
Syntheses, Structures and Reactivity of Metal Complexes of Trindane, Trindene, Truxene, Decacyclene and Related Ring Systems: Manifestations of Three-Fold Symmetry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Organometallic Derivatives of Trindane
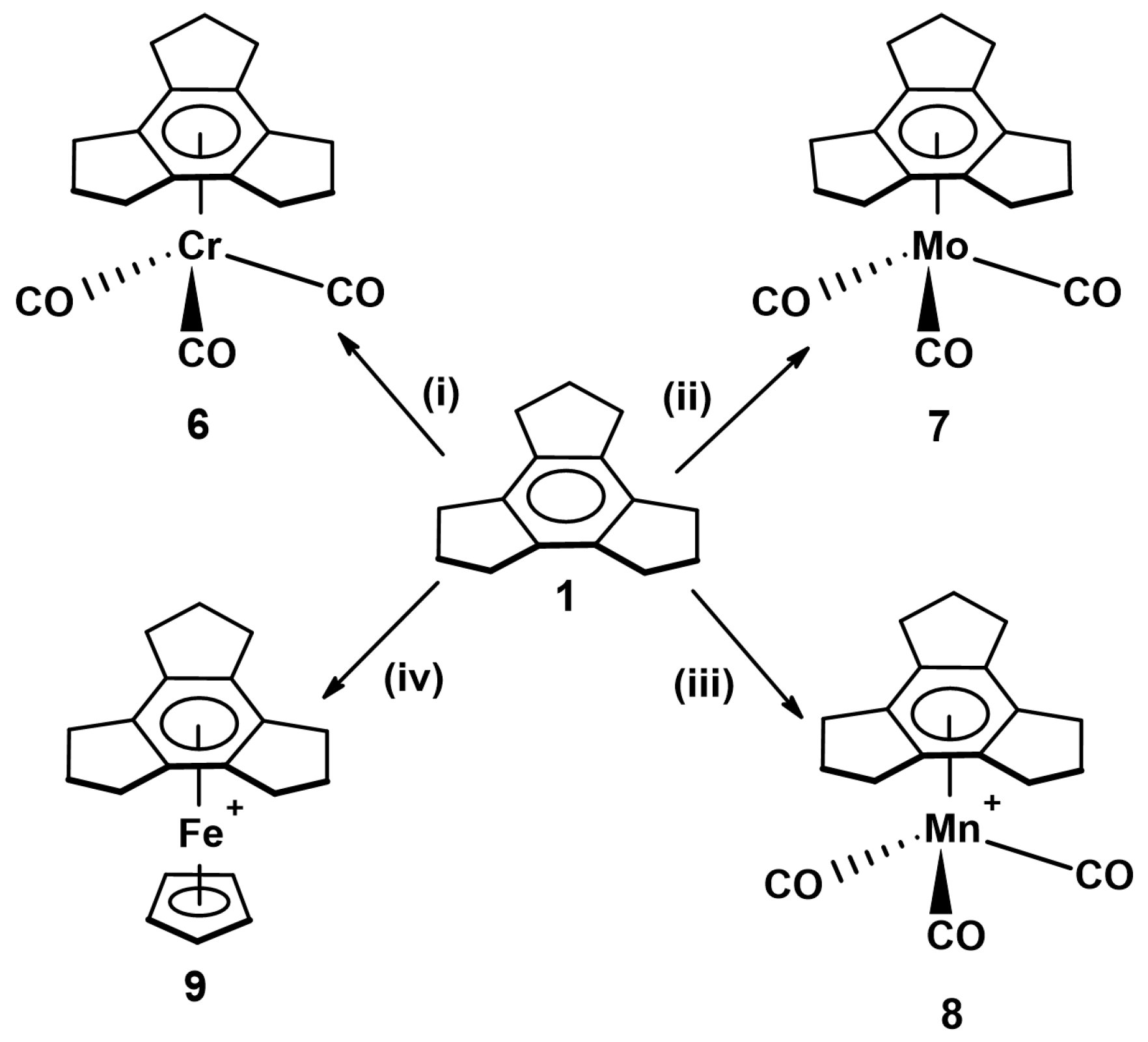
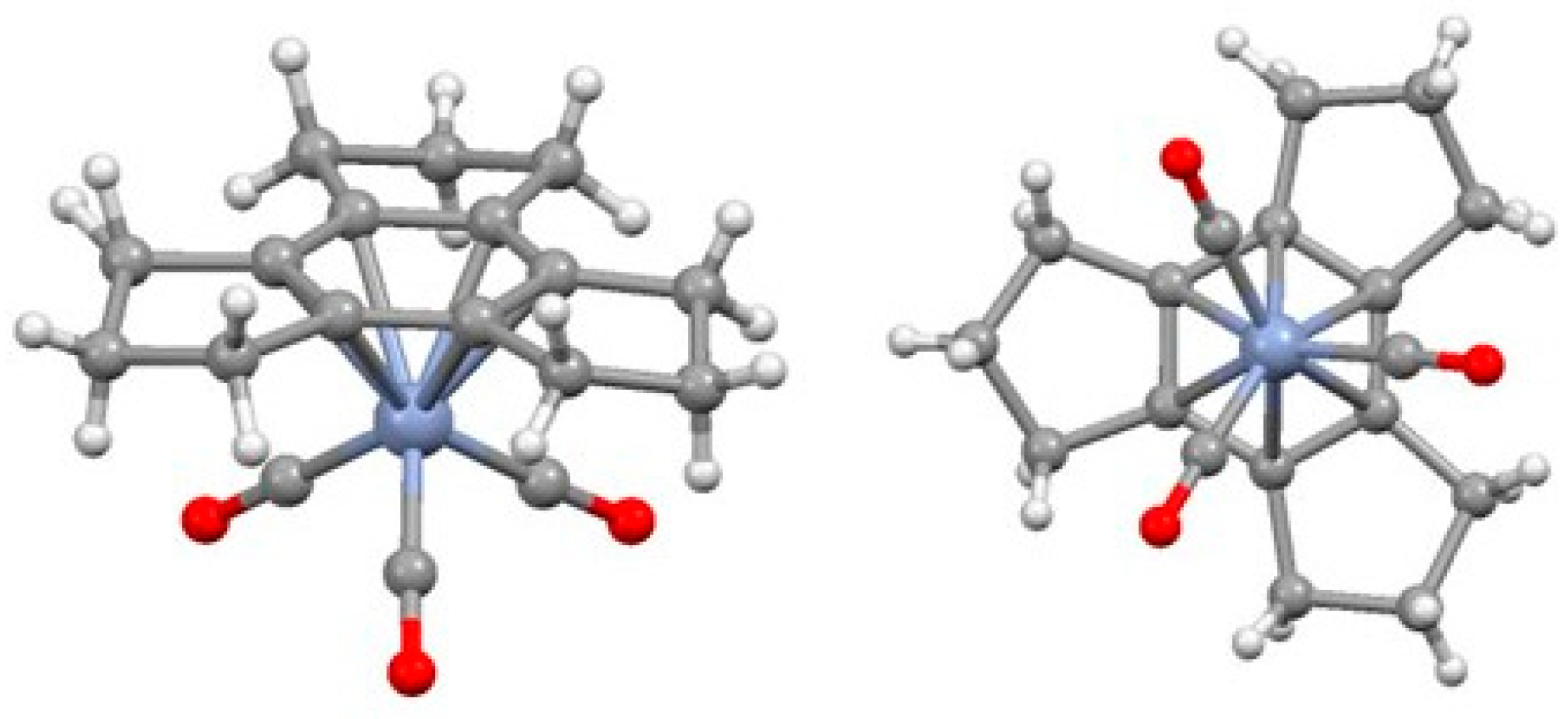
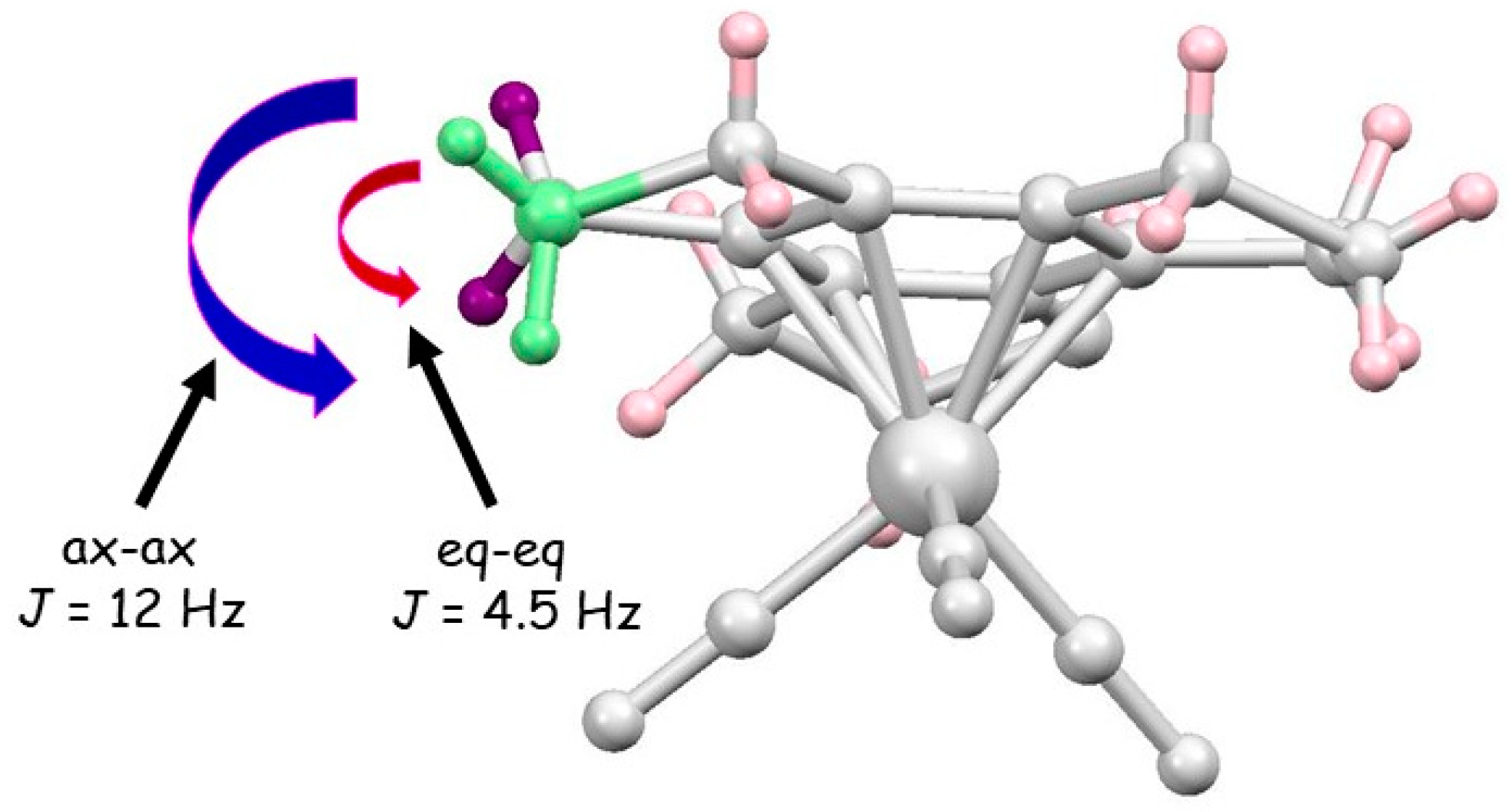
2.1. Trindane Complexes of Chromium, Molybdenum, Manganese and Iron

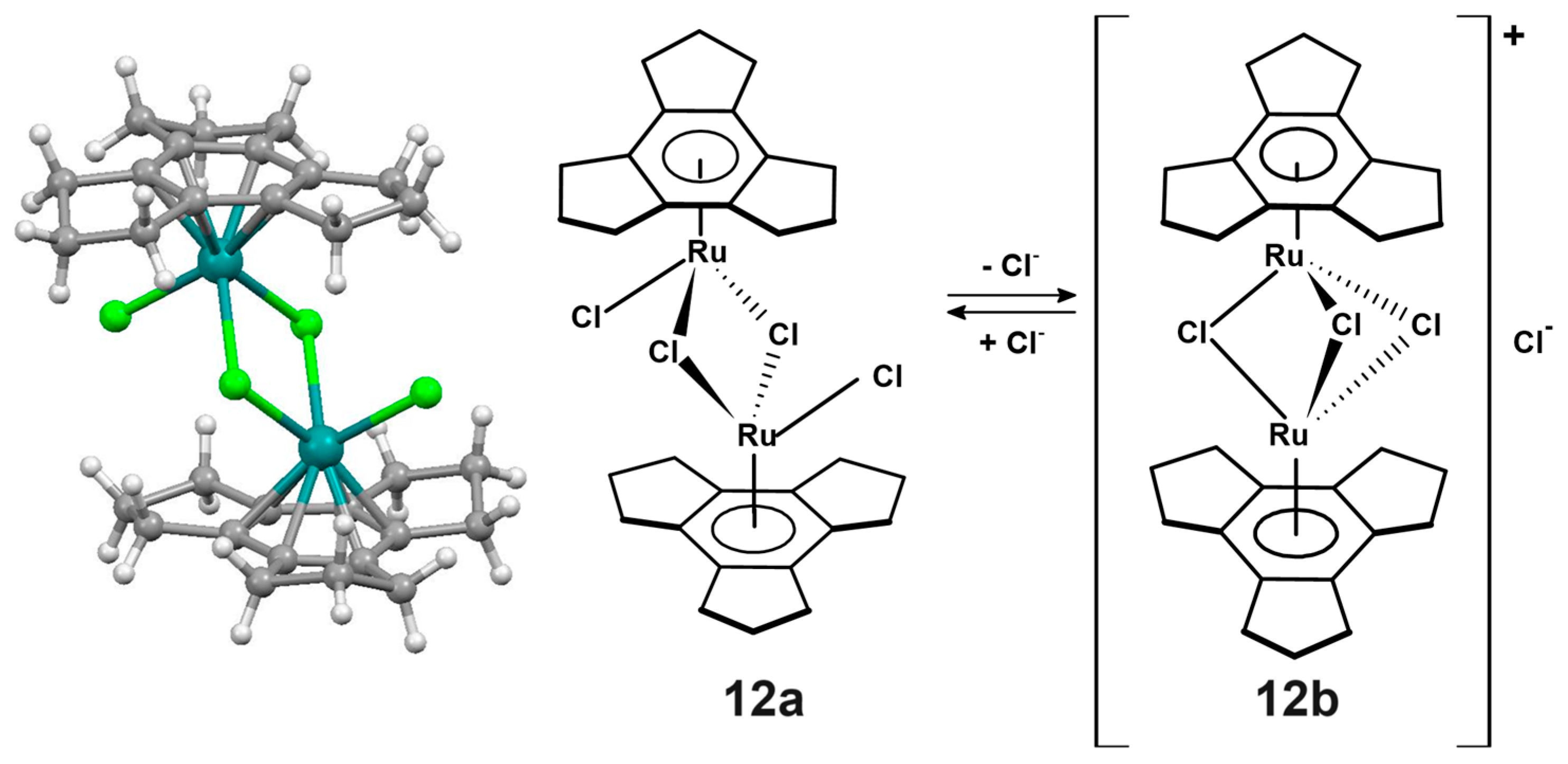
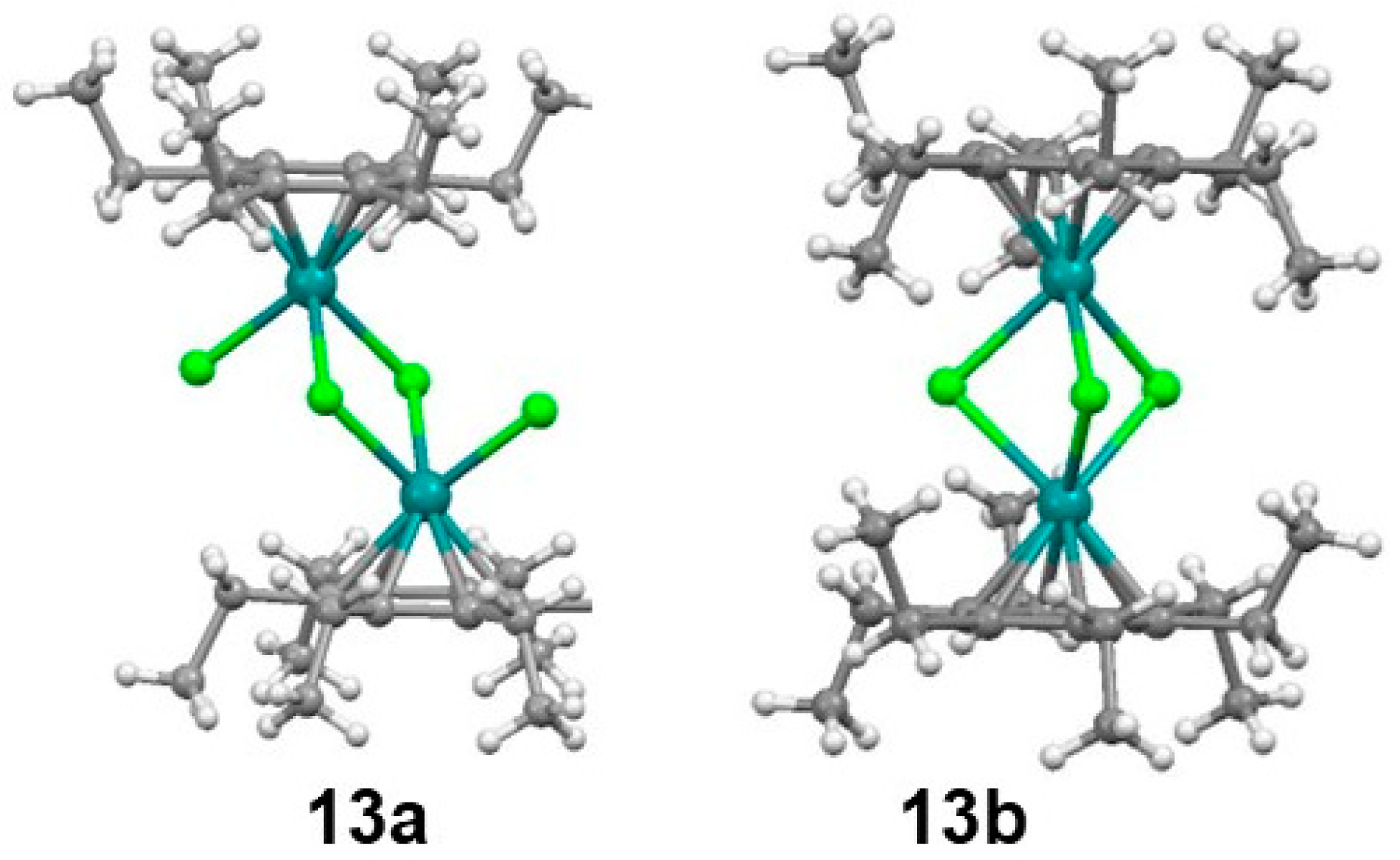
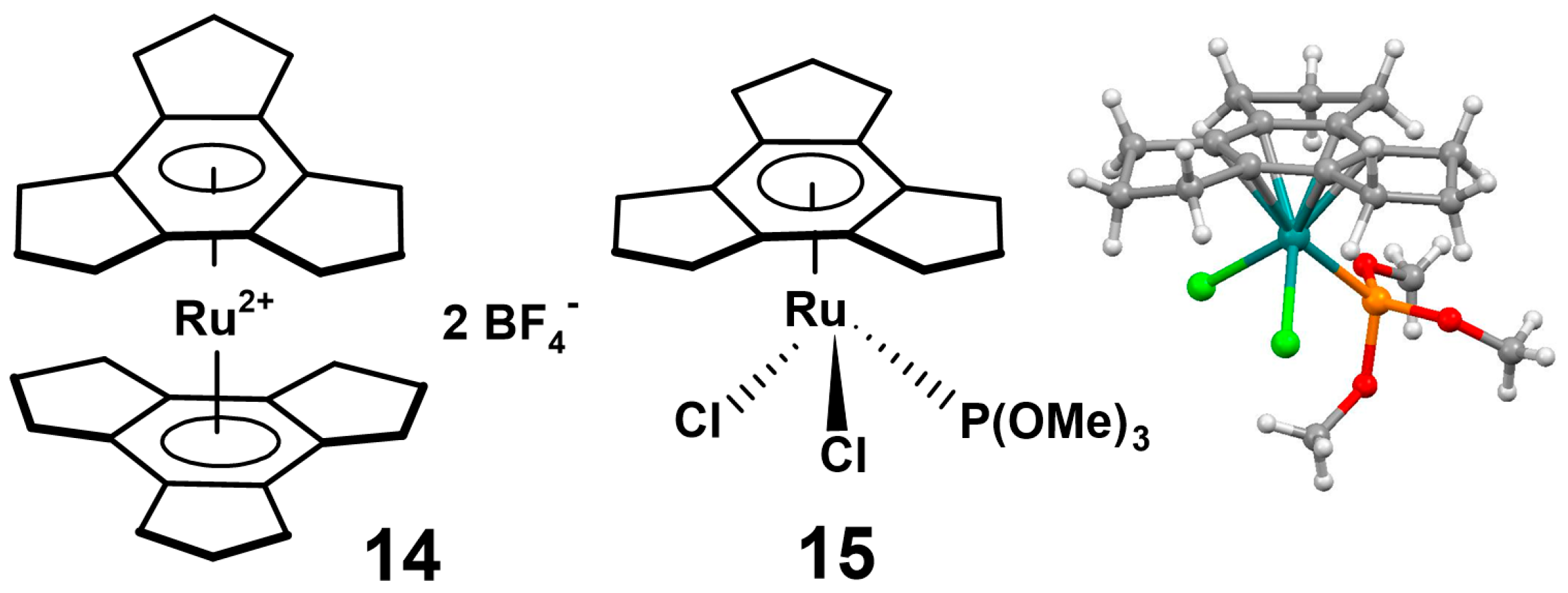
2.2. Trindane Complexes of Ruthenium
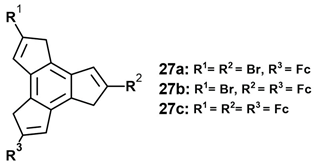
3. Organometallic Derivatives of Trindene
4. Metal Complexes of Truxene
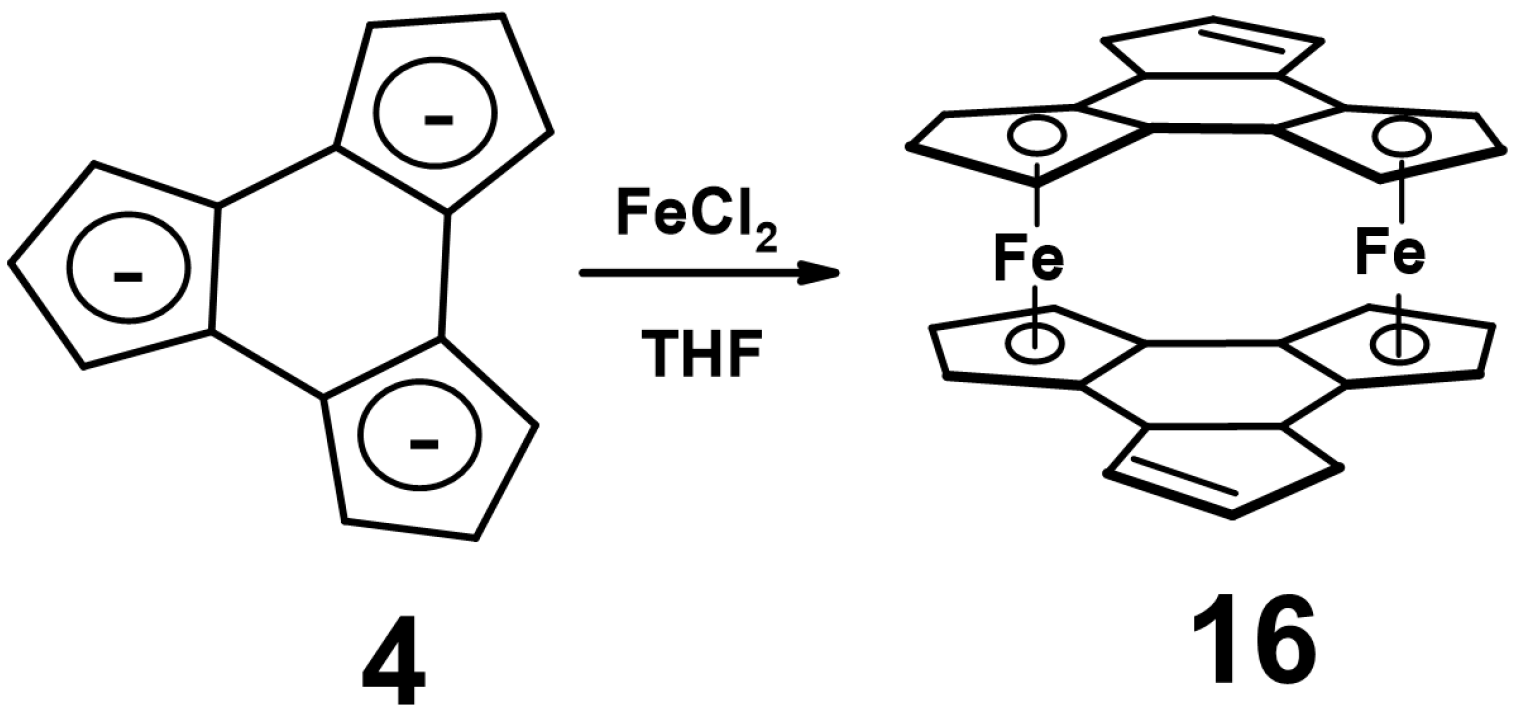
5. Metal Complexes of Decacyclene
6. Towards Sumanene
6.1. The Pyrolytic Approach
6.2. The Proposed Organometallic Approach
7. The Synthesis of Sumanene
8. Towards C60
8.1. From Decacyclene
8.2. The Designed Stepwise Synthesis of C60
9. Organic Chemistry of Trindane
9.1. Oxidation of Trindane
9.2. Synthesis of a Potential Artificial Receptor
10. Applications of Truxene and Truxenone
11. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pammer, F.; Thiel, W.R. Benzannulated homologues of cyclopentadienide as ligands in organometallic chemistry. Coord. Chem. Rev. 2014, 270–271, 14–30. [Google Scholar] [CrossRef]
