
Urea-Functionalized Heterocycles: Structure, Hydrogen Bonding and Applications


Abstract
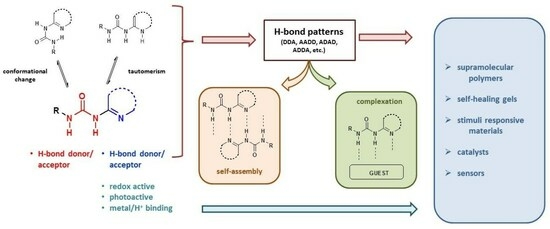
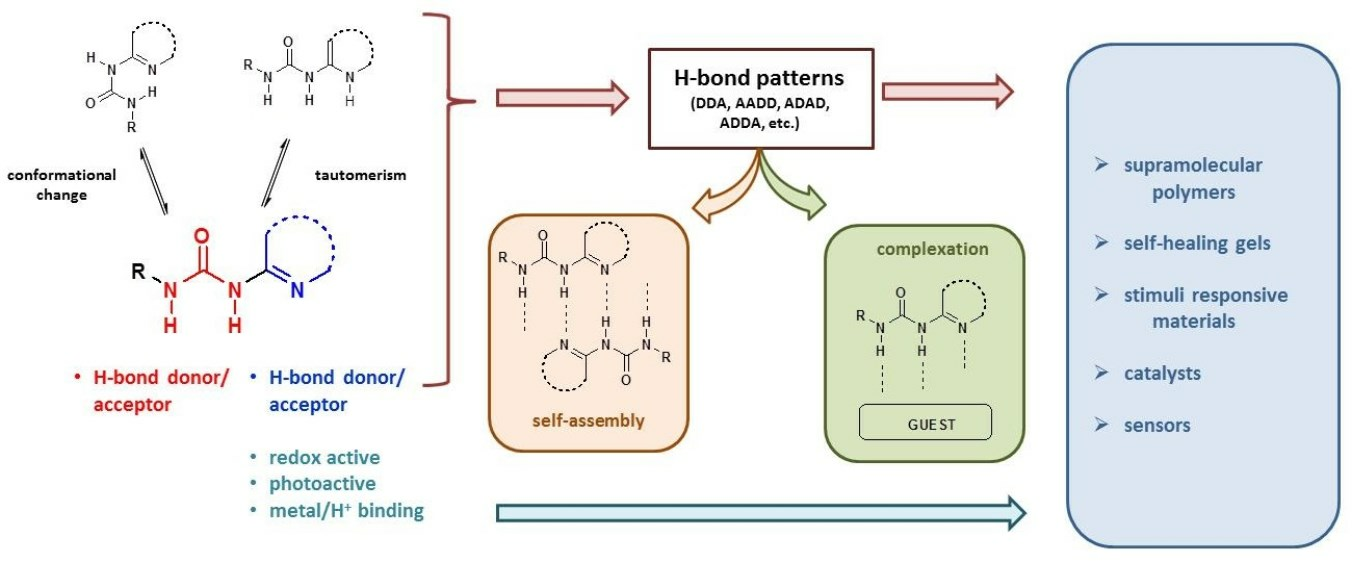
1. Introduction
2. Hydrogen Bonding, Self-Assembly and Complexation with Neutral Guests
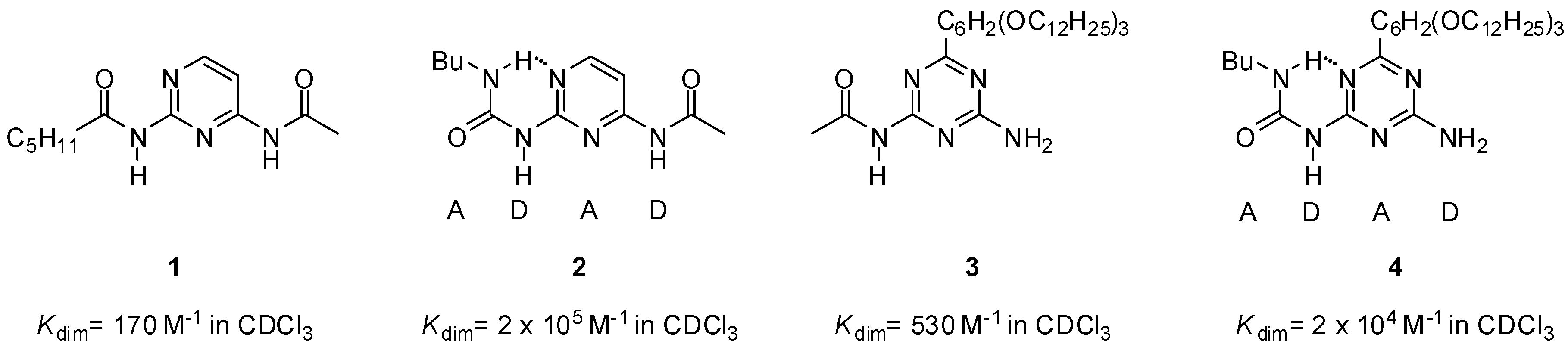
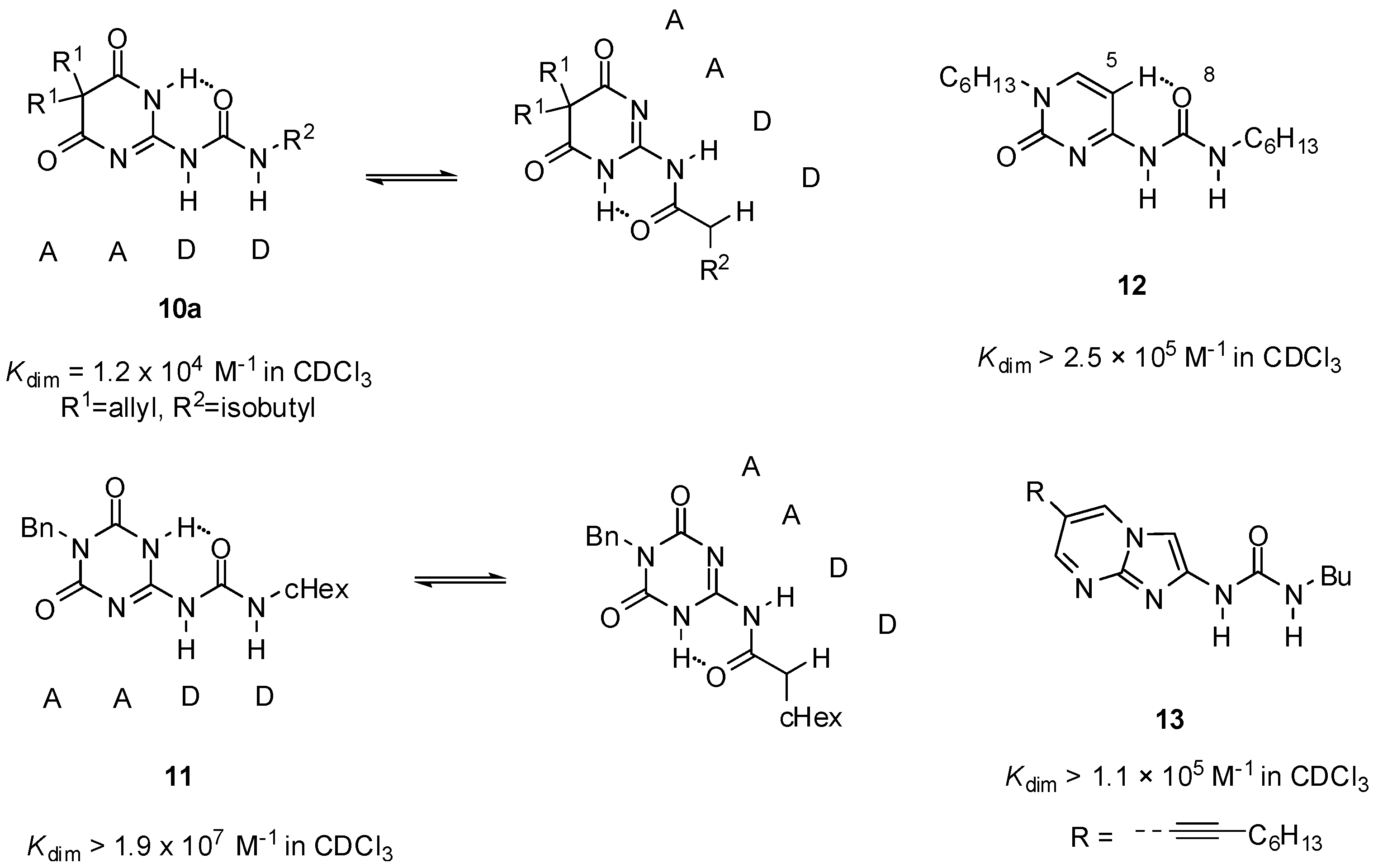
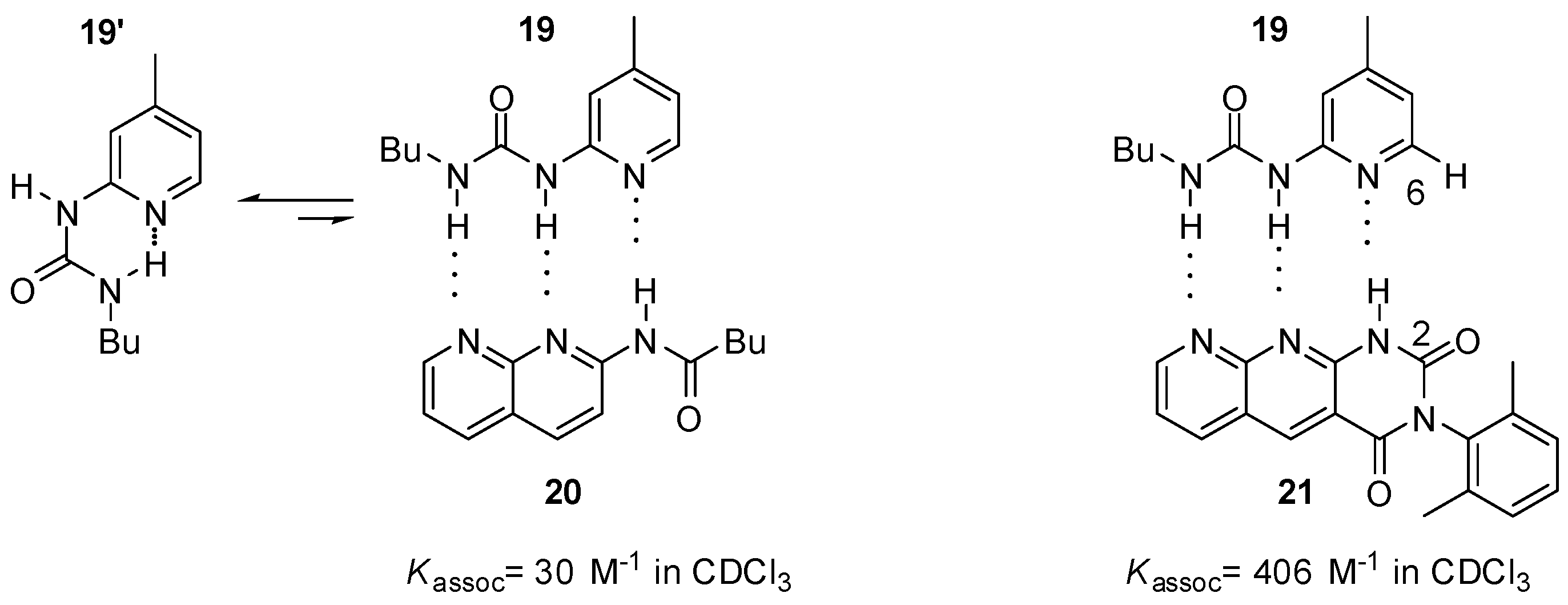
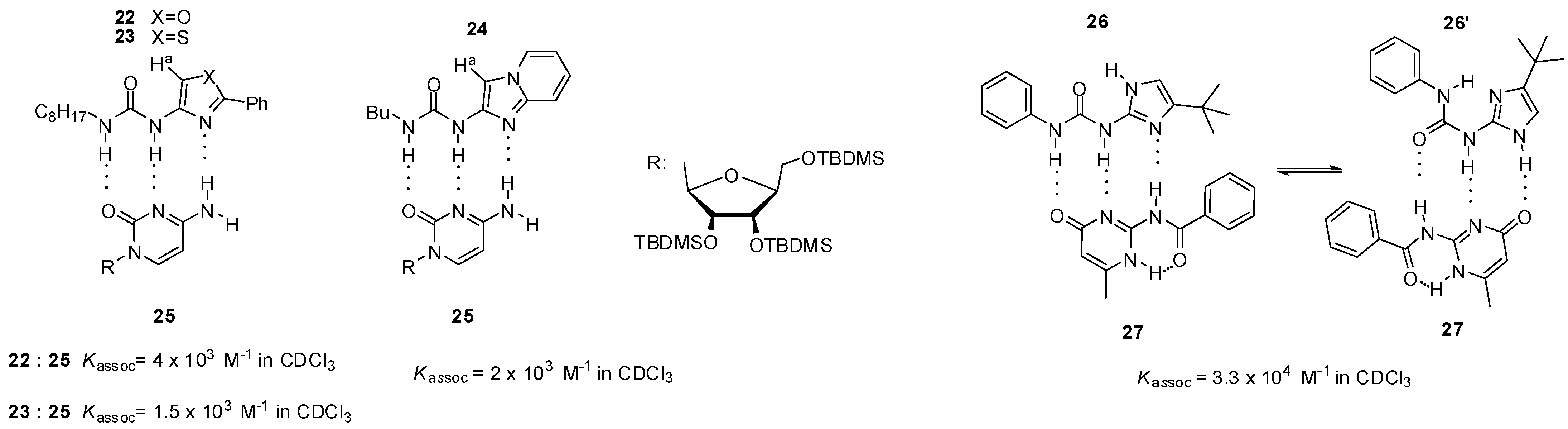
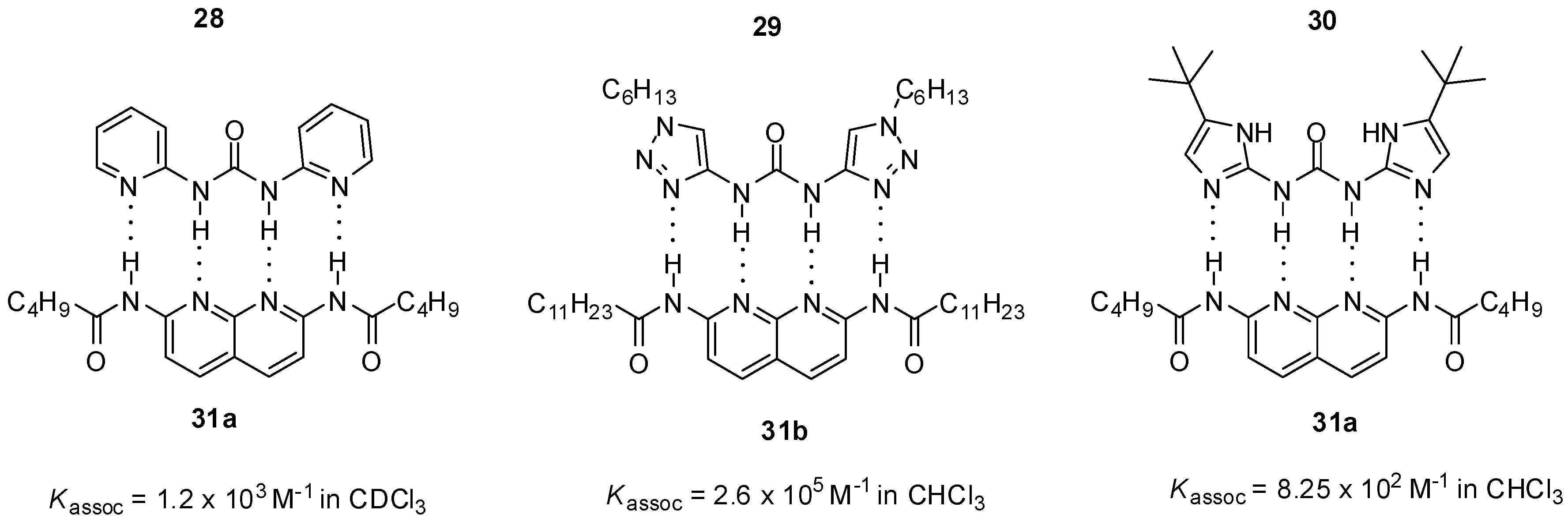
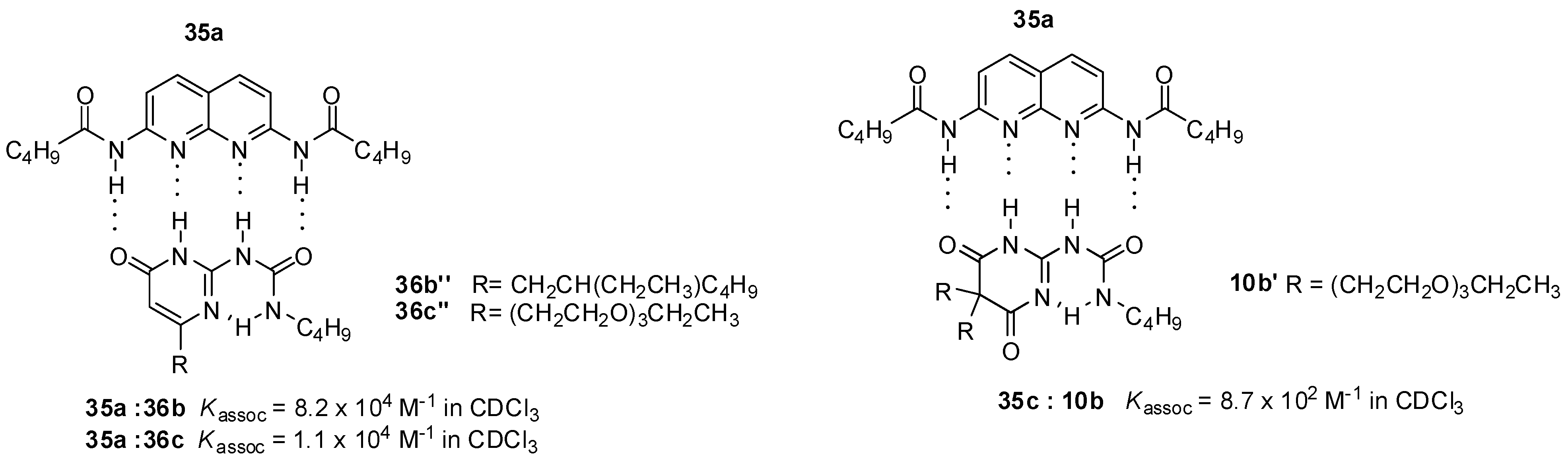
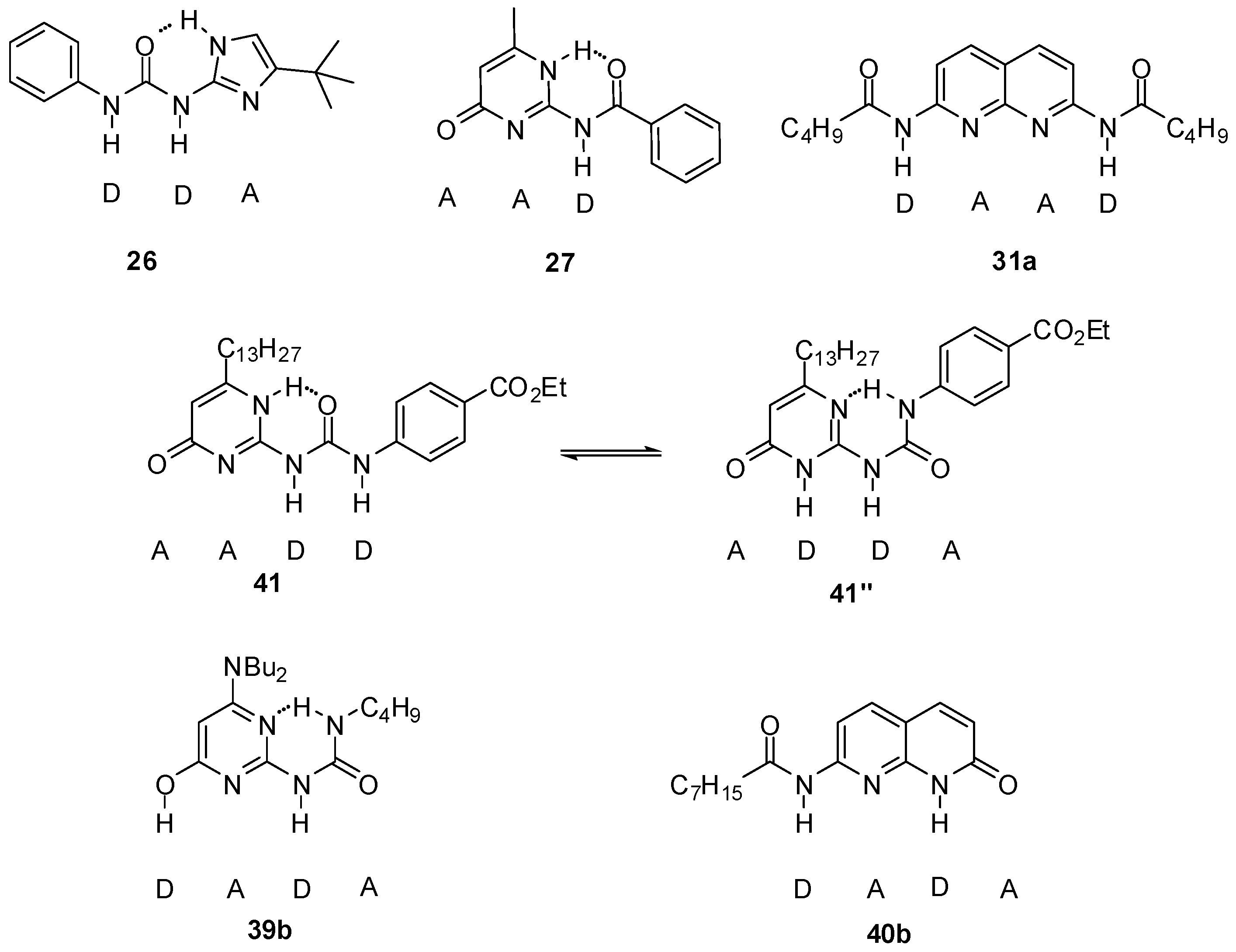
2.1. Formation of Homodimers
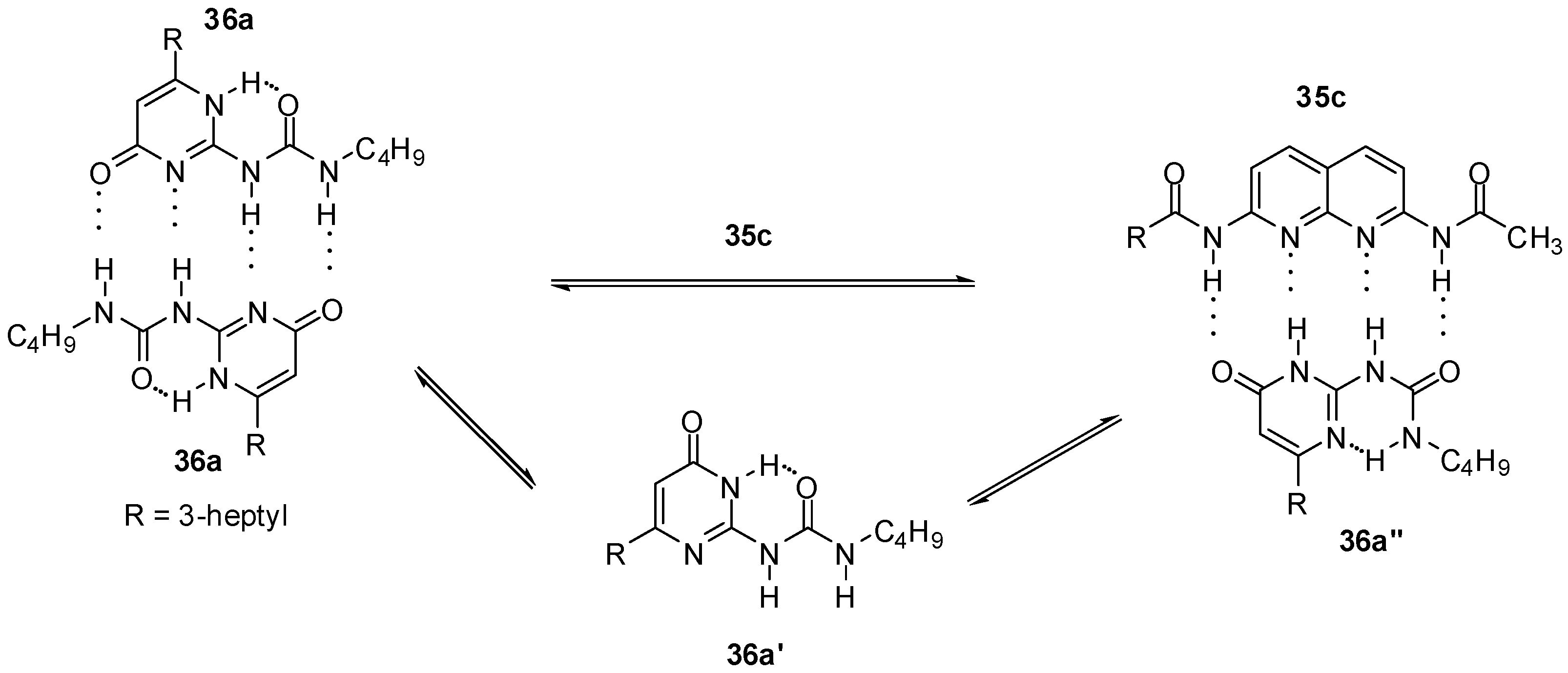
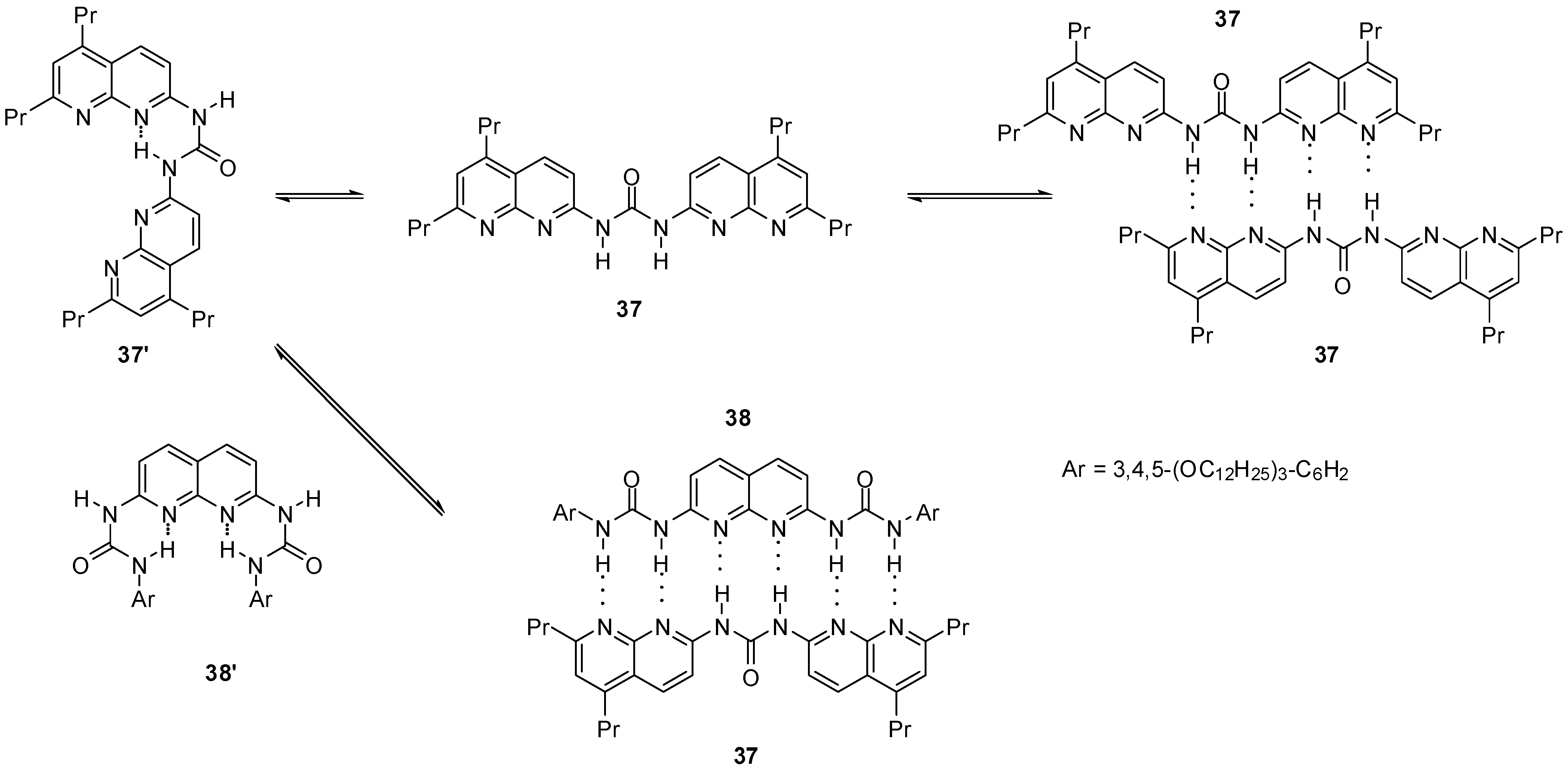
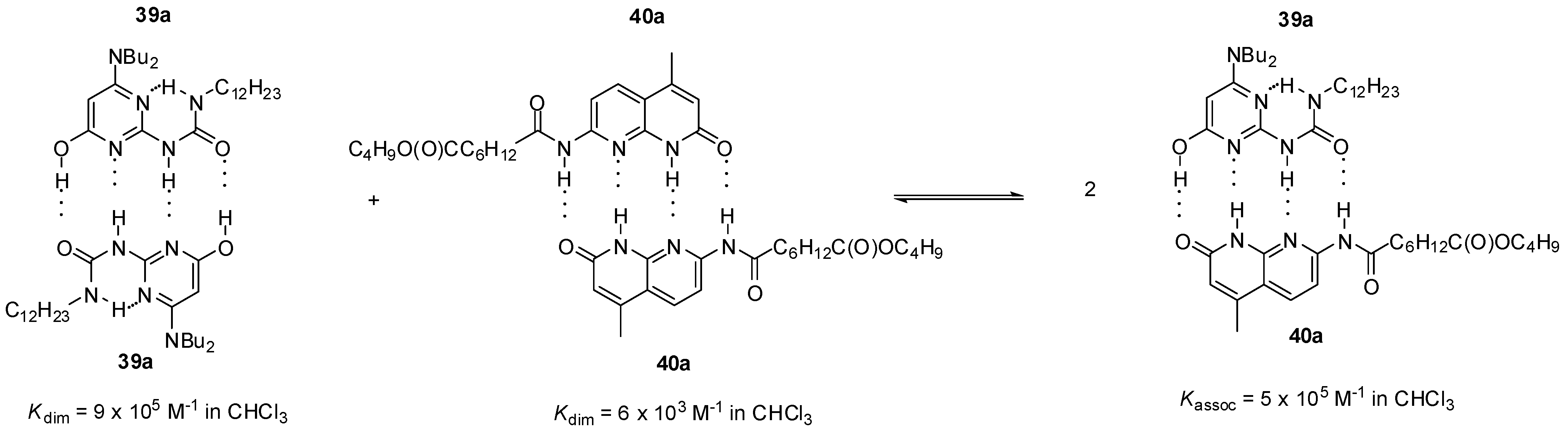
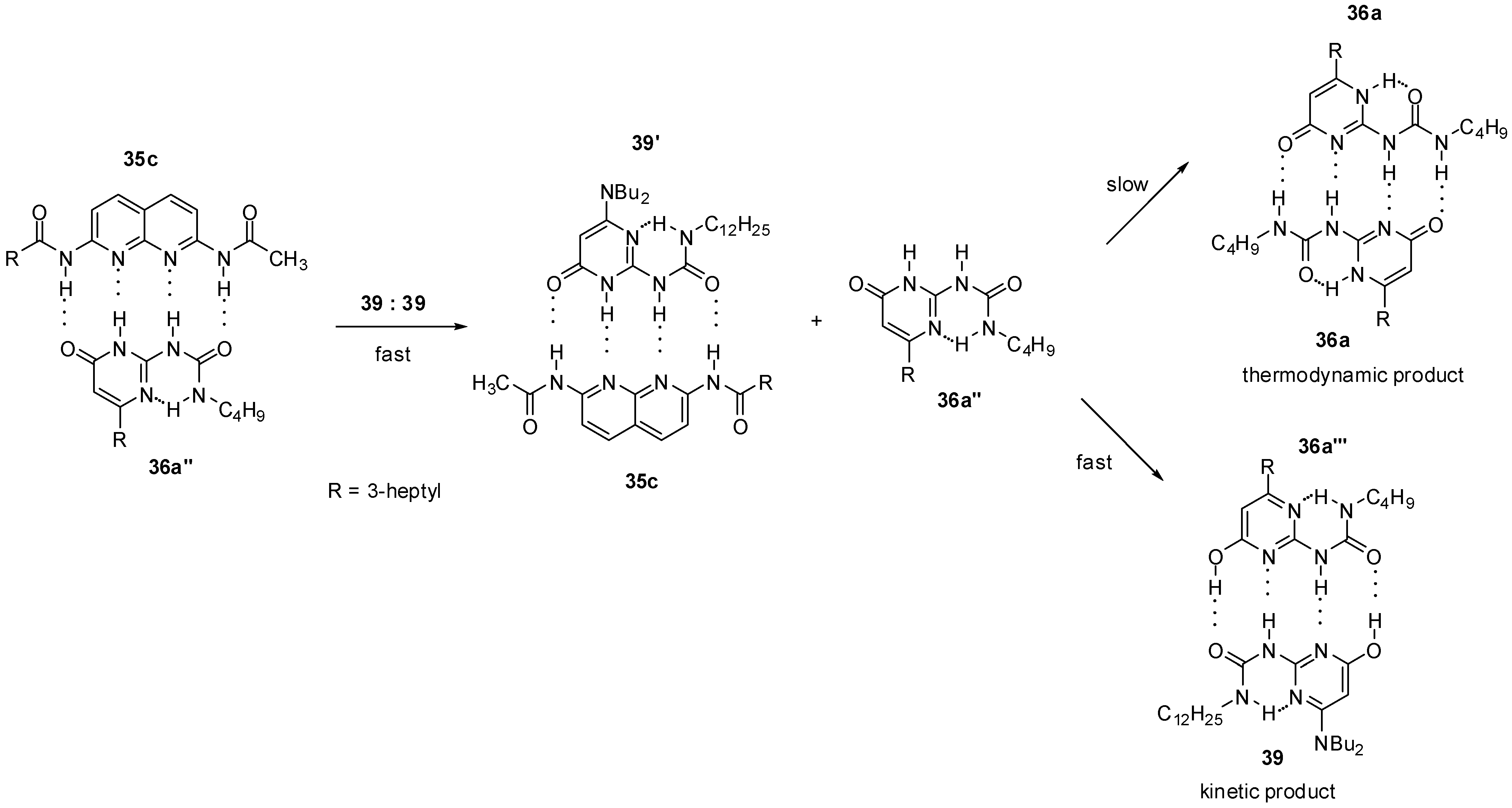
2.2. Formation of Heterodimers
2.3. Incorporation of Stimuli-Responsive Moieties into Urea Derivatives Functionalized by Heteroaromatic Groups
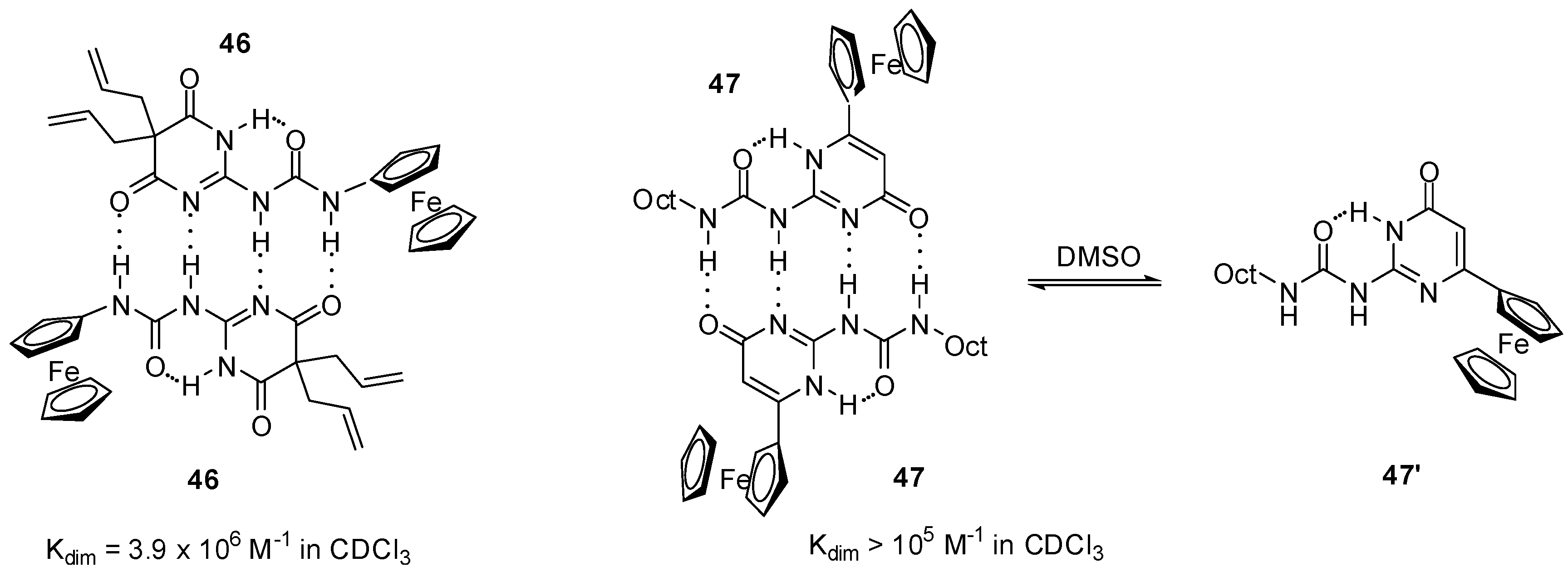
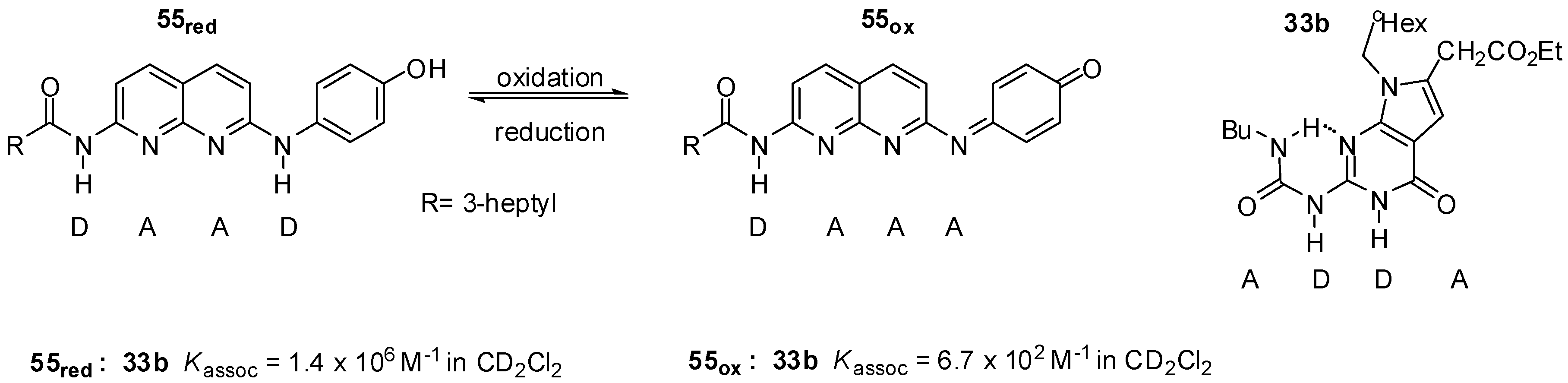
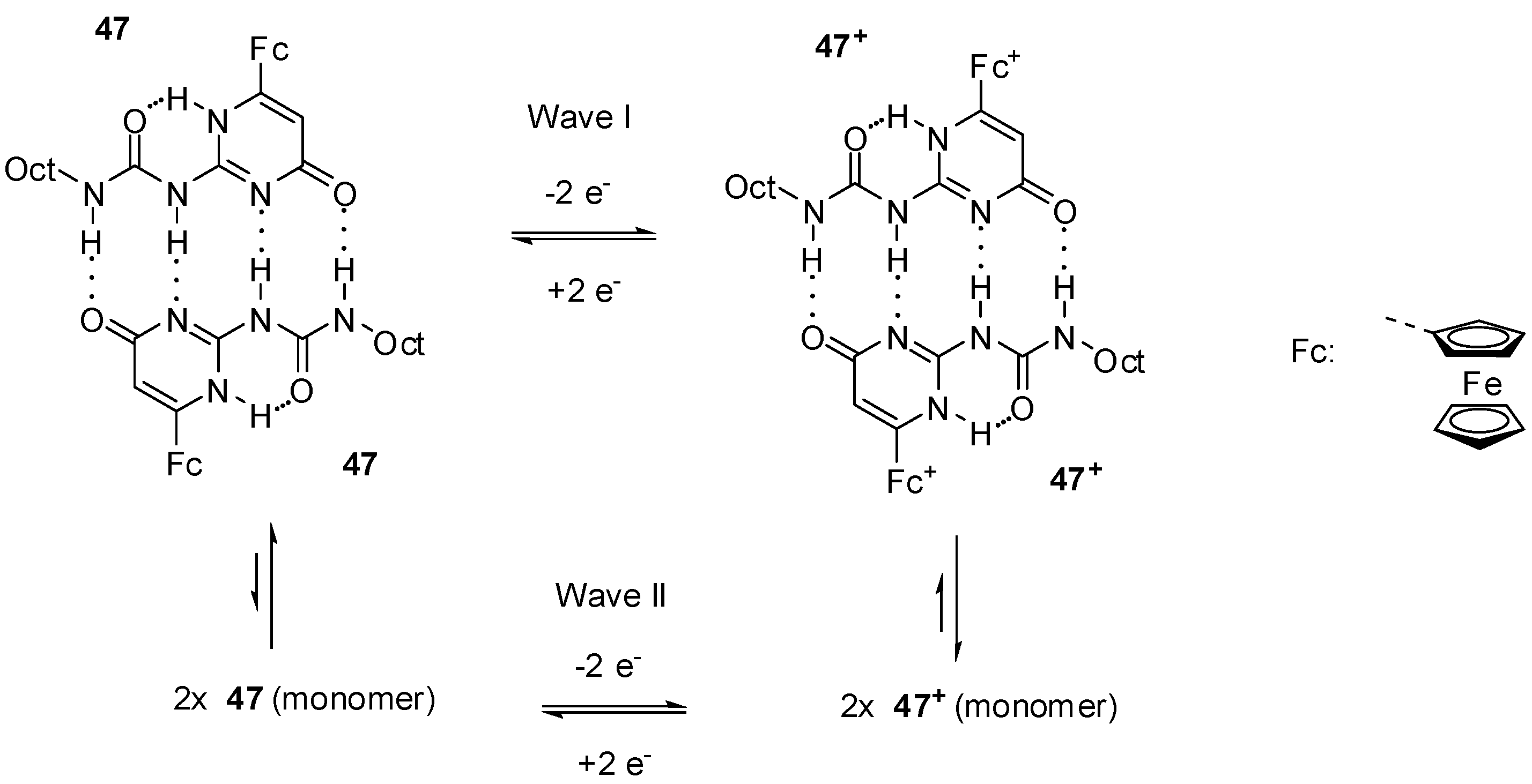
2.3.1. Redox-Active Urea Derivatives
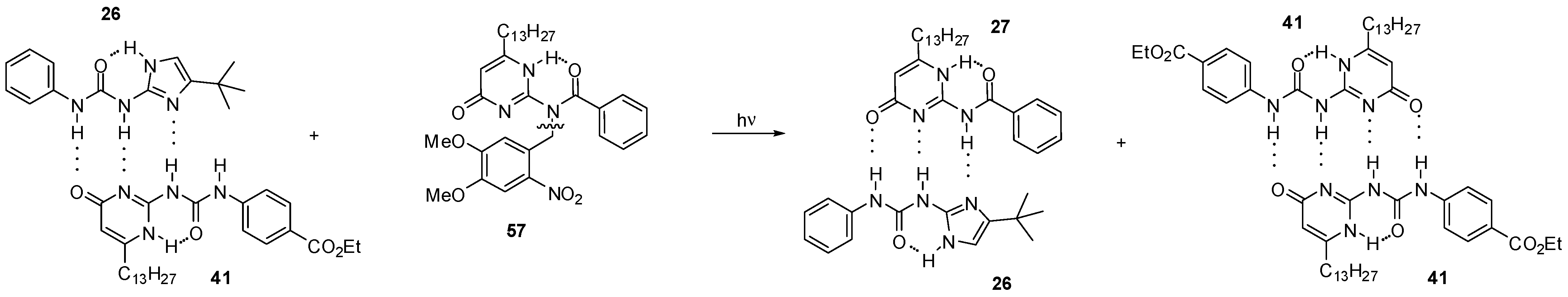
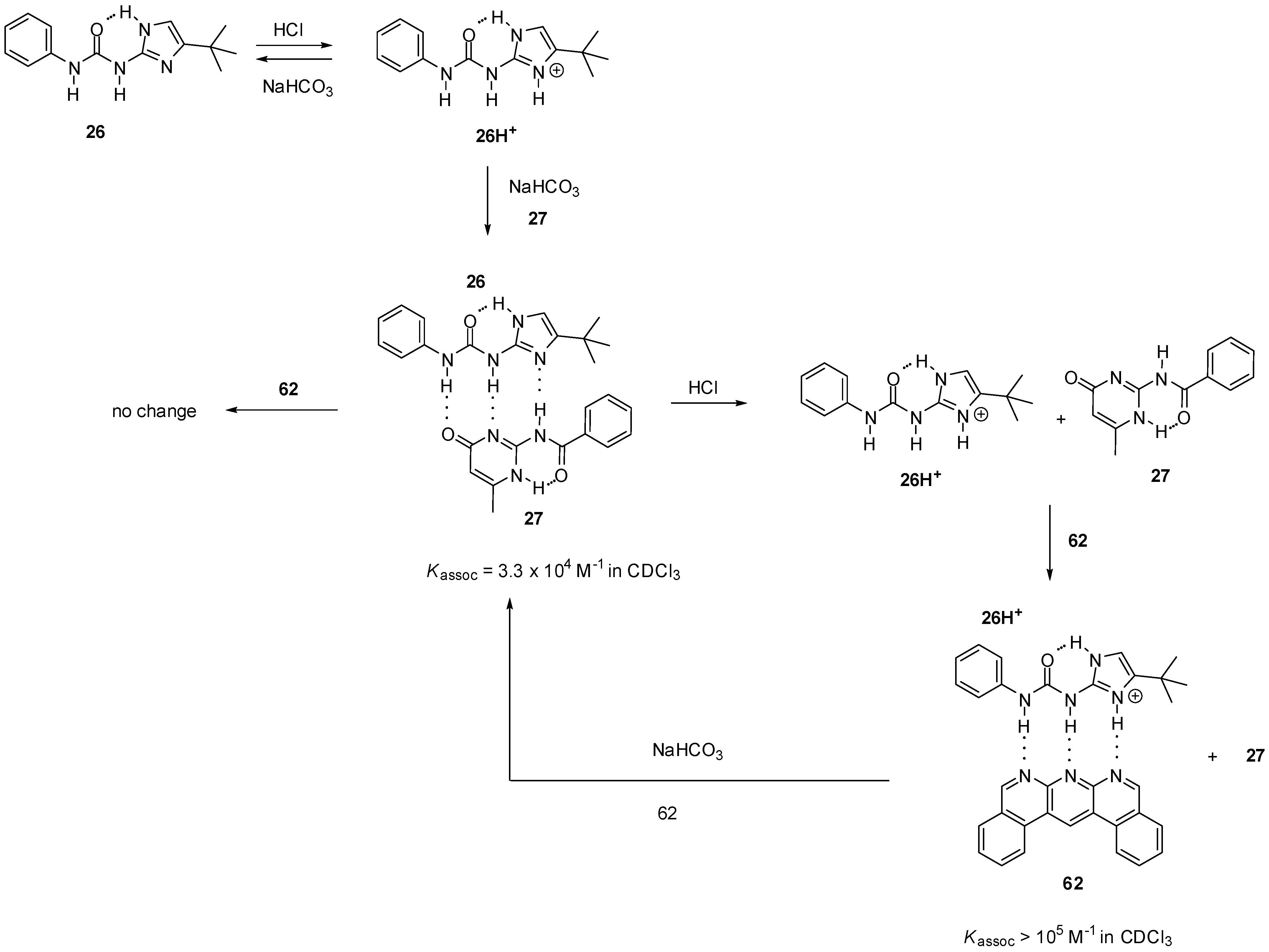
2.3.2. Photo-Switchable Derivatives
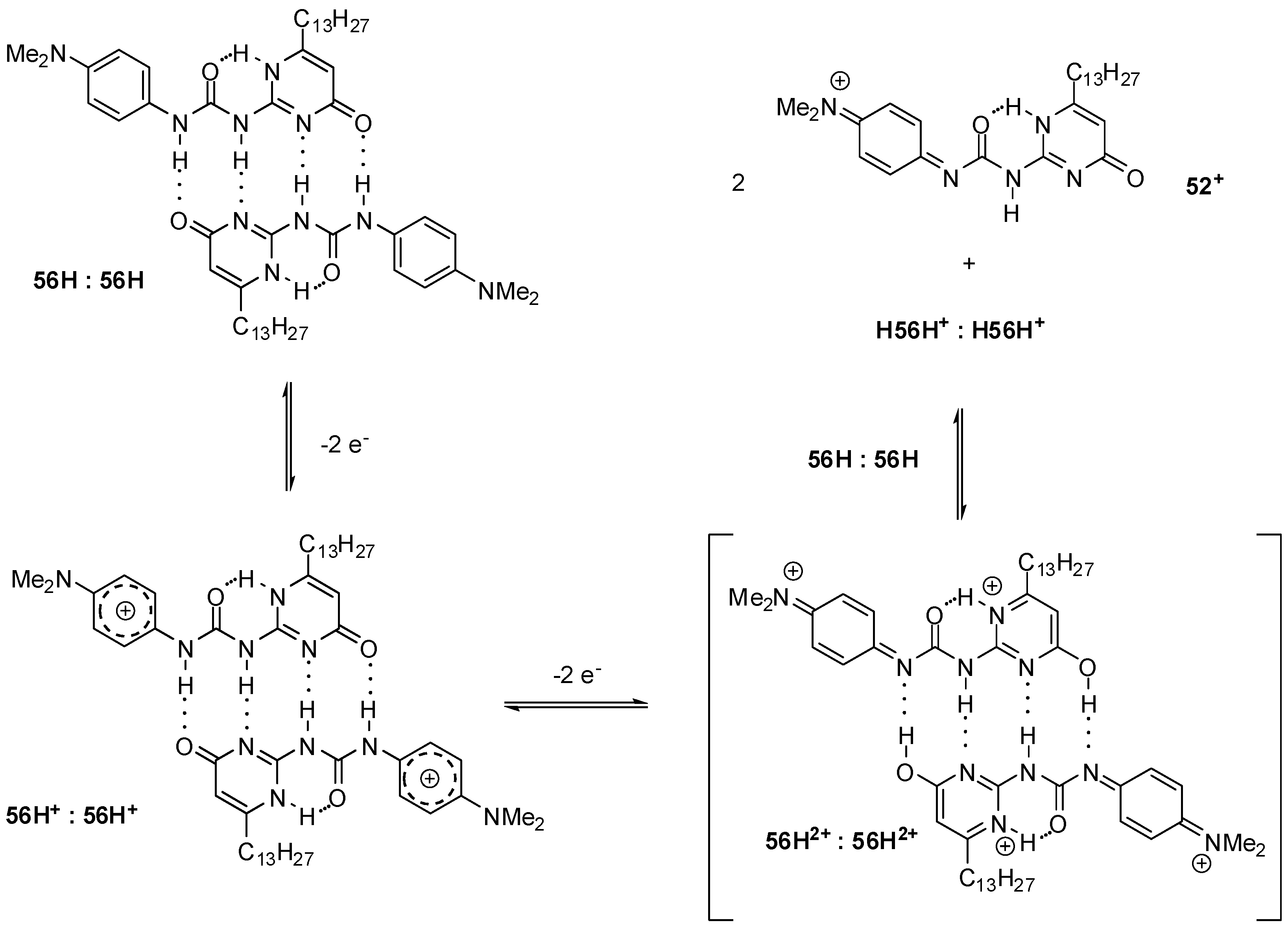
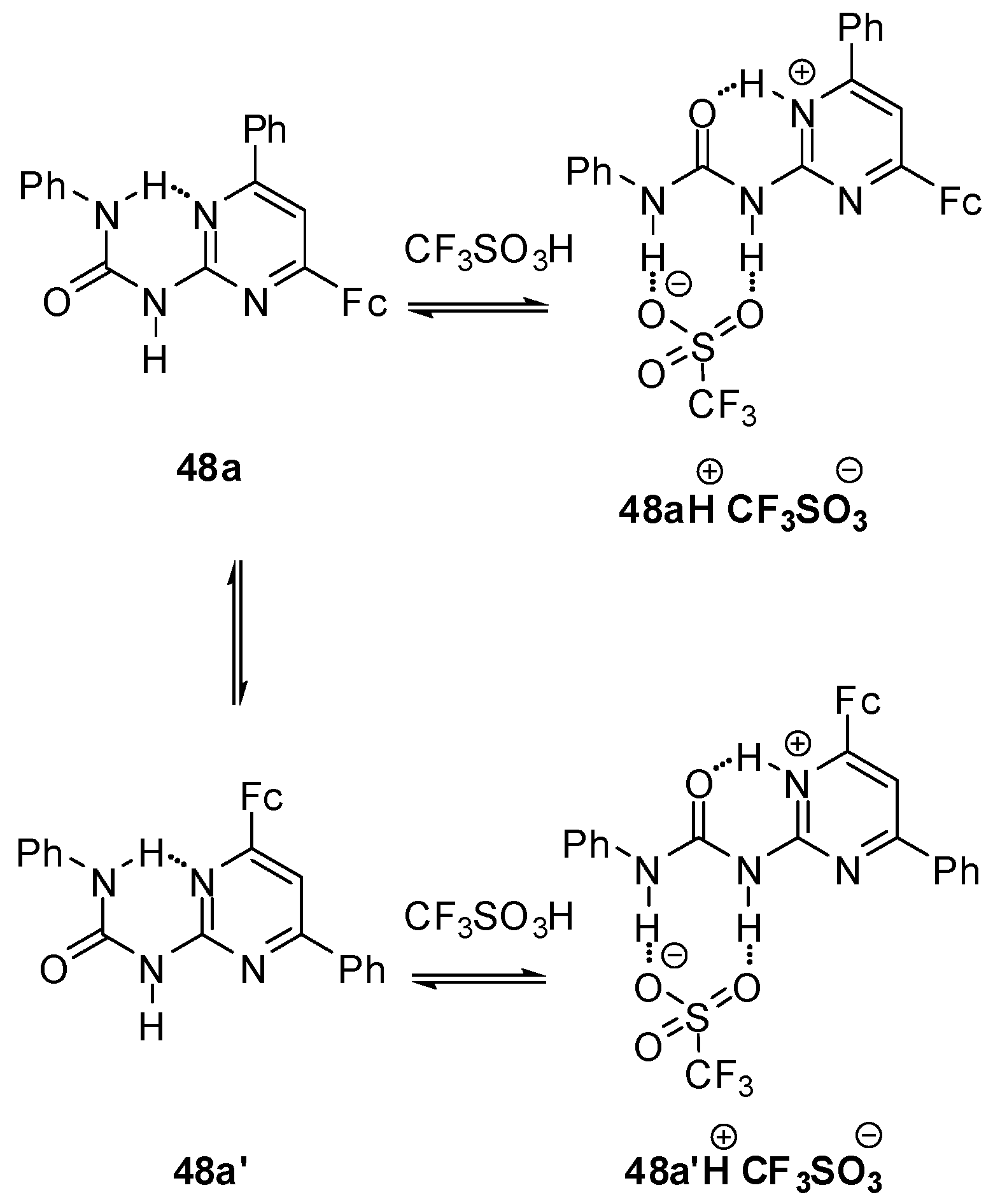
2.3.3. pH-Switchable Derivatives
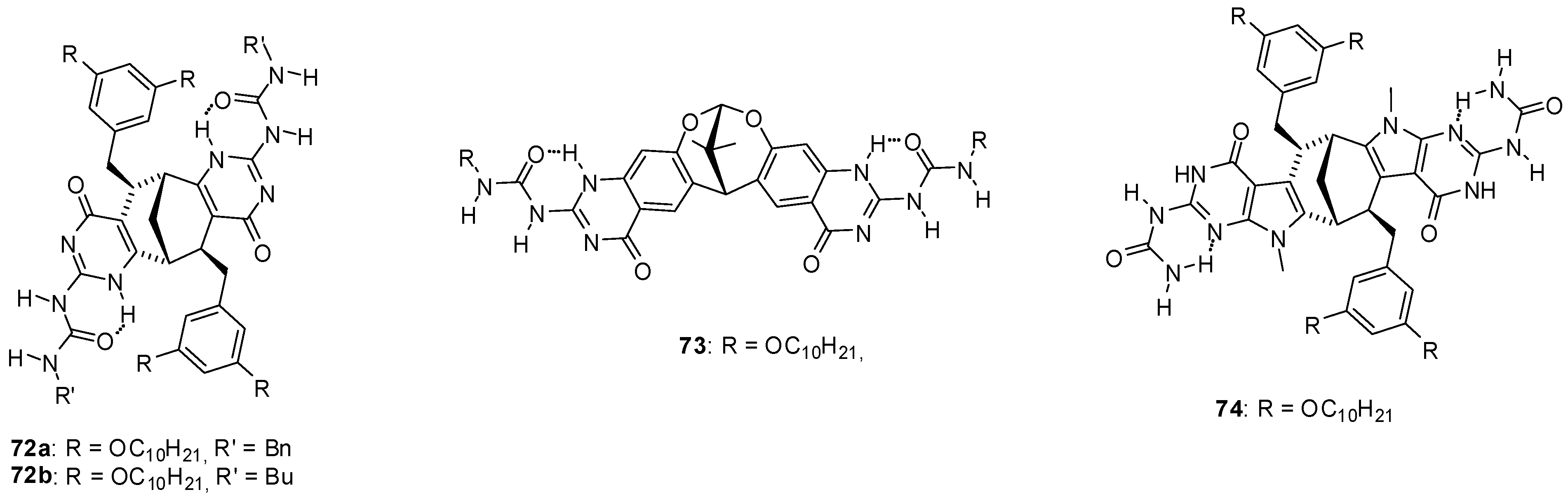
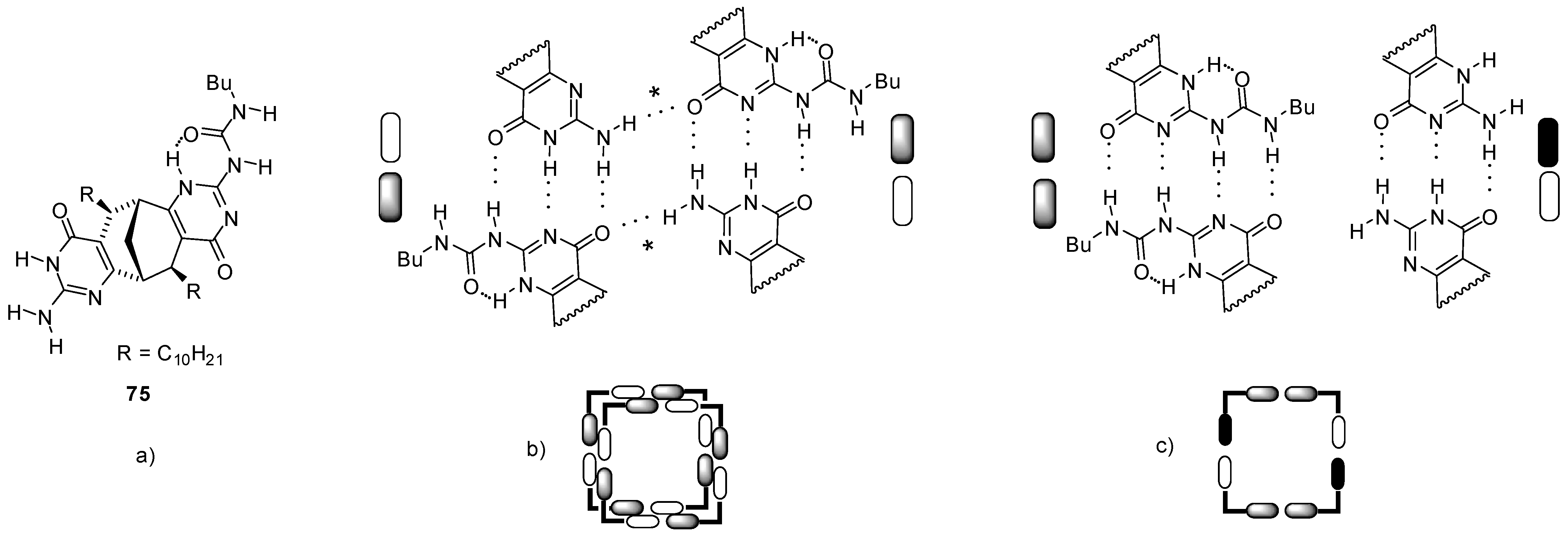
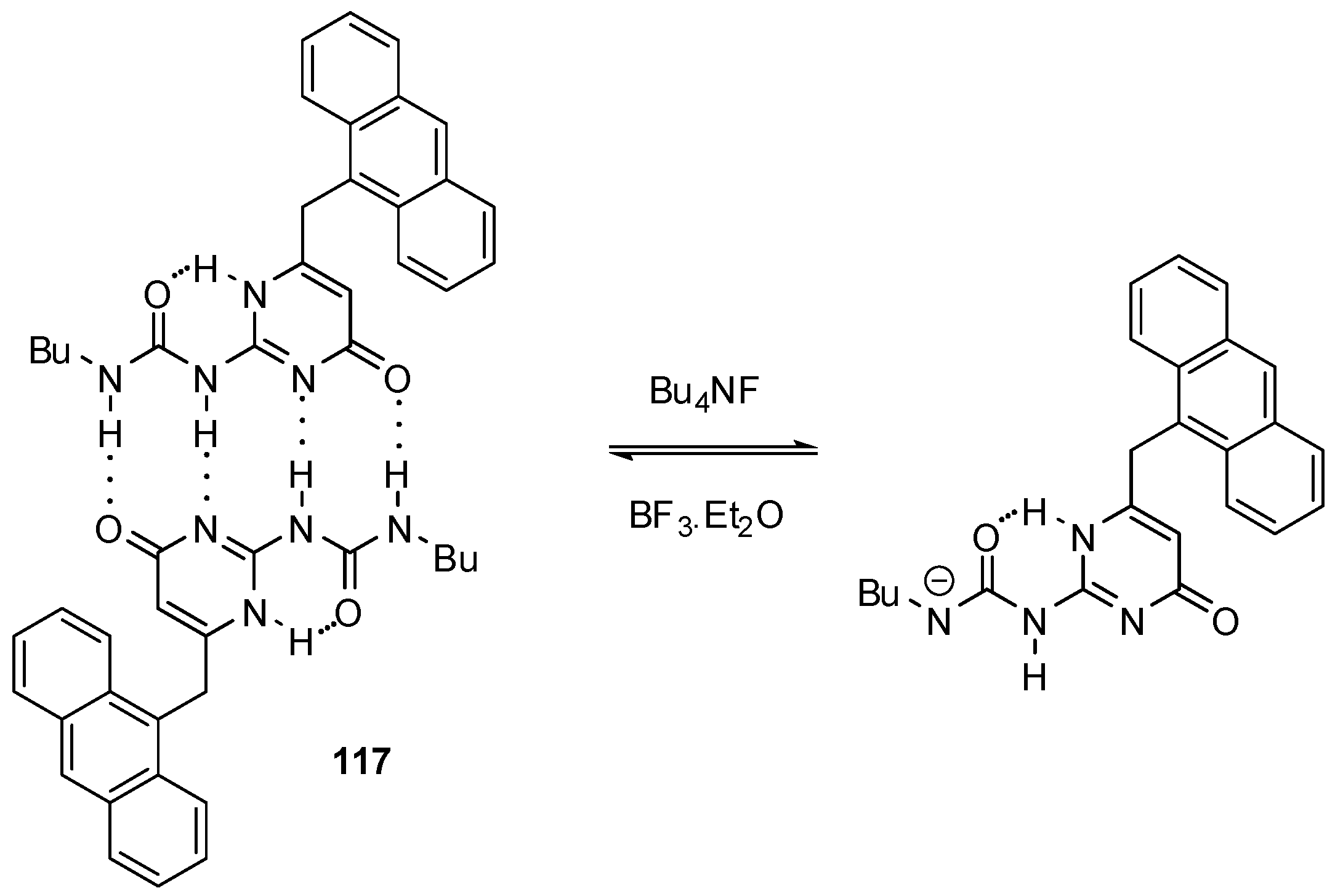
3. Formation of Different Molecular Architectures via Supramolecular Assembly
4. Applications Exploiting the H-Bonding Ability of Ureido-Heterocycles
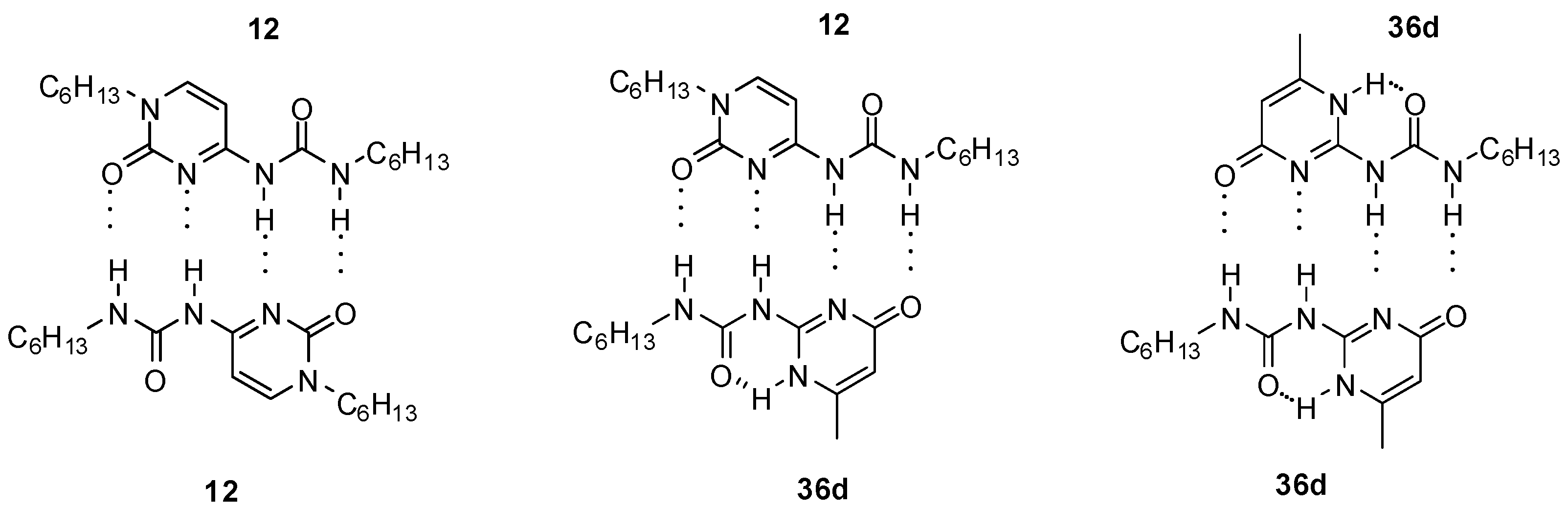
4.1. Supramoleular Polymers
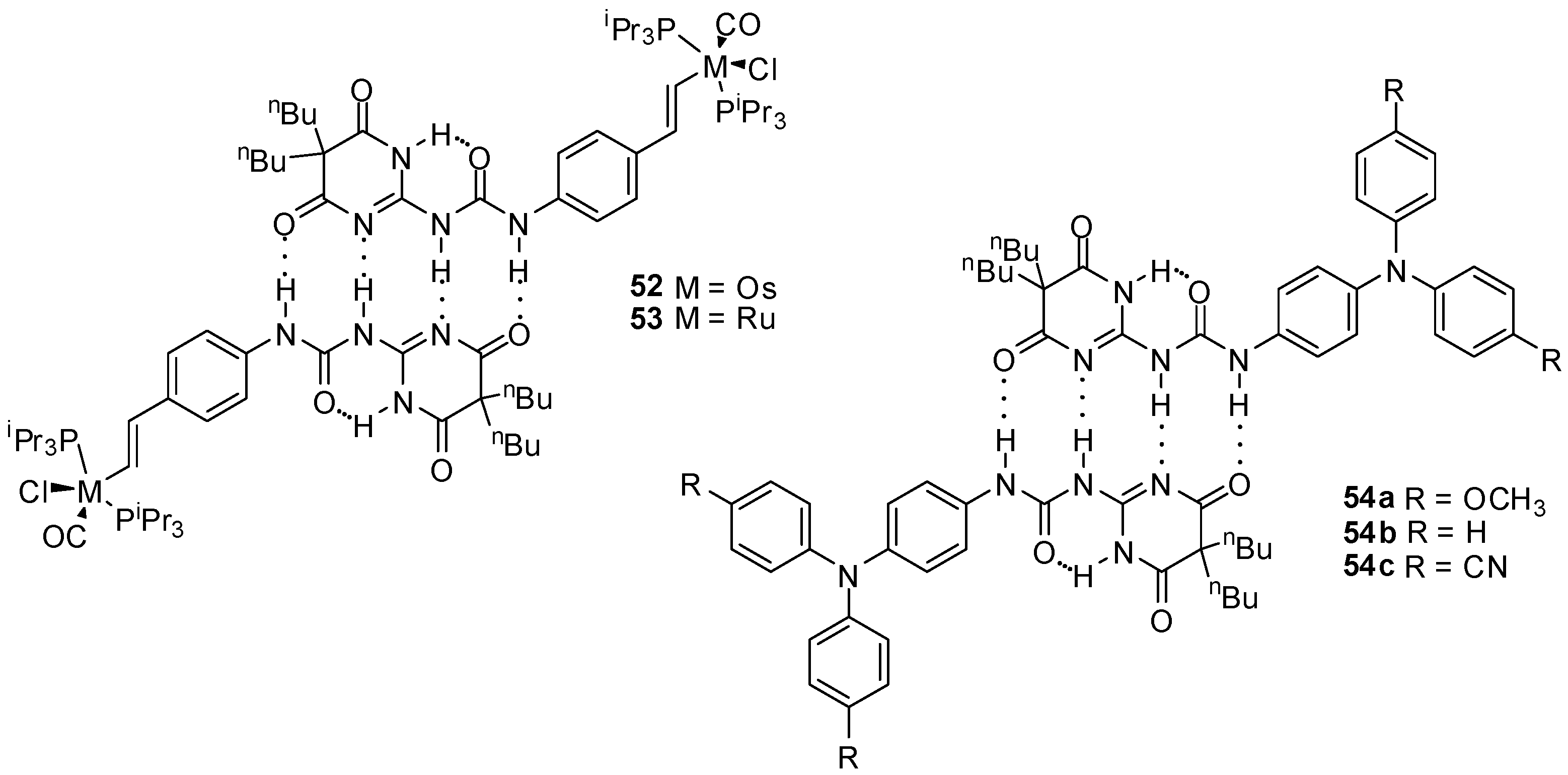
4.2. Electrochemical and Photochemical Applications
4.3. Biomedical Applications
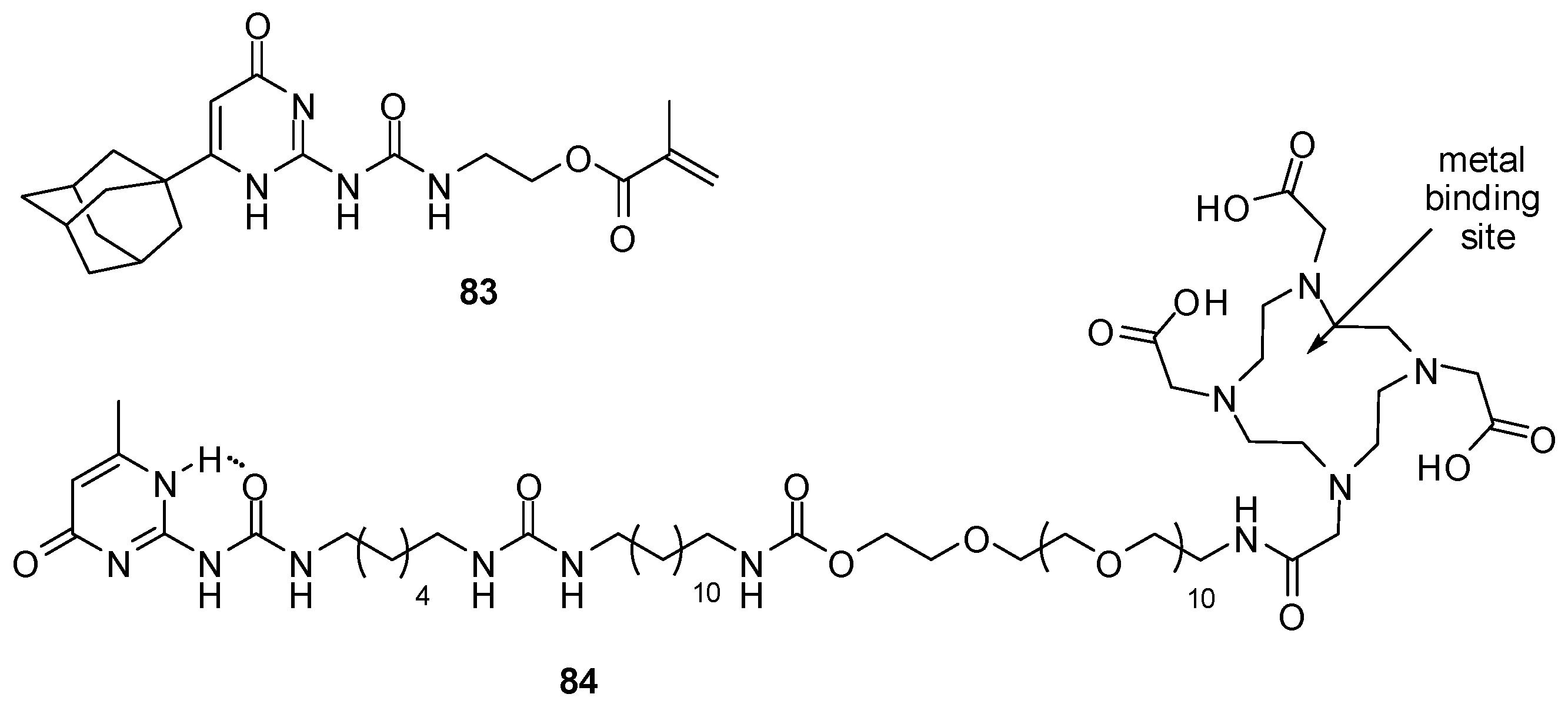
4.4. Applications in Catalysis
4.5. Applications in Sensors
5. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Jorgensen, W.L.; Pranata, J. Importance of secondary interactions in triply hydrogen bonded complexes: Guanine-cytosine vs uracil-2,6-diaminopyridine. J. Am. Chem. Soc. 1990, 112, 2008–2010. [Google Scholar] [CrossRef]
- Pranata, J.; Wierschke, S.G.; Jorgensen, W.L. OPLS potential functions for nucleotide bases. Relative association constants of hydrogen-bonded base pairs in chloroform. J. Am. Chem. Soc. 1991, 113, 2810–2819. [Google Scholar] [CrossRef]
- Murray, T.J.; Zimmerman, S.C. New triply hydrogen bonded complexes with highly variable stabilities. J. Am. Chem. Soc. 1992, 114, 4010–4011. [Google Scholar] [CrossRef]
- Zimmerman, S.C.; Murray, T.J. Hydrogen bonded complexes with the AA·DD, AA·DDD, and AAA·DD motifs: The role of three centered (bifurcated) hydrogen bonding. Tetrahedron Lett. 1994, 35, 4077–4080. [Google Scholar] [CrossRef]
- Quinn, J.R.; Zimmerman, S.C. With regard to the hydrogen bonding in complexes of pyridylureas, less is more. A role for shape complementarity and CH···O interactions? Org. Lett. 2004, 6, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.R.; Zimmerman, S.C.; Del Bene, J.E.; Shavitt, I. Does the A·T or G·C base-pair possess enhanced stability? Quantifying the effects of CH···O interactions and secondary interactions on base-pair stability using a phenomenological analysis and ab initio calculations. J. Am. Chem. Soc. 2007, 129, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.C.; Panunto, T.W. 1,3-Bis(m-nitrophenyl)urea: An exceptionally good complexing agent for proton acceptors. J. Am. Chem. Soc. 1988, 110, 5896–5897. [Google Scholar] [CrossRef]
- Etter, M.C.; Urbanczyk-Lipkowska, Z.; Zia-Ebrahimi, M.; Panunto, T.W. Hydrogen bond-directed cocrystallization and molecular recognition properties of diarylureas. J. Am. Chem. Soc. 1990, 112, 8415–8426. [Google Scholar] [CrossRef]
- Smith, P.J.; Reddington, M.V.; Wilcox, C.S. Ion pair binding by a urea in chloroform solution. Tetrahedron Lett. 1992, 33, 6085–6088. [Google Scholar] [CrossRef]
- Fan, E.; Van Arman, S.A.; Kincaid, S.; Hamilton, A.D. Molecular recognition: Hydrogen-bonding receptors that function in highly competitive solvents. J. Am. Chem. Soc. 1993, 115, 369–370. [Google Scholar] [CrossRef]
- Beijer, F.H.; Sijbesma, R.P.; Kooijman, H.; Spek, A.L.; Meijer, E.W. Strong dimerization of ureidopyrimidones via quadruple hydrogen bonding. J. Am. Chem. Soc. 1998, 120, 6761–6769. [Google Scholar] [CrossRef]
- Sijbesma, R.P.; Meijer, E.W. Quadruple hydrogen bonded systems. Chem. Commun. 2003, 5–16. [Google Scholar] [CrossRef]
- Baruah, P.K.; Khan, S. Self-complementary quadruple hydrogen bonding motifs: From design to function. RSC Adv. 2013, 3, 21202–21217. [Google Scholar] [CrossRef]
- Pei, Q.; Ding, A. The design and application of quadruple hydrogen bonded systems. Prog. Chem. 2019, 31, 258–274. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, Z.; Li, M. Recent developments: Self-healing polymers based on quadruple hydrogen bonds. E3S Web Conf. 2021, 290, 01037. [Google Scholar] [CrossRef]
- Verjans, J.; Hoogenboom, R. Supramolecular polymer materials based on ureidopyrimidinone quadruple hydrogen bonding units. Prog. Polym. Sci. 2023, 142, 101689. [Google Scholar] [CrossRef]
