounds could be considered a potential drug candidate for diabetes treatment.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}