- Wallach, O. Über Condensationsproducte cyclischer Ketone. Chem. Ber. 1897, 30, 1094–1096. [Google Scholar] [CrossRef]
- Mayer, R. Syntheses mit Dicarbonsäuren, Zur Selbstkondensation des Cyclopentanons. Chem. Ber. 1956, 89, 1443–1454. [Google Scholar] [CrossRef]
- Boyko, E.R.; Vaughan, P.A. Crystal structure and molecular statics of trindan C15H18. Acta Cryst. 1964, 17, 152–153. [Google Scholar] [CrossRef]
- Elmorsy, S.S.; Pelter, A.; Smith, K. The direct production of tri-substituted and hexa-substituted benzenes from ketones under mild conditions. Tetrahedron Lett. 1991, 32, 4175–4176. [Google Scholar] [CrossRef]
- Li, Z.; Sun, W.-H.; Jin, X.; Shao, C. Triple self-condensation of ketones yielding aromatics promoted with titanium tetrachloride. Synlett 2001, 12, 1947–1949. [Google Scholar] [CrossRef]
- Kotsuki, H.; Mehta, B.K.; Yanagisawa, K. Novel synthesis of fully-substituted pyridine derivatives via self-condensation of cyclic ketones in aqueous ammonium chloride under hydrothermal conditions. Synlett 2001, 1323–1325. [Google Scholar] [CrossRef]
- Katz, T.J.; Ślusarek, W. The trindene trianion. J. Am. Chem. Soc. 1980, 102, 1058–1063. [Google Scholar] [CrossRef]
- Zhou, S.-J.; Xie, S.-Y.; Huang, R.-B. 1,3,4,6,7,9-Hexabromo2,3,4,5,6,7,8,9-octahydro-1H-trindene. Acta Cryst. Sect. E Struct. Rep. Online 2007, 63, 03819. [Google Scholar] [CrossRef]