- Yokoya, M.; Kimura, S.; Yamanaka, M. Urea Derivatives as functional molecules: Supramolecular capsules, supramolecular polymers, supramolecular gels, artificial hosts, and catalysts. Chem. Eur. J. 2021, 27, 5601–5614. [Google Scholar] [CrossRef]
- Huo, Y.; He, Z.; Wang, C.; Zhang, L.; Xuan, Q.; Wei, S.; Wang, Y.; Pan, D.; Dong, B.; Wen, R.; et al. The recent progress of synergistic supramolecular polymers: Preparation, properties and applications. Chem. Commun. 2021, 57, 1413–1429. [Google Scholar] [CrossRef]
- Sun, P.; Qin, B.; Xu, J.F.; Zhang, X. Supramonomers for controllable supramolecular polymerization and renewable supramolecular polymeric materials. Prog. Polym. Sci. 2022, 124, 101486. [Google Scholar] [CrossRef]
- O’Donnell, A.D.; Salimi, S.; Hart, L.R.; Babra, T.S.; Greenland, B.W.; Hayes, W. Applications of supramolecular polymer networks. React. Funct. Polym. 2022, 172, 105209. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, Q.; Qu, D.H. Emerging hydrogen-bond design for high-performance dynamic polymeric materials. ACS Mater. Lett. 2023, 5, 480–490. [Google Scholar] [CrossRef]
- Peng, H.Q.; Zhu, W.; Guo, W.J.; Li, Q.; Ma, S.; Bucher, C.; Liu, B.; Ji, X.; Huang, F.; Sessler, J.L. Supramolecular polymers: Recent advances based on the types of underlying interactions. Prog. Polym. Sci. 2023, 137, 101635. [Google Scholar] [CrossRef]
- Brunsveld, L.; Folmer, B.J.B.; Meijer, E.W.; Sijbesma, R.P. Supramolecular polymers. Chem. Rev. 2001, 101, 4071–4098. [Google Scholar] [CrossRef]
- Wilson, A.J. Non-covalent polymer assembly using arrays of hydrogen-bonds. Soft Matter 2007, 3, 409–425. [Google Scholar] [CrossRef]
- Dankers, P.Y.W.; Meijer, E.W. Supramolecular biomaterials. A modular approach towards tissue engineering. Bull. Chem. Soc. Jpn. 2007, 80, 2047–2073. [Google Scholar] [CrossRef]
- Beijer, F.H.; Kooijman, H.; Spek, A.L.; Sijbesma, R.P.; Meijer, E.W. Self-complementarity achieved through quadruple hydrogen bonding. Angew. Chem. Int. Ed. 1998, 37, 75–78. [Google Scholar] [CrossRef]
- Sijbesma, R.P.; Beijer, F.H.; Brunsveld, L.; Folmer, B.J.; Hirschberg, J.H.; Lange, R.F.; Lowe, J.K.; Meijer, E.W. Reversible polymers formed from self-complementary monomers using quadruple hydrogen bonding. Science 1997, 278, 1601–1604. [Google Scholar] [CrossRef]
- Söntjens, S.H.M.; Sijbesma, R.P.; van Genderen, M.H.P.; Meijer, E.W. Stability and lifetime of quadruply hydrogen bonded 2-ureido-4[1H]-pyrimidinone dimers. J. Am. Chem. Soc. 2000, 122, 7487–7493. [Google Scholar] [CrossRef]
- de Greef, T.F.A.; Ercolani, G.; Ligthart, G.B.W.L.; Meijer, E.W.; Sijbesma, R.P. Influence of selectivity on the supramolecular polymerization of AB-type polymers capable of both A·A and A·B interactions. J. Am. Chem. Soc. 2008, 130, 13755–13764. [Google Scholar] [CrossRef] [PubMed]
- de Greef, T.F.A.; Ligthart, G.W.B.L.; Lutz, M.; Spek, A.L.; Meijer, E.W.; Sijbesma, R.P. The Mechanism of Ureido-Pyrimidinone: 2,7-Diamido-naphthyridine complexation and the presence of kinetically controlled pathways in multicomponent hydrogen-bonded systems. J. Am. Chem. Soc. 2008, 130, 5479–5486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- de Greef, T.F.A.; Nieuwenhuizen, M.M.L.; Sijbesma, R.P.; Meijer, E.W. Competitive intramolecular hydrogen bonding in oligo(ethylene oxide) substituted quadruple hydrogen bonded systems. J. Org. Chem. 2010, 75, 598–610. [Google Scholar] [CrossRef]
- Kheria, S.; Rayavarapu, S.; Kotmaleb, A.S.; Sanjayan, G.J. Three in one: Prototropy-free highly stable AADD-type self-complementary quadruple hydrogen-bonded molecular duplexes with a built-in fluorophore. Chem. Commun. 2017, 53, 2689–2692. [Google Scholar] [CrossRef]
- Corbin, P.S.; Zimmerman, S.C. Self-association without regard to prototropy. A heterocycle that forms extremely stable quadruply hydrogen-bonded dimers. J. Am. Chem. Soc. 1998, 120, 9710–9711. [Google Scholar] [CrossRef]
- Corbin, P.S.; Zimmerman, S.C.; Thiessen, P.A.; Hawryluk, N.A.; Murray, T.J. Complexation-induced unfolding of heterocyclic ureas. simple foldamers equilibrate with multiply hydrogen-bonded sheetlike structures. J. Am. Chem. Soc. 2001, 123, 10475–10488. [Google Scholar] [CrossRef] [PubMed]
- Lüning, U.; Kühl, C.; Uphoff, A. Four Hydrogen Bonds—DDAA, DADA, DAAD and ADDA hydrogen bond motifs. Eur. J. Org. Chem. 2002, 23, 4063–4070. [Google Scholar] [CrossRef]
- Baruah, P.K.; Gonnade, R.; Phalgune, U.D.; Sanjayan, G.J. Self-assembly with degenerate prototropy. J. Org. Chem. 2005, 70, 6461–6467. [Google Scholar] [CrossRef]
- Kheria, S.; Rayavarapu, S.; Kotmale, A.S.; Gonnade, R.G.; Sanjayan, G.J. Triazine-based highly stable AADD-type self-complementary quadruple hydrogen-bonded systems devoid of prototropy. Chem. Eur. J. 2017, 23, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Lafitte, V.G.H.; Aliev, A.E.; Horton, P.N.; Hursthouse, M.B.; Bala, K.; Golding, P.; Hailes, H.C. Quadruply hydrogen bonded cytosine modules for supramolecular applications. J. Am. Chem. Soc. 2006, 128, 6544–6545. [Google Scholar] [CrossRef]
- Greco, E.; Aliev, A.E.; Lafitte, V.G.H.; Bala, K.; Duncan, D.; Pilon, L.; Golding, P.; Hailes, H.C. Cytosine modules in quadruple hydrogen bonded arrays. New J. Chem. 2010, 34, 2634–2642. [Google Scholar] [CrossRef]