- Wester, D.W.; Coveney, J.R.; Nosco, D.L.; Robbins, M.S.; Dean, R.T. Synthesis, characterization and myocardial uptake of cationic bis(arene)technetium(I) complexes. J. Med. Chem. 1991, 34, 3284–3290. [Google Scholar] [CrossRef]
- Gupta, H.K.; Lock, P.E.; McGlinchey, M.J. Metal complexes of trindane: Possible precursors of sumanene. Organometallics 1997, 16, 3628–3634. [Google Scholar] [CrossRef]
- Gommans, L.H.P.; Main, L.; Nicholson, B.K. Metal-carbonyl complexes of dodecahydrotriphenylene; a comparison of the structures of isoelectronic [(η6-C18H24)Mn(CO)3]+ and (η6-C18H24)Cr(CO)3. J. Organomet. Chem. 1988, 346, 385–395. [Google Scholar] [CrossRef]
- Hoyano, J.K.; Graham, W.A.G. Photochemistry of (η-C5Me5)Re(CO)3: Preparation and X-ray crystal structure of (η-C5Me5)2Re2(CO)3. J. Chem. Soc. Chem. Commun. 1982, 27–28. [Google Scholar] [CrossRef]
- Gupta, H.K.; Lock, P.E.; Hughes, D.W.; McGlinchey, M.J. Trindane-ruthenium sandwich complexes: An NMR and X-ray crystallographic study of [(trindane)RuCl2]2, (trindane)RuCl2[P(OMe)3], and [(trindane)2Ru][BF4]2. Organometallics 1997, 16, 4355–4361. [Google Scholar] [CrossRef]
- Baldwin, R.; Bennett, M.A.; Hockless, D.C.R.; Pertici, P.; Verrazzani, A.; Uccello, B.G.; Marchetti, F.; Salvadori, P. Synthesis, structures and dynamic NMR spectra of η6-hexaethylbenzene complexes of ruthenium(0) and ruthenium(II). J. Chem. Soc. Dalton Trans. 2002, 23, 4488–4496. [Google Scholar] [CrossRef]