- Hisamatsu, Y.; Shirai, N.; Ikeda, S.; Odashima, K. A new quadruple hydrogen-bonding module with a DDAA array: Formation of a stable homodimer without competition from undesired hydrogen-bonded dimers. Org. Lett. 2009, 11, 4342–4345. [Google Scholar] [CrossRef]
- Cui, L.; Gadde, S.; Shukla, A.D.; Sun, H.; Mague, J.T.; Kaifer, A.E. Dimerization of aromatic ureido pyrimidinedione derivatives: Observation of an unexpected tautomer in the solid state. Chem. Commun. 2008, 12, 1446–1448. [Google Scholar] [CrossRef]
- Lafitte, V.G.H.; Aliev, A.E.; Hailes, H.C.; Bala, K.; Golding, P. Ureidopyrimidinones incorporating a functionalizablep-aminophenyl electron-donating group at C-6. J. Org. Chem. 2005, 70, 2701–2707. [Google Scholar] [CrossRef]
- Zhang, J.; Qi, S.; Yu, H.; Lin, Z.; Li, B.; Wang, M.; Dong, Z. Dimensionally confined nanosheets self-assembled through self-shielding multiple hydrogen bonding interactions in aqueous media. Chin. Chem. Lett. 2022, 33, 4856–4859. [Google Scholar] [CrossRef]
- Brammer, S.; Lüning, U.; Kühl, C. A new quadruply bound heterodimer DDAD·AADA and investigations into the association process. Eur. J. Org. Chem. 2002, 2002, 4054–4062. [Google Scholar] [CrossRef]
- Hisamatsu, Y.; Fukumi, Y.; Shirai, N.; Ikeda, S.; Odashima, K. Five-membered heterocyclic ureas suitable for the donor–donor–acceptor hydrogen-bonding modules. Tetrahedron Lett. 2008, 49, 2005–2009. [Google Scholar] [CrossRef]
- Pellizzaro, M.L.; McGhee, A.M.; Renton, L.C.; Nix, M.G.; Fisher, J.; Turnbull, W.B.; Wilson, A.J. Conformer-independent ureidoimidazole motifs—Tools to probe conformational and tautomeric effects on the molecular recognition of triply hydrogen-bonded heterodimers. Chem. Eur. J. 2011, 17, 14508–14517. [Google Scholar] [CrossRef] [PubMed]
- Gooch, A.; Barrett, S.; Fisher, J.; Lindsay, C.I.; Wilson, A.J. Ditopic triply hydrogen-bonded heterodimers. Org. Biomol. Chem. 2011, 9, 5938–5940. [Google Scholar] [CrossRef] [PubMed]
- Hisamatsu, Y.; Shirai, N.; Ikeda, S.; Odashima, K. A new quadruple hydrogen-bonding module based on five-membered heterocyclic urea structure. Org. Lett. 2010, 12, 1776–1779. [Google Scholar] [CrossRef] [PubMed]
- Pellizzaro, M.L.; Barrett, S.A.; Fisher, J.; Wilson, A.J. Design, synthesis and binding studies of a novel quadruple ADDA hydrogen-bond array. Org. Biomol. Chem. 2012, 10, 4899–4906. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Todd, E.M.; Nakashima, S.; Zimmerman, S.C. A Quadruply hydrogen bonded heterocomplex displaying high-fidelity recognition. J. Am. Chem. Soc. 2005, 127, 18133–18142. [Google Scholar] [CrossRef]
- Ong, H.C.; Zimmerman, S.C. Higher Affinity Quadruply Hydrogen-bonded complexation with 7-deazaguanine urea. Org. Lett. 2006, 8, 1589–1592. [Google Scholar] [CrossRef]
- Kuykendall, D.W.; Anderson, C.A.; Zimmerman, S.C. Hydrogen-bonded DeUG·DAN heterocomplex: Structure and stability and a scalable synthesis of DeUG with reactive functionality. Org. Lett. 2009, 11, 61–64. [Google Scholar] [CrossRef]
- Taubitz, J.; Lüning, U. On the importance of the nature of hydrogen bond donors in multiple hydrogen bond systems. Eur. J. Org. Chem. 2008, 2008, 5922–5927. [Google Scholar] [CrossRef]
- Otte, P.; Taubitz, J.; Lüning, U. Lipophilicity enhancing substituents for ADDA recognition domains of DAAD·ADDA heterodimers with quadruple hydrogen bonds. Eur. J. Org. Chem. 2013, 2013, 2130–2139. [Google Scholar] [CrossRef]
- Corbin, P.S.; Zimmerman, S.C. Complexation-induced unfolding of heterocyclic ureas: A hydrogen-bonded, sheetlike heterodimer. J. Am. Chem. Soc. 2000, 122, 3779–3780. [Google Scholar] [CrossRef]
- Felder, T.; de Greef, T.F.A.; Nieuwenhuizen, M.M.L.; Sijbesma, R.P. Alternation and tunable composition in hydrogen bonded supramolecular copolymers. Chem. Commun. 2014, 50, 2455–2457. [Google Scholar] [CrossRef]
- Pellizzaro, M.L.; Houton, K.A.; Wilson, A.J. Sequential and phototriggered supramolecular selfsorting cascades using hydrogen-bonded motifs. Chem. Sci. 2013, 4, 1825–1829. [Google Scholar] [CrossRef]
- Coubrough, H.M.; van der Lubbe, S.C.C.; Hetherington, K.; Minard, A.; Pask, C.; Howard, M.J.; Guerra, C.F.; Wilson, A.J. Supramolecular self-sorting networks using hydrogen-bonding motifs. Chem. Eur. J. 2019, 25, 785–795. [Google Scholar] [CrossRef]
- Kwiatkowski, A.; Grela, I.; Ośmiałowski, B. Conformational change in the association of a heterocyclic urea derivative forming two intramolecular hydrogen bonds in polar solvent. New J. Chem. 2017, 41, 1073–1081. [Google Scholar] [CrossRef]
- Cooke, G.; Rotello, V.M. Methods of modulating hydrogen bonded interactions in synthetic host–guest systems. Chem. Soc. Rev. 2002, 31, 275–286. [Google Scholar] [CrossRef]
- Smith, D.K. Electrochemically controlled hydrogen bonding for supramolecular assembly: Challenges and opportunities. Curr. Opin. Electrochem. 2017, 2, 76–81. [Google Scholar] [CrossRef]
- Smith, D.K. Exploring the role of H-bonding in organic electrochemistry—From supramolecular applications to mechanistic investigations. Chem. Rec. 2021, 21, 2488–2501. [Google Scholar] [CrossRef]
- Sun, H.; Steeb, J.; Kaifer, A.E. Efficient electronic communication between two identical ferrocene centers in a hydrogen-bonded dimer. J. Am. Chem. Soc. 2006, 128, 2820–2821. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Bria, M.; Brunklaus, G.; Caldwell, S.; Cooke, G.; Garety, J.F.; Hewage, S.G.; Hocquel, Y.; McDonald, N.; Rabani, G.; et al. Probing the solvent-induced tautomerism of a redox-active ureidopyrimidinone. Chem. Commun. 2007, 22, 2246–2248. [Google Scholar] [CrossRef][Green Version]
- Wang, S.M.; Zhao, R.N.; Wang, Q.G.; Guo, H.; Li, J.H.; Zhang, W.H. Ureidopyrimidinone quadruple hydrogen-bonded ferrocene dimer: Control of electronic communication. Acta Phys.-Chim. Sin. 2016, 32, 611–616. [Google Scholar] [CrossRef]
- Cedano, M.R.; Smith, D.K. Redox-responsive dimerization in a ferrocene-ureidopyrimidone supramolecular assembly. J. Org. Chem. 2018, 83, 11595–11603. [Google Scholar] [CrossRef]
- Fehér, C.; Papp, M.; Gömöry, Á.; Nagy, L.; Wouters, J.; Lendvay, G.; Skoda-Földes, R. Synthesis of 2-ureido-4-ferrocenyl pyrimidine guests. Investigation of complementary molecular recognition of 2,6-diaminopyridine. Organometallics 2016, 35, 4023–4032. [Google Scholar] [CrossRef]
- Rispens, M.T.; Sánchez, L.; Knol, J.; Hummelen, J.C. Supramolecular organization of fullerenes by quadruple hydrogen bonding. Chem. Commun. 2001, 2, 161–162. [Google Scholar] [CrossRef][Green Version]
- González, J.J.; González, S.; Priego, E.M.; Luo, C.; Guldi, D.M.; de Mendoza, J.; Martín, N. A new approach to supramolecular C60-dimers based in quadruple hydrogen bonding. Chem. Commun. 2001, 2, 163–164. [Google Scholar] [CrossRef][Green Version]
- Sánchez, L.; Rispens, M.T.; Hummelen, J.C. A supramolecular array of fullerenes by quadruple hydrogen bonding. Angew. Chem. Int. Ed. 2002, 41, 838–840. [Google Scholar] [CrossRef]
- Pichlmaier, M.; Winter, R.F.; Zabel, M.; Záliš, S. Electron transfer across multiple hydrogen bonds: The case of ureapyrimidinedione-substituted vinyl ruthenium and osmium complexes. J. Am. Chem. Soc. 2009, 131, 4892–4903. [Google Scholar] [CrossRef]
- Tahara, K.; Nakakita, T.; Katao, S.; Kikuchi, J. Organic mixed valency in quadruple hydrogen-bonded triarylamine dimers bearing ureido pyrimidinedione moieties. Chem. Commun. 2014, 50, 15071–15074. [Google Scholar] [CrossRef]
- Li, Y.; Park, T.; Quansah, J.K.; Zimmerman, S.C. Synthesis of a redox-responsive quadruple hydrogen-bonding unit for applications in supramolecular chemistry. J. Am. Chem. Soc. 2011, 133, 17118–17121. [Google Scholar] [CrossRef] [PubMed]
- Clarea, L.A.; Smith, D.K. Use of an electrochemically-induced proton-coupled electron transfer reaction to control dimerization in a ureidopyrimidone 4 H-bond array. Chem. Commun. 2016, 52, 7253–7256. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Han, S.; Jin, T.; Zhan, T.; Liu, L.; Cui, J.; Zhang, K. Towards photoswitchable quadruple hydrogen bonds via a reversible “photolocking” strategy for photocontrolled self-assembly. Chem. Sci. 2021, 12, 1762–1771. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Duan, H.; Chen, L.; Zhan, T.; Liu, L.; Kong, L.; Zhang, K. Photo-controlled macroscopic self-assembly based on photo-switchable hetero-complementary quadruple hydrogen bonds. Chem. Asian J. 2021, 16, 3886–3889. [Google Scholar] [CrossRef]
- Chen, L.; Wei, L.; Hai, Y.; Liu, L.; Zhang, K.; Zhan, T. Construction of photoswitchable urea-based multiple H-bonding motifs. Tetrahedron 2023, 136, 133343. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Y.; Wu, D.; Wu, L.; Tung, C.; Yang, Q. Photoresponsive hydrogen-bonded supramolecular polymers based on a stiff stilbene unit. Angew. Chem. Int. Ed. 2013, 52, 9738–9742. [Google Scholar] [CrossRef]
- Coubrough, H.M.; Balonova, B.; Pask, C.M.; Blight, B.A.; Wilson, A.J. A pH-switchable triple hydrogen-bonding motif. ChemistryOpen 2020, 9, 40–44. [Google Scholar] [CrossRef]
- ten Cate, A.T.; Kooijman, H.; Spek, A.L.; Sijbesma, R.P.; Meijer, E.W. Conformational control in the cyclization of hydrogen-bonded supramolecular polymers. J. Am. Chem. Soc. 2004, 126, 3801–3808. [Google Scholar] [CrossRef]
- Yang, Y.; Xue, M.; Marshall, L.J.; de Mendoza, J. Hydrogen-bonded cyclic tetramers based on ureidopyrimidinones attached to a 3,6-carbazolyl spacer. Org. Lett. 2011, 13, 3186–3189. [Google Scholar] [CrossRef]
- Ye, C.; Yuan, W.; Wei, X.; Liang, R.; Jozeliūnaitė, A.; de Mendoza, J.; Orentas, E.; Shi, O. Metal coordination guided formation of discrete neutral three-component hydrogen-bonded architectures. Org. Lett. 2020, 22, 9215–9219. [Google Scholar] [CrossRef]
- Shi, Q.; Zhou, X.; Yuan, W.; Su, X.; Neniškis, A.; Wei, X.; Taujenis, L.; Snarskis, G.; Ward, J.S.; Rissanen, K.; et al. Selective formation of S4- and T-symmetric supramolecular tetrahedral cages and helicates in polar media assembled via cooperative action of coordination and hydrogen bonds. J. Am. Chem. Soc. 2020, 142, 3658–3670. [Google Scholar] [CrossRef]
- Xu, J.; Niu, L.; Chen, Y.; Wu, L.; Tung, C.; Yang, Q. Hydrogen bonding directed self-assembly of small-molecule amphiphiles in water. Org. Lett. 2014, 16, 4016–4019. [Google Scholar] [CrossRef]
- Myllymäki, T.T.T.; Nonappa, N.; Yang, H.; Liljeström, V.; Kostiainen, M.A.; Malho, J.M.; Zhub, X.X.; Ikkala, O. Hydrogen bonding asymmetric star-shape derivative of bile acid leads to supramolecular fibrillar aggregates that wrap into micrometer spheres. Soft Matter 2016, 12, 7159–7165. [Google Scholar] [CrossRef]