- Gupta, H.K.; Lock, P.E.; Reginato, N.; Britten, J.F.; McGlinchey, M.J. Hexaethylbenzene complexes of ruthenium and manganese—X-ray crystal structures and NMR spectra of [(HEB)2 Ru2(μ-Cl)3][C5(CO2Me)5], [(HEB)Ru(H2O)3][BF4]2, (HEB)RuCl2[P(OMe)3], trans-RuCl2(PMe3)4, and (HEB)Mn(CO)2Br. Can. J. Chem. 2006, 84, 277–287. [Google Scholar] [CrossRef]
- McGlinchey, M.J. Hexaethylbenzene: A sterically crowded arene and conformationally versatile ligand. ChemPlusChem 2018, 83, 480–499. [Google Scholar] [CrossRef]
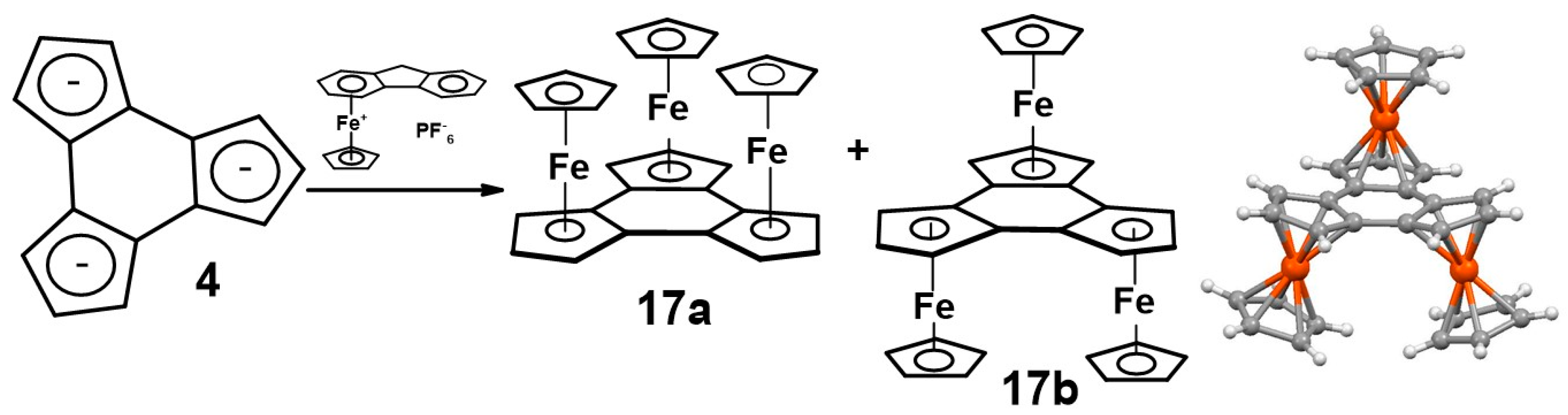
- Santi, S.; Orian, L.; Donoli, A.; Bisello, A.; Scapinello, M.; Bentollo, F.; Ganis, P.; Ceccon, A. Synthesis of the prototypical cyclic metallocene triad: Mixed-valence properties of [(FeCp)3(trindenyl)] isomers. Angew. Chem. Int. Ed. 2008, 47, 5331–5334. [Google Scholar] [CrossRef]
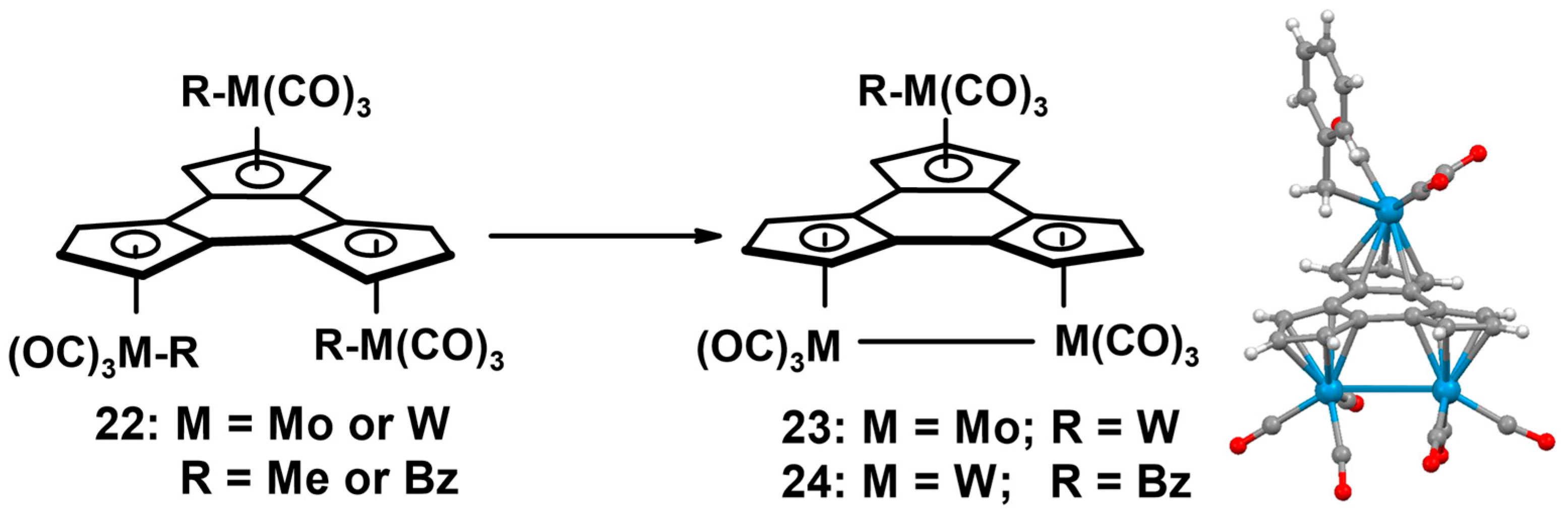
- Lynch, T.J.; Helvenston, M.C.; Rheingold, A.L.; Staley, D.L. Metal carbonyl complexes of the trindenyl ligand: Crystal structure of trans-[Re(CO)3]3(trindenyl). Organometallics 1989, 8, 1959–1963. [Google Scholar] [CrossRef]
- Lynch, T.J.; Carroll, J.M.; Helvenston, M.C.; Tisch, T.L.; Rheingold, A.L.; Staley, D.L.; Mahmoudkhani, A. Trindenyl trimolybdenum and tritungsten complexes: Crystal structure of (trindenyl)[(OC)3W-W(CO)3]WCO)3Bn. Organometallics 2012, 31, 3300–3307. [Google Scholar] [CrossRef]