- Bertula, K.; Nonappa; Myllymäki, T.T.T.; Yang, H.; Zhu, X.X.; Ikkala, O. Hierarchical self-assembly from nanometric micelles to colloidal spherical superstructures. Polymer 2017, 126, 177–187. [Google Scholar] [CrossRef]
- Huerta, E.; Metselaar, G.A.; Fragoso, A.; Santos, E.; Bo, C.; de Mendoza, J. Selective binding and easy separation of C70 by nanoscale self-assembled capsules. Angew. Chem. Int. Ed. 2007, 46, 202–205. [Google Scholar] [CrossRef]
- Shi, Q.; Bergquist, K.; Huo, R.; Li, J.; Lund, M.; Vácha, R.; Sundin, A.; Butkus, E.; Orentas, E.; Wärnmark, K. Composition- and size-controlled cyclic self-assembly by solvent- and C60-responsive self-sorting. J. Am. Chem. Soc. 2013, 135, 15263–15268. [Google Scholar] [CrossRef]
- Chen, Q.; Su, X.; Orentas, E.; Shi, Q. Supramolecular crowns: A new class of cyclic hydrogen-bonded cavitands. Org. Chem. Front. 2019, 6, 611–617. [Google Scholar] [CrossRef]
- Račkauskaitė, D.; Gegevičius, R.; Matsuo, Y.; Wärnmark, K.; Orentas, E. An enantiopure hydrogen-bonded octameric tube: Self-sorting and guest-induced rearrangement. Angew. Chem. Int. Ed. 2016, 55, 208–212. [Google Scholar] [CrossRef]
- Shi, Q.; Javorskis, T.; Bergquist, K.E.; Ulčinas, A.; Niaura, G.; Matulaitienė, I.; Orentas, E.; Wärnmark, K. Stimuli-controlled self-assembly of diverse tubular aggregates from one single small monomer. Nat. Commun. 2017, 8, 14943. [Google Scholar] [CrossRef]
- Xiao, T.; Zhou, L.; Sun, X.; Huang, F.; Lin, C.; Wang, L. Supramolecular polymers fabricated by orthogonal self-assembly based on multiple hydrogen bonding and macrocyclic host–guest interactions. Chin. Chem. Lett. 2020, 31, 1–9. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, H.; Mitsuishi, M. Nanosheets based on hard 2-ureido-4[1H]-pyrimidone units and soft cyclosiloxane units as membrane materials. ACS Appl. Nano Mater. 2023, 6, 5653–5663. [Google Scholar] [CrossRef]
- Bertrand, A.; Lortie, F.; Bernard, J. Routes to hydrogen bonding chain-end functionalized polymers. Macromol. Rapid Commun. 2012, 33, 2062–2091. [Google Scholar] [CrossRef]
- Myllymäki, T.T.T.; Lemetti, L.; Nonappa; Ikkala, O. Hierarchical supramolecular cross-linking of polymers for biomimetic fracture energy dissipating sacrificial bonds and defect tolerance under mechanical loading. ACS Macro Lett. 2017, 6, 210–214. [Google Scholar] [CrossRef]
- Gooch, A.; Murphy, N.S.; Thomson, N.H.; Wilson, A.J. Side-chain supramolecular polymers employing conformer independent triple hydrogen bonding arrays. Macromolecules 2013, 46, 9634–9641. [Google Scholar] [CrossRef]
- Tellers, J.; Canossa, S.; Pinalli, R.; Soliman, M.; Vachon, J.; Dalcanale, E. Dynamic cross-linking of polyethylene via sextuple hydrogen bonding array. Macromolecules 2018, 51, 7680–7691. [Google Scholar] [CrossRef]
- Schmuck, C.; Wienand, W. Self-complementary quadruple hydrogen-bonding motifs as a functional principle: From dimeric supramolecules to supramolecular polymers. Angew. Chem. Int. Ed. 2001, 40, 4363–4369. [Google Scholar] [CrossRef]
- Aida, T.; Meijer, E.W. Supramolecular polymers—We’ve come full circle. Isr. J. Chem. 2020, 60, 33–47. [Google Scholar] [CrossRef]
- Xie, Z.; Hu, B.; Li, R.; Zhang, Q. Hydrogen bonding in self-healing elastomers. ACS Omega 2021, 6, 9319–9333. [Google Scholar] [CrossRef]
- Xie, J.; Yu, P.; Wang, Z.; Li, J. Recent advances of self-healing polymer materials via supramolecular forces for biomedical applications. Biomacromolecules 2022, 23, 641–660. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Jones, A.R.; Briggs, E.M.; Novitsky, E.J.; Kuykendall, D.W.; Sottos, N.R.; Zimmerman, S.C. High-affinity DNA base analogs as supramolecular, nanoscale promoters of macroscopic adhesion. J. Am. Chem. Soc. 2013, 135, 7288–7295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Anderson, C.A.; Zimmerman, S.C. Quadruply hydrogen bonding modules as highly selective nanoscale adhesive agents. Org. Lett. 2013, 15, 3506–3509. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yu, M.; Feng, K.; Li, X.; Chen, Y.; Chen, B.; Tung, C.; Wu, L. Efficient electronic communication-driven photoinduced charge-separation in 2-ureido-4[1H]-pyrimidinone quadruple hydrogen-bonded N,N-dimethylaniline-anthracene assemblies. J. Photochem. Photobiol. 2018, 355, 457–466. [Google Scholar] [CrossRef]
- Lian, Z.; Qiao, F.; Jiang, M.; Wang, R.; Xing, L.; Liu, S.; Wang, S. Quadruple hydrogen bonded hyperbranched supramolecular polymers with aggregation-induced emission for artificial light-harvesting. Dyes Pigm. 2019, 171, 107774. [Google Scholar] [CrossRef]
- Peng, H.; Xu, J.; Chen, Y.; Wu, L.; Tung, C.; Yang, Q. Water-dispersible nanospheres of hydrogen-bonded supramolecular polymers and their application for mimicking light-harvesting systems. Chem. Commun. 2014, 50, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qian, H.; Wu, Z.; Zhang, Q.; Li, S.; Cheng, M.; Xiao, T. Non-covalent dimer as donor chromophore for constructing artificial light-harvesting system in water. Molecules 2022, 27, 8876. [Google Scholar] [CrossRef]