- Winter, R.; Pierce, D.T.; Geiger, W.E.; Lynch, T.J. Stepwise oxidation of three communicating metal centres: Electrochemistry of trinuclear trindenyl complexes of manganese or rhodium. J. Chem. Soc. Chem. Commun. 1994, 17, 1949–1950. [Google Scholar] [CrossRef]
- Rheingold, A.L.; (Cambridge Crystallographic Data Centre, Cambridge, UK); Geiger, W.E.; (Cambridge Crystallographic Data Centre, Cambridge, UK). CSD Private communication, 2015.
- Donoli, A.; Bisello, A.; Cardena, R.; Prinzivalli, C.; Santi, S. Charge transfer properties of multi(ferrocenyl)trindenes. Organometallics 2013, 32, 1029–1036. [Google Scholar] [CrossRef]
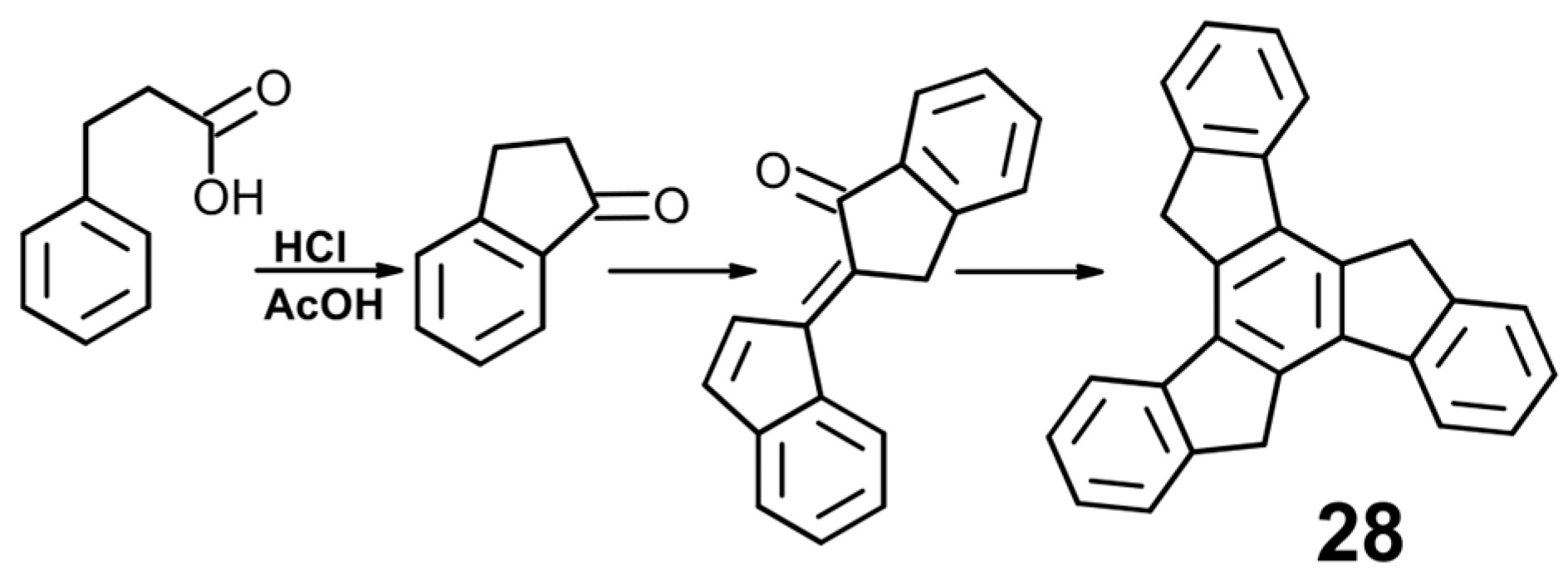
- Hausmann, J. Einwirkung von o-Cyanbenzylchlorid auf natriummalonester. Untersuchung des α-Hydrindons. Chem. Ber. 1889, 22, 2019–2026. [Google Scholar] [CrossRef]
- Górski, K.; Mech-Piskorz, J.; Pietraszkiewicz, M. From truxenes to heterotruxenes: Playing with heteroatoms and the symmetry of molecules. New J. Chem. 2022, 46, 8939–8966. [Google Scholar] [CrossRef]
- Tisch, T.L.; Lynch, T.J.; Dominguez, R. Polymetallic compounds of extended polyaromatic ligands. Rhenium and manganese carbonyl derivatives of truxene. J. Organomet. Chem. 1989, 377, 265–273. [Google Scholar] [CrossRef]
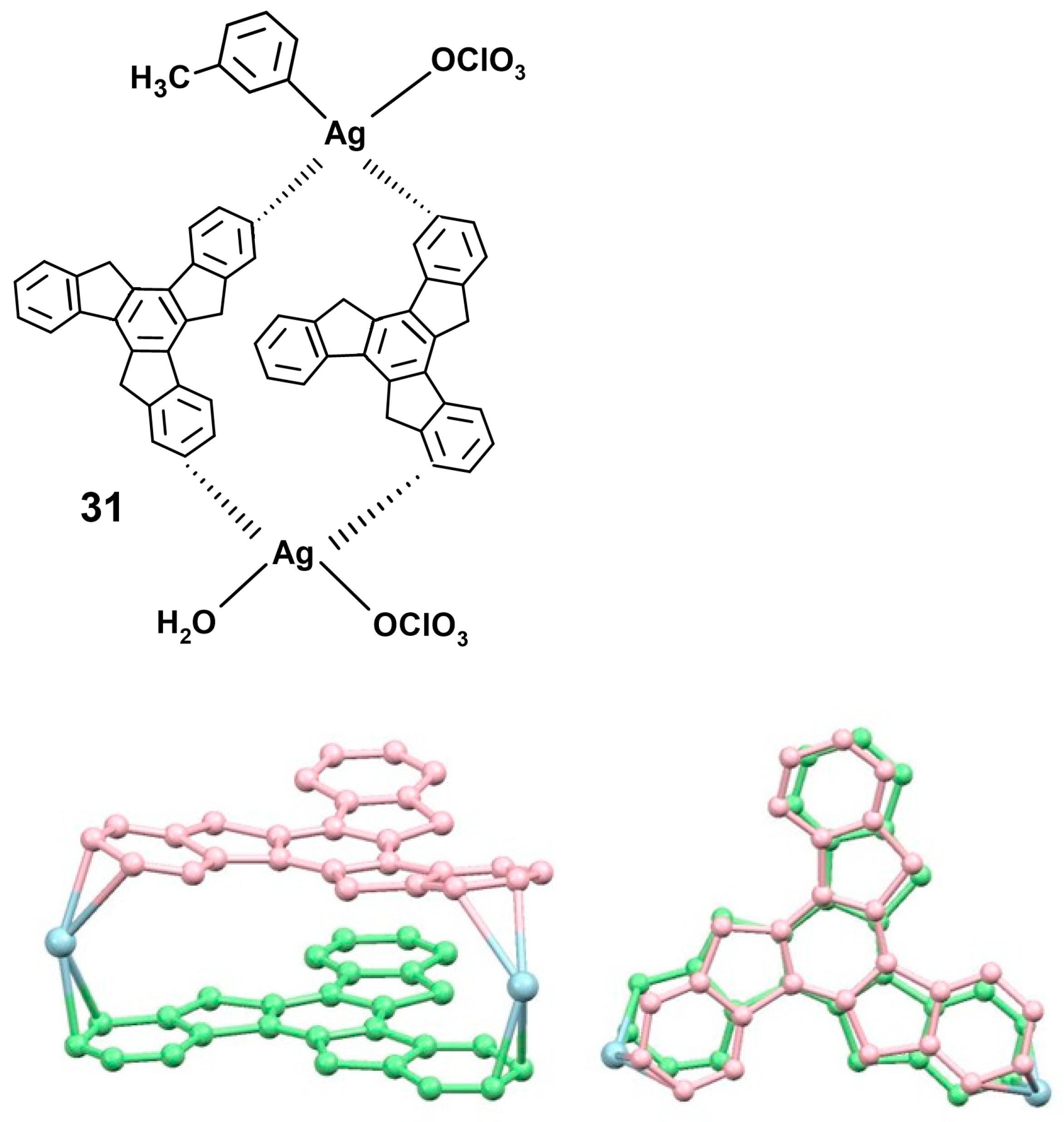