- Kailasa, S.K.; Joshi, D.J.; Kateshiya, M.R.; Koduru, J.R.; Malek, N.I. Review on the biomedical and sensing applications of nanomaterial-incorporated hydrogels. Mater. Today Chem. 2022, 23, 100746. [Google Scholar] [CrossRef]
- Sánchez-Cid, P.; Jiménez-Rosado, M.; Romero, A.; Pérez-Puyana, V. Novel trends in hydrogel development for biomedical applications: A review. Polymers 2022, 14, 3023. [Google Scholar] [CrossRef]
- Omar, J.; Ponsford, D.; Dreiss, C.A.; Lee, T.; Loh, X.J. Supramolecular hydrogels: Design strategies and contemporary biomedical applications. Chem. Asian J. 2022, 17, e202200081. [Google Scholar] [CrossRef]
- Wintjens, A.G.W.E.; Fransen, P.K.H.; Lenaerts, K.; Liu, H.; van Almen, G.C.; van Steensel, S.; Gijbels, M.J.; de Hingh, I.H.J.T.; Dankers, P.Y.W.; Bouvy, N.D. Development of a supramolecular hydrogel for intraperitoneal injections. Macromol. Biosci. 2023, 2300005. [Google Scholar] [CrossRef]
- Dankers, P.; Harmsen, M.; Brouwer, L.; van Luyn, M.J.A.; Meijer, E.W. A modular and supramolecular approach to bioactive scaffolds for tissue engineering. Nat. Mater. 2005, 4, 568–574. [Google Scholar] [CrossRef]
- Gilpin, A.; Zeng, Y.; Hoque, J.; Ryu, J.H.; Yang, Y.; Zauscher, S.; Eward, W.; Varghese, S. Self-healing of hyaluronic acid to improve in vivo retention and function. Adv. Healthc. Mater. 2021, 10, 2100777. [Google Scholar] [CrossRef] [PubMed]
- Chirila, T.V.; Lee, H.H.; Oddon, M.; Nieuwenhuizen, M.M.L.; Blakey, I.; Nicholson, T.M. Hydrogen-bonded supramolecular polymers as self-healing hydrogels: Effect of a bulky adamantyl substituent in the ureido-pyrimidinone monomer. J. Appl. Polym. Sci. 2014, 131, 39932. [Google Scholar] [CrossRef]
- Lu, L.; Zhou, W.; Chen, Z.; Hu, Y.; Yang, Y.; Zhang, G.; Yang, Z. A supramolecular hydrogel enabled by the synergy of hydrophobic interaction and quadruple hydrogen bonding. Gels 2022, 8, 244. [Google Scholar] [CrossRef] [PubMed]
- Bastings, M.M.C.; Koudstaal, S.; Kieltyka, R.E.; Nakano, Y.; Pape, A.C.H.; Feyen, D.A.M.; van Slochteren, F.J.; Doevendans, P.A.; Sluijter, J.P.G.; Meijer, E.W.; et al. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv. Healthc. Mater. 2014, 3, 70–78. [Google Scholar] [CrossRef]
- Ilhami, F.B.; Yang, Y.; Lee, A.; Chiao, Y.; Chen, J.; Lee, D.; Lai, J.; Cheng, C. Hydrogen bond strength-mediated self-assembly of supramolecular nanogels for selective and effective cancer treatment. Biomacromolecules 2021, 22, 4446–4457. [Google Scholar] [CrossRef] [PubMed]
- Alavijeh, R.Z.; Shokrollahi, P.; Barzin, J. A thermally and water activated shape memory gelatin physical hydrogel, with a gel point above the physiological temperature, for biomedical applications. J. Mater. Chem. B 2017, 5, 2302–2314. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, H.; Zhu, H.; Jiang, L.; Yang, H. Self-healing gelatin-based shape memory hydrogels via quadruple hydrogen bonding and coordination crosslinking for controlled delivery of 5-fluorouracil. J. Biomater. Sci. Polym. Ed. 2020, 31, 712–728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Lv, L.; Deng, Y.; Wang, C. Self-healing gelatin hydrogels cross-linked by combining multiple hydrogen bonding and ionic coordination. Macromol. Rapid Commun. 2017, 38, 1700018. [Google Scholar] [CrossRef]
- Schotman, M.J.G.; Peters, M.M.C.; Krijger, G.C.; van Adrichem, I.; de Roos, R.; Bemelmans, J.L.M.; Pouderoijen, M.J.; Rutten, M.G.T.A.; Neef, K.; Chamuleau, S.A.J.; et al. In vivo retention quantification of supramolecular hydrogels engineered for cardiac delivery. Adv. Healthc. Mater. 2021, 10, 2001987. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Peng, Q.; Thundat, T.; Zeng, H. Stretchable, injectable, and self-healing conductive hydrogel enabled by multiple hydrogen bonding toward wearable electronics. Chem. Mater. 2019, 31, 4553–4563. [Google Scholar] [CrossRef]
- Dai, X.; Huang, L.; Du, Y.; Han, J.; Zheng, Q.; Kong, J.; Hao, J. Self-healing, flexible, and tailorable triboelectric nanogenerators for self-powered sensors based on thermal effect of infrared radiation. Adv. Funct. Mater. 2020, 30, 1910723. [Google Scholar] [CrossRef]
- Dai, X.; Huang, L.; Du, Y.; Han, J.; Kong, J. Self-healing flexible strain sensors based on dynamically cross-linked conductive nanocomposites. Compos. Commun. 2021, 24, 100654. [Google Scholar] [CrossRef]
- Kieltyka, R.E.; Bastings, M.M.C.; van Almen, G.C.; Besenius, P.; Kemps, E.W.L.; Dankers, P.Y.W. Modular synthesis of supramolecular ureidopyrimidinone–peptide conjugates. Chem. Commun. 2012, 48, 1452–1454. [Google Scholar] [CrossRef]
- Mollet, B.B.; Comellas-Aragonès, M.; Spiering, A.J.H.; Söntjens, S.H.M.; Meijer, E.W.; Dankers, P.Y.W. A modular approach to easily processable supramolecular bilayered scaffolds with tailorable properties. J. Mater. Chem. B 2014, 2, 2483–2493. [Google Scholar] [CrossRef]
- Mollet, B.B.; Bogaerts, I.L.J.; van Almen, G.C.; Dankers, P.Y.W. A bioartificial environment for kidney epithelial cells based on a supramolecular polymer basement membrane mimic and an organotypical culture system. J. Tissue Eng. Regen. 2015, 11, 1820–1834. [Google Scholar] [CrossRef]
- Atashkar, B.; Zolfigol, M.A.; Mallakpour, S. Applications of biological urea-based catalysts in chemical processes. Mol. Catal. 2018, 452, 192–246. [Google Scholar] [CrossRef]