- Wombacher, T.; Foro, S.; Schneider, J.J. Synthesis and molecular structures of the cationic silver complexes of the polycondensed aromatics fluorene, pseudorubrene, and truxene. Eur. J. Inorg. Chem. 2016, 32, 5152–5160. [Google Scholar] [CrossRef]
- Dziewonski, K. Über Dekacyclen (Trinaphtylenbenzol) einen neuen hochmolekuren aromatischen Kohlenwasserstoff, und über Dinaphtylenthiophen, einen rothen Thiokörper. Chem. Ber. 1903, 36, 962–971. [Google Scholar] [CrossRef]
- Ho, D.M.; Pascal, R.A., Jr. Decacyclene: A molecular propeller with helical crystals. Chem. Mat. 1993, 5, 1358–1361. [Google Scholar] [CrossRef]
- Iglesias, B.; Peña, D.; Pérez, D.; Guitián, E.; Castedo, L. Palladium-catalyzed trimerization of strained cycloalkynes: Synthesis of decacyclene. Synlett 2002, 3, 486–488. [Google Scholar] [CrossRef]
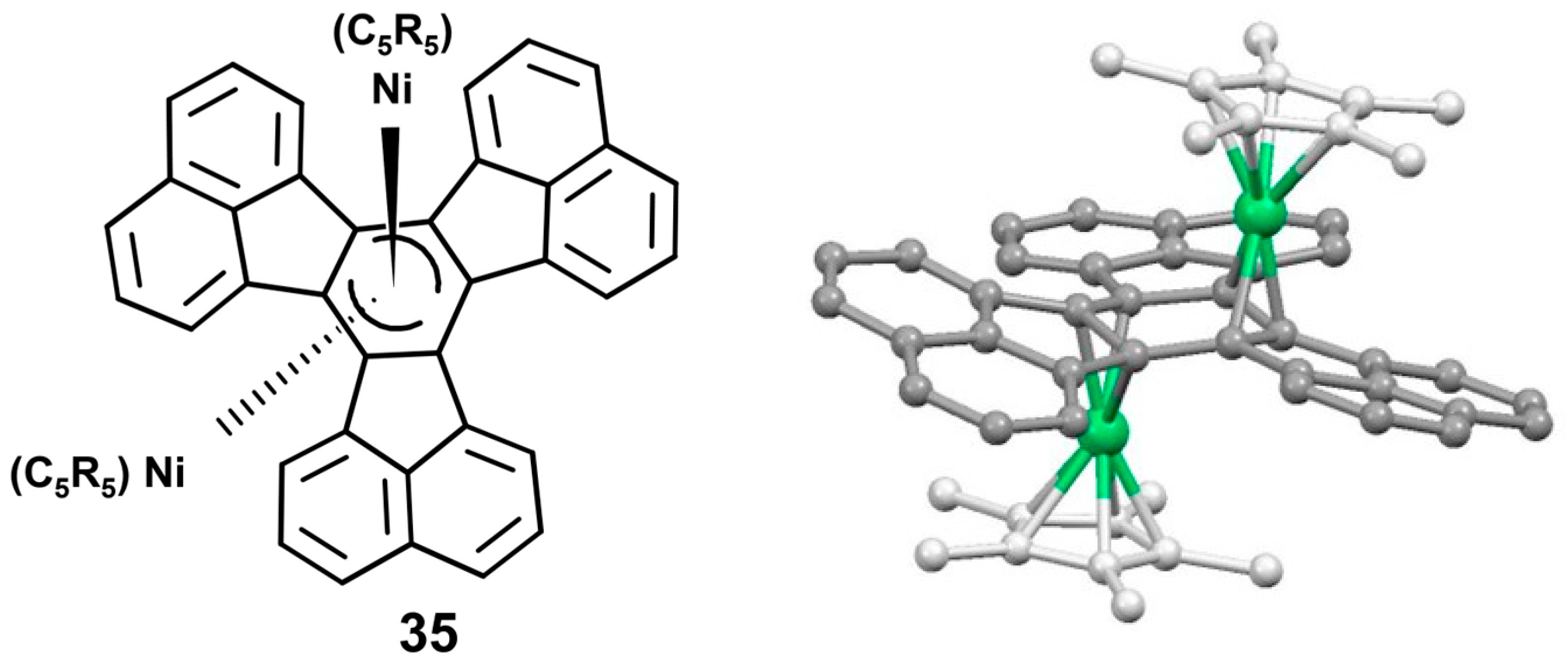
- Schneider, J.J.; Spickermann, D.; Bläser, D.; Boese, R.; Rademacher, P.; Labahn, T.; Magull, J.; Janiak, C.; Seidel, N.; Jakob, K. A π-stacked organometallic propeller: Experimental and theoretical studies on reactivity and bonding in the π-arene-bridged nickel triple-decker [{η5-Me4EtC5)Ni}2(μ-η3:η3-decacyclene)]. Eur. J. Inorg. Chem. 2001, 2001, 1371–1382. [Google Scholar] [CrossRef]
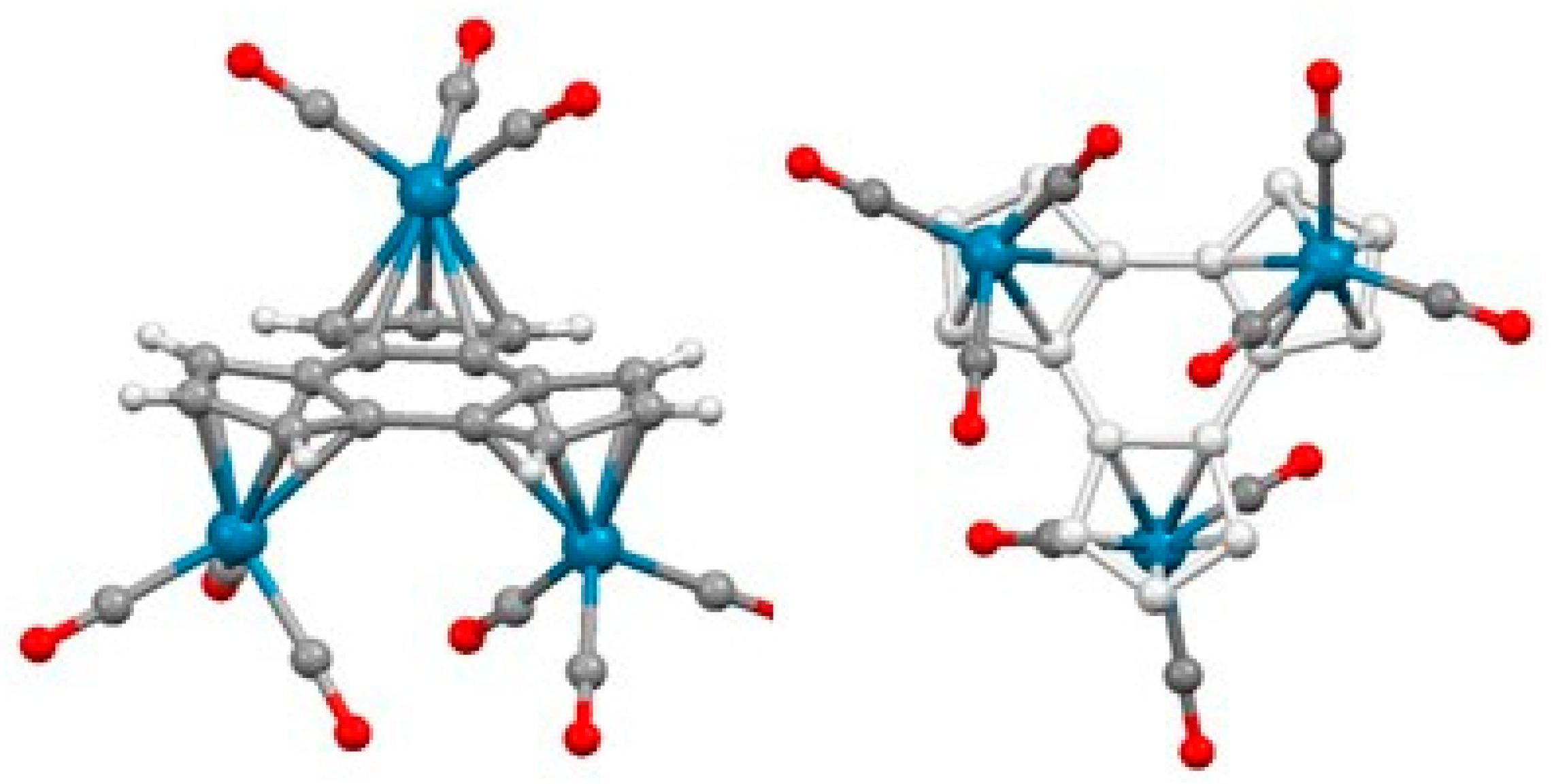
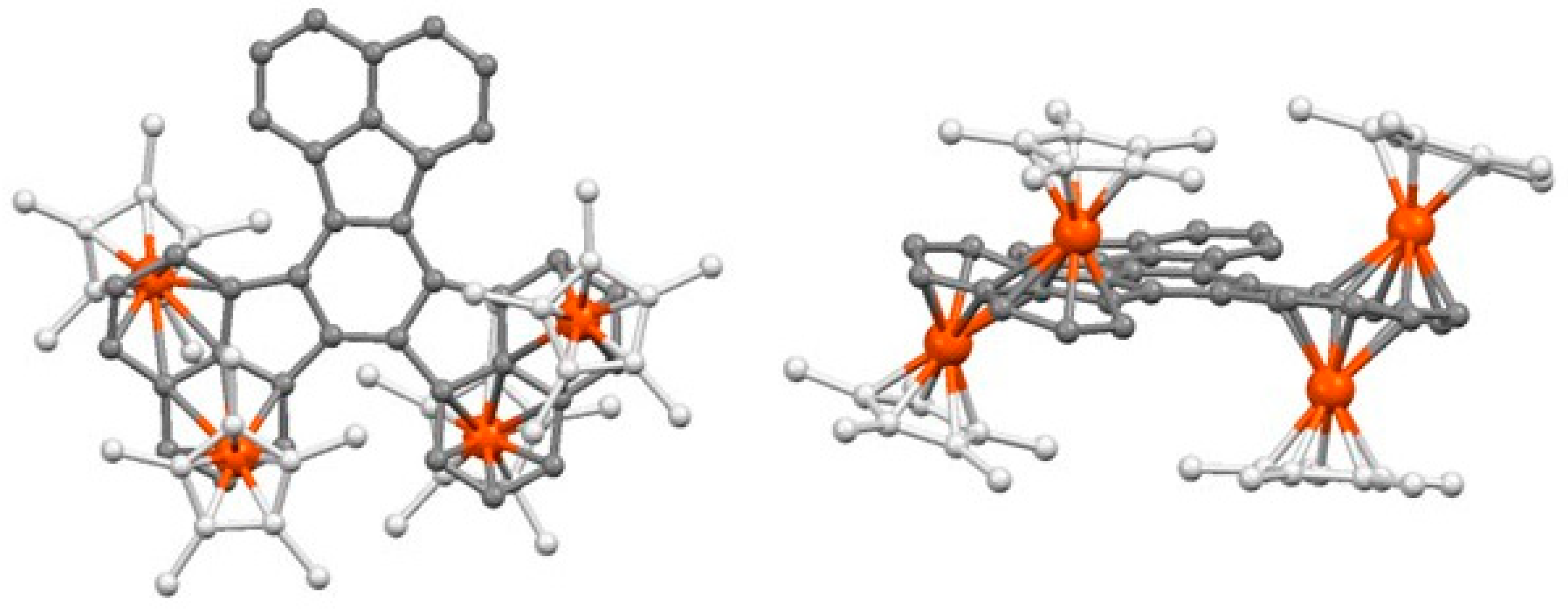
- Schneider, J.J.; Spickermann, D.; Lehmann, C.W.; Magull, J.; Krüger, H.-J.; Ensling, J.; Gütlich, P. Decacyclene as complexation manifold: Synthesis, structure and properties of its Fe2 and Fe4 slipped triple-decker complexes. Chem. Eur. J. 2006, 12, 1427–1435. [Google Scholar] [CrossRef]
- Scott, L.T.; Hashemi, M.M.; Meyer, D.T.; Warren, H.B. Corannulene, A three-step synthesis. J. Am. Chem. Soc. 1997, 119, 10963–10968. [Google Scholar] [CrossRef]
- Butterfield, A.M.; Gilomen, B.; Siegel, J.S. Kilogram-scale production of corannulene. Org. Process. Res. Dev. 2012, 16, 664–676. [Google Scholar] [CrossRef]
- Yu, Y.T.; Siegel, J.S. Aromatic molecular-bowl hydrocarbons: Synthetic derivatives, their structures and physical properties. Chem. Rev. 2016, 106, 4843–4867. [Google Scholar] [CrossRef]
- Scott, L.T. Methods for the chemical synthesis of fullerenes. Angew. Chem. Int. Ed. 2004, 43, 4994–5007. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Shah, S.R.; Ravikumar, K. Towards the design of tricyclopenta[def,jkl,pqr] triphenylene (Sumanene): A bowl-shaped hydrocarbon featuring a structural motif present in C60 (Buckminsterfullerene). J. Chem. Soc. Chem. Commun. 1993, 12, 1006–1008. [Google Scholar] [CrossRef]
- Sastry, G.N.; Jemmis, E.D.; Mehta, G.; Shah, S.R. Synthetic strategies towards C60. Molecular mechanics and MNDO study on sumanene and related structures. J. Chem. Soc. Perkin Trans. 1993, 2, 1867–1871. [Google Scholar] [CrossRef]