- Fan, Y.; Kass, S.R. Electrostatically enhanced thioureas. Org. Lett. 2016, 18, 188–191. [Google Scholar] [CrossRef]
- Yan, C.; Wu, R.; Lu, K.; Yang, F.; Yang, X.; Wang, R.; Yang, X.; Zhou, P.; Shao, X. Why electrostatically enhanced thiourea is better than Schreiner’s thiourea in both catalytic activity and regioselectivity? Org. Chem. Front. 2019, 6, 1821–1831. [Google Scholar] [CrossRef]
- Ganesh, M.; Seidel, D. Catalytic enantioselective additions of indoles to nitroalkenes. J. Am. Chem. Soc. 2008, 130, 16464–16465. [Google Scholar] [CrossRef]
- Borah, P.; Mondal, J.; Zhao, Y. Urea-pyridine bridged periodic mesoporous organosilica: An efficient hydrogen-bond donating heterogeneous organocatalyst for Henry reaction. J. Catal. 2015, 330, 129–134. [Google Scholar] [CrossRef]
- Gruijters, B.W.T.; Broeren, M.A.C.; van Delft, F.L.; Sijbesma, R.P.; Hermkens, P.H.H.; Rutjes, F.P.J.T. Catalyst recycling via hydrogen-bonding-based affinity tags. Org. Lett. 2006, 8, 3163–3166. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Llansola, F.; Meijer, E.W. supramolecular autoregulation. J. Am. Chem. Soc. 2013, 135, 6549–6553. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, A.J.P.; van der Haas, R.J.C.; Vekemans, J.A.J.M.; Palmans, A.R.A.; Meijer, E.W. Scope and limitations of supramolecular autoregulation. Bull. Chem. Soc. Jpn. 2016, 89, 308–314. [Google Scholar] [CrossRef]
- Teunissen, A.J.P.; Paffen, T.F.E.; Filot, I.A.W.; Lanting, M.D.; van der Haas, R.J.C.; de Greef, T.F.A.; Meijer, E.W. Supramolecular interactions between catalytic species allow rational control over reaction kinetics. Chem. Sci. 2019, 10, 9115–9124. [Google Scholar] [CrossRef] [PubMed]
- Bregović, V.B.; Basarić, N.; Mlinarić-Majerski, K. Anion binding with urea and thiourea derivatives. Coord. Chem. Rev. 2015, 295, 80–124. [Google Scholar] [CrossRef]
- Kundu, S.; Egboluche, T.K.; Hossain, M.A. Urea- and thiourea-based receptors for anion binding. Acc. Chem. Res. 2023, 56, 1320–1329. [Google Scholar] [CrossRef]
- Caltagirone, C.; Hiscock, J.R.; Hursthouse, M.B.; Light, M.E.; Gale, P.A. 1,3-Diindolylureas and 1,3-diindolylthioureas: Anion complexation studies in solution and the solid state. Chem. Eur. J. 2008, 14, 10236–10243. [Google Scholar] [CrossRef]
- Bates, G.W.; Triyanti; Light, M.E.; Albrecht, M.; Gale, P.A. 2,7-Functionalized indoles as receptors for anions. J. Org. Chem. 2007, 72, 8921–8927. [Google Scholar] [CrossRef]
- Makuc, D.; Triyanti, T.; Albrecht, M.; Plavec, J.; Rissanen, K.; Valkonen, A.; Schalley, C.A. The halide binding behavior of 2-carbamoyl-7-ureido-1H-indoles: Conformational aspects. Eur. J. Org. Chem. 2009, 28, 4854–4866. [Google Scholar] [CrossRef]
- Andrews, N.J.; Haynes, C.J.E.; Light, M.E.; Moore, S.J.; Tong, C.C.; Davis, J.T.; Harrell, W.A.; Gale, P.A. Structurally simple lipid bilayer transport agents for chloride and bicarbonate. Chem. Sci. 2011, 2, 256–260. [Google Scholar] [CrossRef]
- Mendonça, J.G.P.; Silla, J.M.; Andrade, L.A.F.; Fernandes, S.A.; Cormanich, R.A.; Freitas, M.P. Theoretical and NMR experimental insights on urea, thiourea and diindolyurea as fluoride carriers. J. Mol. Struct. 2016, 1114, 13–20. [Google Scholar] [CrossRef]
- Gale, P.A.; Hiscock, J.R.; Moore, S.J.; Caltagirone, C.; Hursthouse, M.B.; Light, M.E. Anion–anion proton transfer in hydrogen bonded complexes. Chem. Asian J. 2010, 5, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Gale, P.A.; Hiscock, J.R.; Jie, C.Z.; Hursthouse, M.B.; Light, M.E. Acyclic indole and carbazole-based sulfate receptors. Chem. Sci. 2010, 1, 215–220. [Google Scholar] [CrossRef][Green Version]
- Dydio, P.; Zieliński, T.; Jurczak, J. 7,7′-Diureido-2,2′-diindolylmethanes: Anion receptors effective in a highly competitive solvent, methanol. Org. Lett. 2010, 12, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Hiscock, J.R.; Caltagirone, C.; Light, M.E.; Hursthouse, M.B.; Gale, P.A. Fluorescent carbazolylurea anion receptors. Org. Biomol. Chem. 2009, 7, 1781–1783. [Google Scholar] [CrossRef]
- Hiscock, J.R.; Gale, P.A.; Caltagirone, C.; Hursthouse, M.B.; Light, M.E. Fluorescent carbazolylurea- and carbazolylthiourea-based anion receptors and sensors. Supramol. Chem. 2010, 22, 647–652. [Google Scholar] [CrossRef]
- Martin, K.; Nõges, J.; Haav, K.; Kadam, S.A.; Pung, A.; Leito, I. Exploring selectivity of 22 acyclic urea-, carbazole- and indolocarbazole-based receptors towards 11 monocarboxylates. Eur. J. Org. Chem. 2017, 2017, 5231–5237. [Google Scholar] [CrossRef]
- Gale, P.A.; Hiscock, J.R.; Lalaoui, N.; Light, M.E.; Wells, N.J.; Wenzel, M. Benzimidazole-based anion receptors: Tautomeric switching and selectivity. Org. Biomol. Chem. 2012, 10, 5909–5915. [Google Scholar] [CrossRef]
- Ragusa, A.; Rossi, S.; Hayes, J.M.; Stein, M.; Kilburn, J.D. Novel enantioselective receptors for N-protected glutamate and aspartate. Chem. Eur. J. 2005, 11, 5674–5688. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.; Triyanti; Schiffers, S.; Osetska, O.; Raabe, G.; Wieland, T.; Russo, L.; Rissanen, K. Anion receptors based on a quinoline backbone. Eur. J. Org. Chem. 2007, 2007, 2850–2858. [Google Scholar] [CrossRef]
- Otón, F.; Tárraga, A.; Espinosa, A.; Velasco, M.D.; Molina, P. Heteroditopic ferrocene-based ureas as receptors for anions and cations. Dalton Trans. 2006, 30, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Merckx, T.; Verwilst, P.; Dehaen, W. Preorganization in bistriazolyl anion receptors. Tetrahedron Lett. 2013, 54, 4237–4240. [Google Scholar] [CrossRef]
- Keszei, S.J.; Balogh, S.; Fehér, C.; Nagy, L.; Tumanov, N.; Wouters, J.; Lendvay, G.; Skoda-Földes, R. Molecular recognition of strong acids by using a 2-ureido-4-ferrocenyl pyrimidine receptor. Eur. J. Inorg. Chem. 2019, 2019, 4095–4104. [Google Scholar] [CrossRef]
- Turner, D.R.; Spencer, E.C.; Howard, J.A.K.; Tocher, D.A.; Steed, J.W. A modular, self-assembled, separated ion pair binding system. Chem. Commun. 2004, 1352–1353. [Google Scholar] [CrossRef]
- Russell, J.M.; Parker, A.D.M.; Radosavljevic-Evans, I.; Howard, J.A.K.; Steed, J.W. Simultaneous anion and cation binding by a simple polymer-bound ureidopyridyl ligand. Chem. Commun. 2006, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.T.X.; Hicks, J.; Wallace, K.J.; Turner, D.R. Binding of mono- and dianions within silver thiazolylurea tweezers and capsules. Inorg. Chem. 2017, 56, 12535–12541. [Google Scholar] [CrossRef]
- Custelcean, R.; Bosano, J.; Bonnesen, P.V.; Kertesz, V.; Hay, B.P. Computer-aided design of a sulfate-encapsulating receptor. Angew. Chem. Int. Ed. 2009, 48, 4025–4029. [Google Scholar] [CrossRef]
- Custelcean, R.; Bonnesen, P.V.; Duncan, N.C.; Zhang, X.; Watson, L.A.; Berkel, G.V.; Parson, W.B.; Hay, B.P. Urea-functionalized M4L6 cage receptors: Anion-templated self-assembly and selective guest exchange in aqueous solutions. J. Am. Chem. Soc. 2012, 134, 8525–8534. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Ganguly, B.; Das, A. Urea-based ruthenium(II)- polypyridyl complex as an optical sensor for anions: Synthesis, characterization, and binding studies. Inorg. Chem. 2007, 46, 9912–9918. [Google Scholar] [CrossRef]
- Ghosh, A.; Verma, S.; Ganguly, B.; Ghosh, H.N.; Das, A. Influence of urea N–H acidity on receptor–anionic and neutral analyte binding in a ruthenium(ii)–polypyridyl-based colorimetric sensor. Eur. J. Inorg. Chem. 2009, 2009, 2496–2507. [Google Scholar] [CrossRef]
- Kitchen, J.A.; Boyle, E.M.; Gunnlaugsson, T. Synthesis, structural characterisation and luminescent anion sensing studies of a Ru(II)polypyridyl complex featuring an aryl urea derivatised 2,2′-bpy auxiliary ligand. Inorganica Chim. Acta 2012, 381, 236–242. [Google Scholar] [CrossRef]
- Baggi, G.; Boiocchi, M.; Ciarrocchi, C.; Fabbrizzi, L. Enhancing the anion affinity of urea-based receptors with a Ru(terpy)22+ chromophore. Inorg. Chem. 2013, 52, 5273–5283. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.K.; Chakraborty, S.; Chowdhury, B.; Ghosh, P. Bis-heteroleptic ruthenium(ii) complex of pendant urea functionalized pyridyl triazole and phenathroline for recognition, sensing, and extraction of oxyanions. Inorg. Chem. 2017, 56, 5371–5382. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Mondal, S.; Bej, S.; Nandi, M.; Ghosh, P. An integrated urea and halogen bond donor based receptor for superior and selective sensing of phosphates. Dalton Trans. 2019, 48, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.M.G.; Gunnlaugsson, T. Exploring the luminescent sensing of anions by the use of an urea functionalised 1,10-phenanthroline (phen)-based (3:1) Eu(III) complex. Supramol. Chem. 2009, 21, 173–180. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, C.; Wu, L.; Zhang, L.; Tung, C.; Pan, Y. First fluorescent sensor for fluoride based on 2-ureido-4[1H]-pyrimidinone quadruple hydrogen-bonded AADD supramolecular assembly. J. Org. Chem. 2006, 71, 2143–2146. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
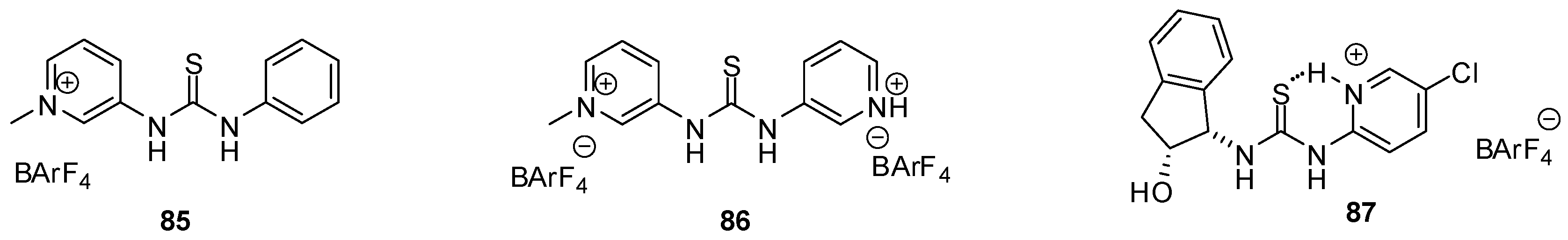{kind=link}
{kind=link}
{kind=link}
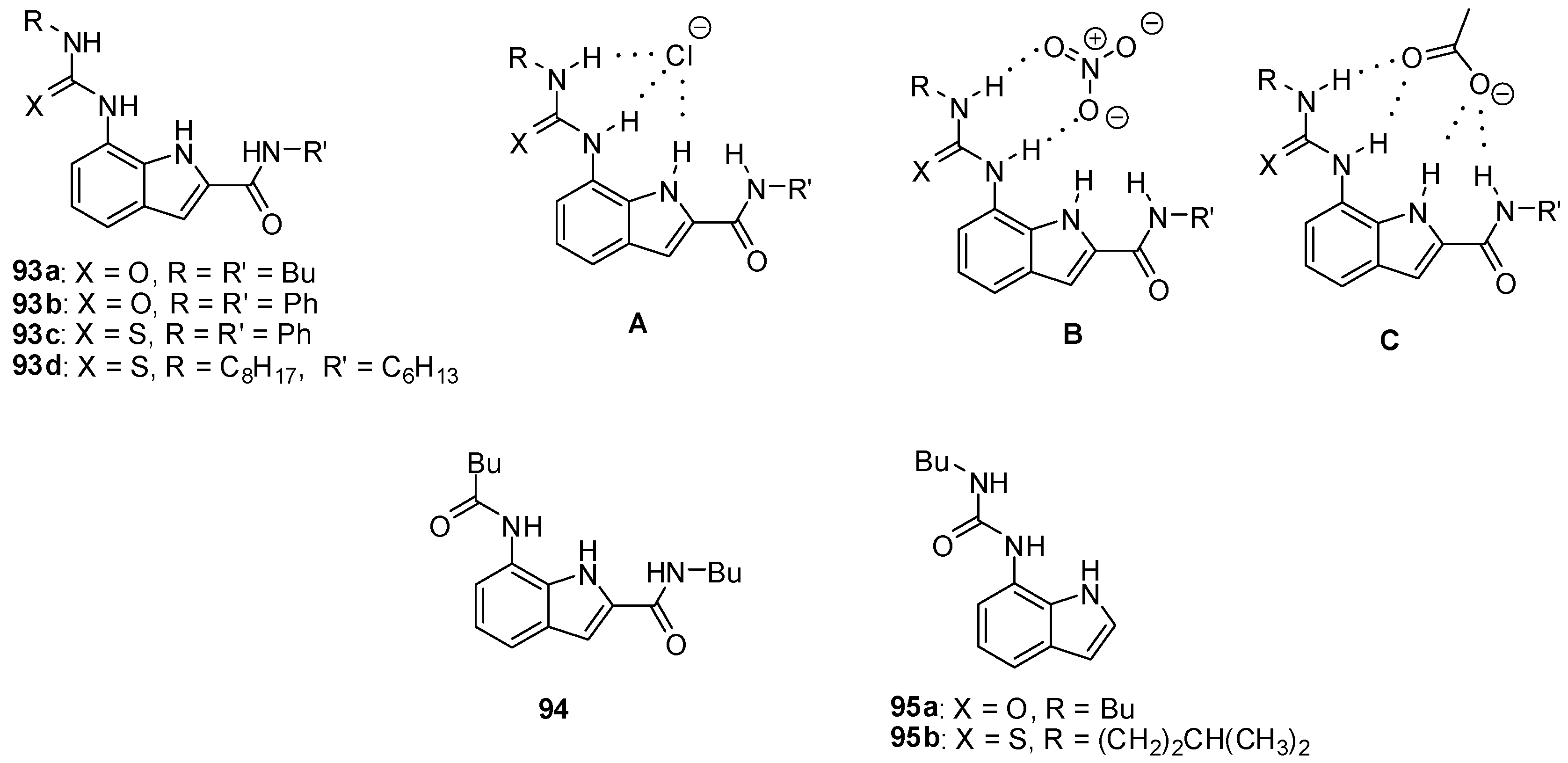{kind=link}
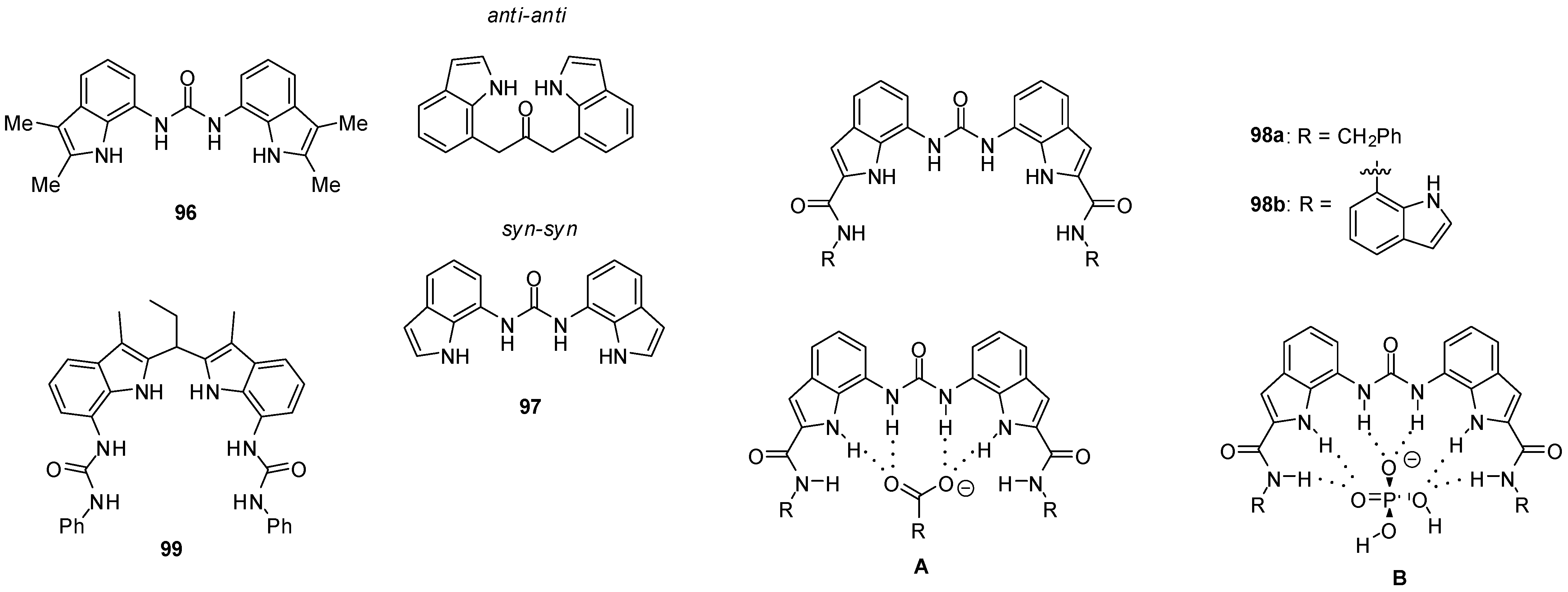{kind=link}
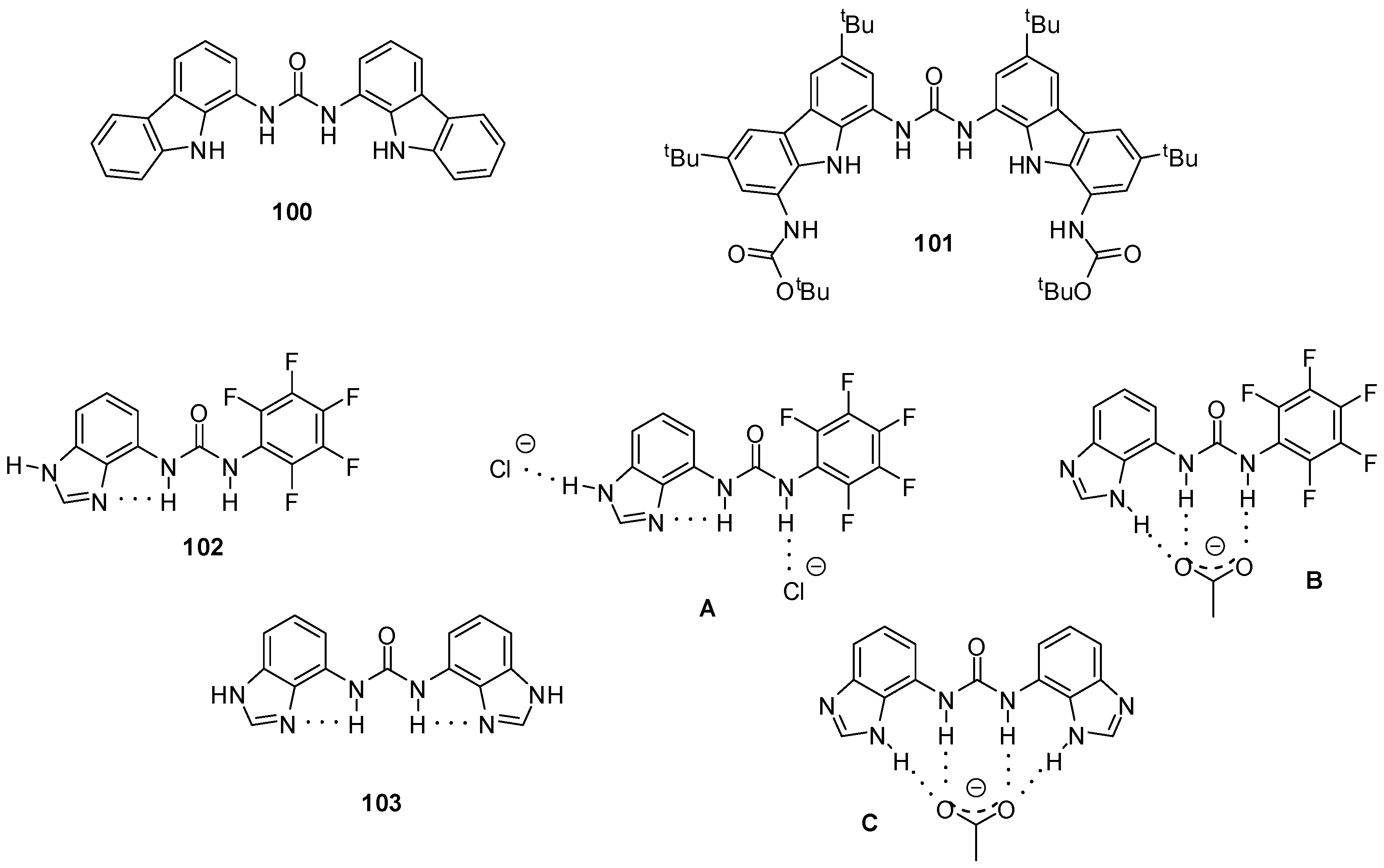{kind=link}
{kind=link}
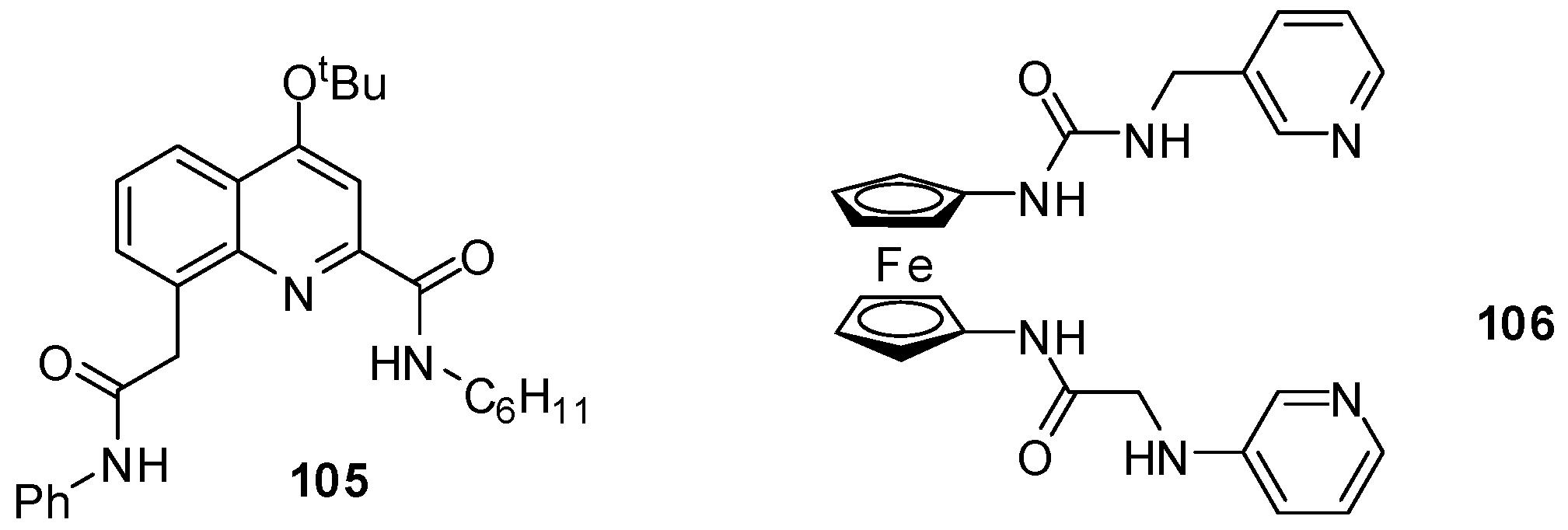{kind=link}
{kind=link}
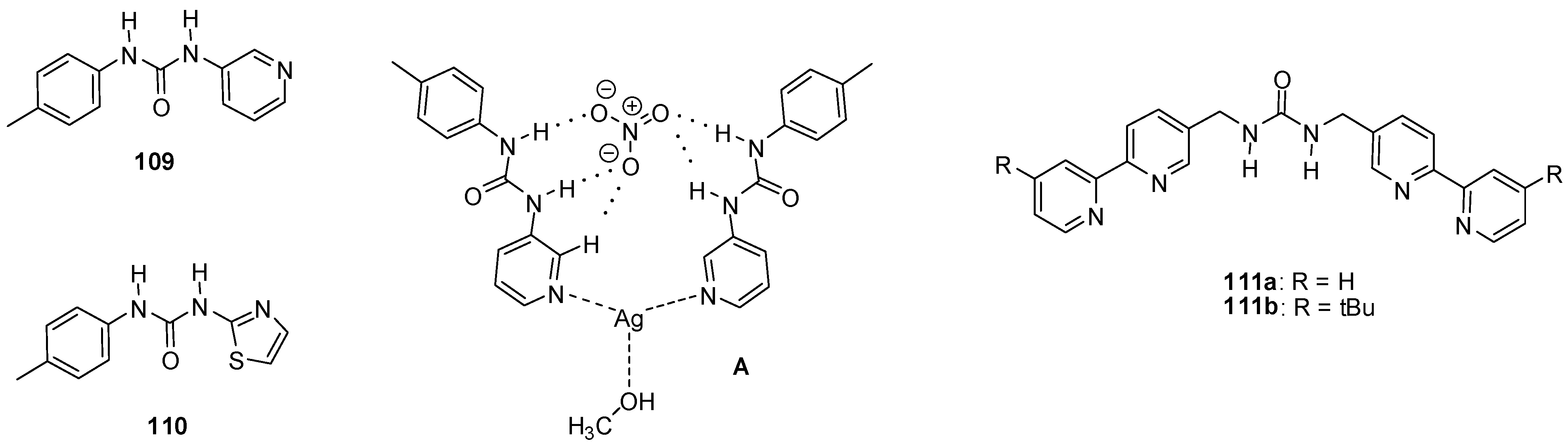{kind=link}
{kind=link}
{kind=link}
{kind=link}
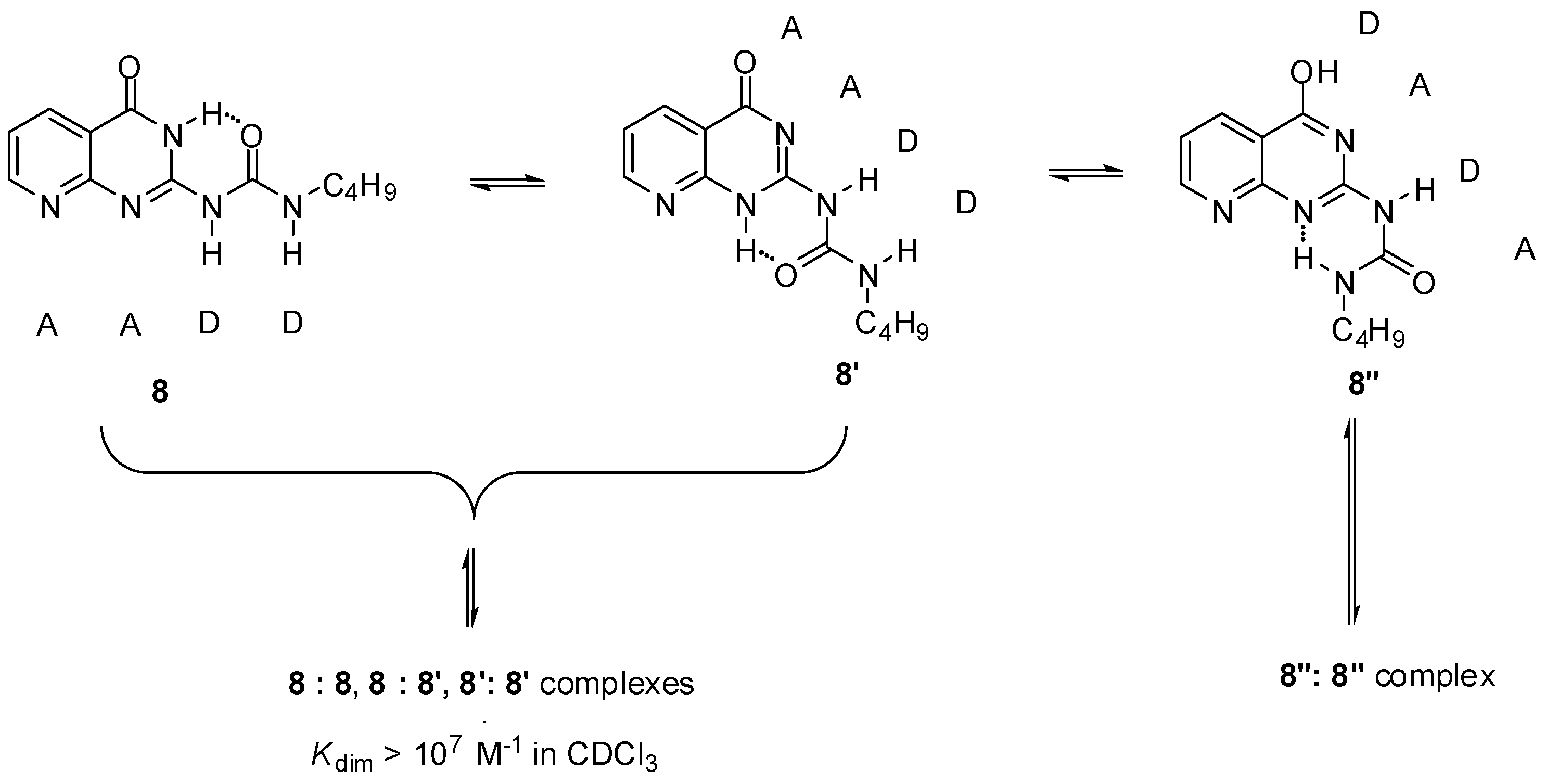{kind=link}
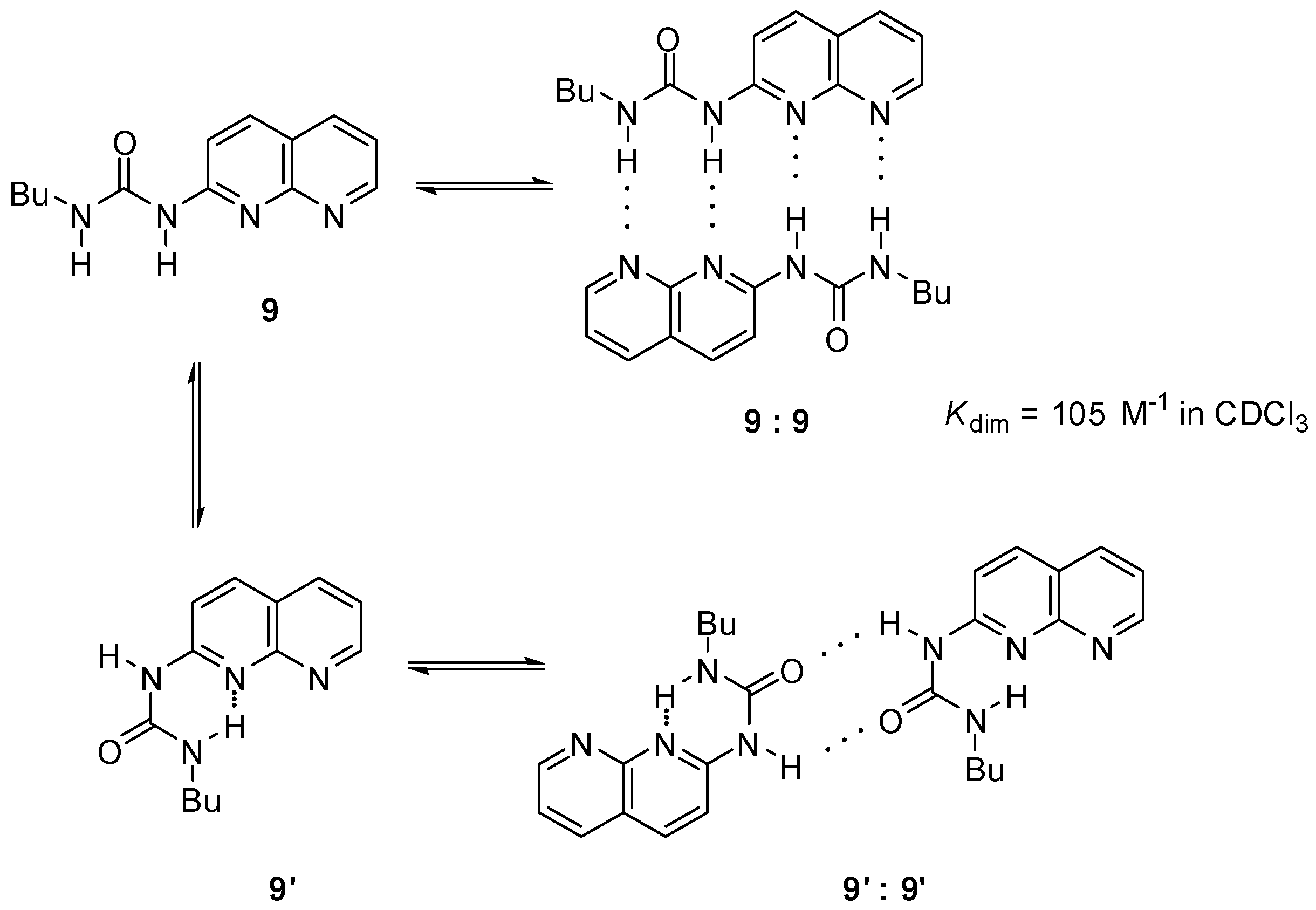{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keszei, S.J.; Váradi, M.; Skoda-Földes, R. Urea-Functionalized Heterocycles: Structure, Hydrogen Bonding and Applications. Molecules 2023, 28, 7757. https://doi.org/10.3390/molecules28237757
Keszei SJ, Váradi M, Skoda-Földes R. Urea-Functionalized Heterocycles: Structure, Hydrogen Bonding and Applications. Molecules. 2023; 28(23):7757. https://doi.org/10.3390/molecules28237757
Chicago/Turabian StyleKeszei, Soma J., Márk Váradi, and Rita Skoda-Földes. 2023. "Urea-Functionalized Heterocycles: Structure, Hydrogen Bonding and Applications" Molecules 28, no. 23: 7757. https://doi.org/10.3390/molecules28237757
APA StyleKeszei, S. J., Váradi, M., & Skoda-Földes, R. (2023). Urea-Functionalized Heterocycles: Structure, Hydrogen Bonding and Applications. Molecules, 28(23), 7757. https://doi.org/10.3390/molecules28237757