- Priyakumar, U.D.; Sastry, G.N. Theory provides a clue to accomplish the synthesis of sumanene, C21H12, the prototypical C3v-buckyball. Tetrahedron Lett. 2001, 42, 1379–1381. [Google Scholar] [CrossRef]
- March, J. Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 4th ed.; John Wiley & Sons: New York, NY, USA, 1992; pp. 1030–1032. [Google Scholar]
- Hamon, J.-R.; Saillard, J.-Y.; Le Beuze, A.; McGlinchey, M.J.; Astruc, D. Organometallic electron reservoirs. 7. One-step multiple formation of C-C bonds in CpFe(arene)+ sandwiches and unusual C6Et6 geometry in the X-ray crystal structure of CpFe+(η6-C6Et6)BF4−. J. Am. Chem. Soc. 1982, 104, 7549–7555. [Google Scholar] [CrossRef]
- LaBrush, D.M.; Eyman, D.P.; Baenziger, N.C.; Mallis, L.M. Preparation, characterization, and reactivity of (η5-pentamethylbenzyl)manganese tricarbonyl. Organometallics 1991, 10, 1026–1033. [Google Scholar] [CrossRef]
- Moler, J.L.; Eyman, D.P.; Neilson, J.M.; Morken, A.M.; Schauer, S.J.; Snyder, D.B. (η5-Benzyl) manganese complexes. 2. Preparation, characterization and reactivity of [η5-C6Me5(CH2)] Mn(CO)2PR3 (R = n-Bu, Me, Ph, OMe, OPh). Structure of [η5-C6Me5(CH2)]Mn(CO)2PMe3. Organometallics 1993, 12, 3304–3315. [Google Scholar] [CrossRef]
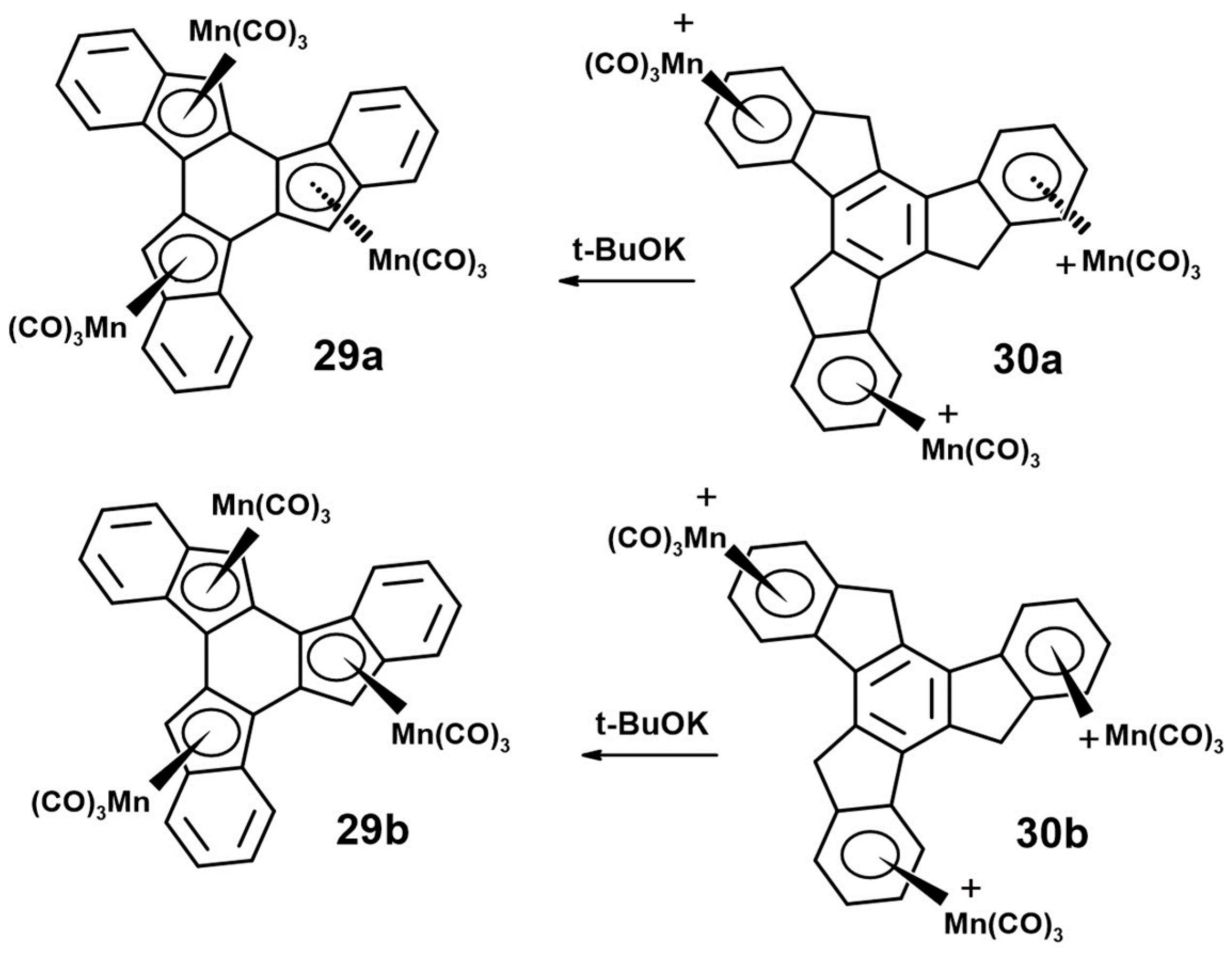
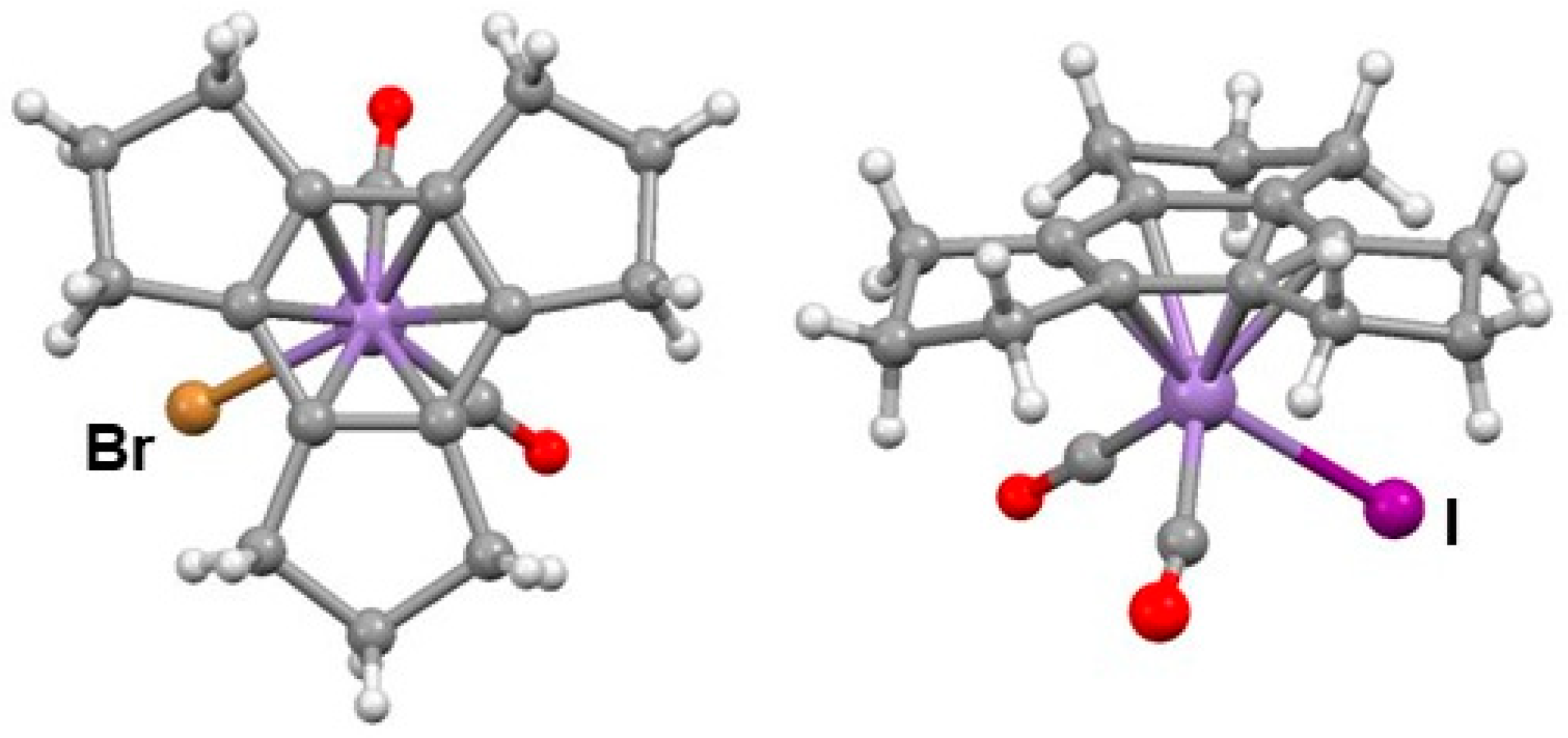
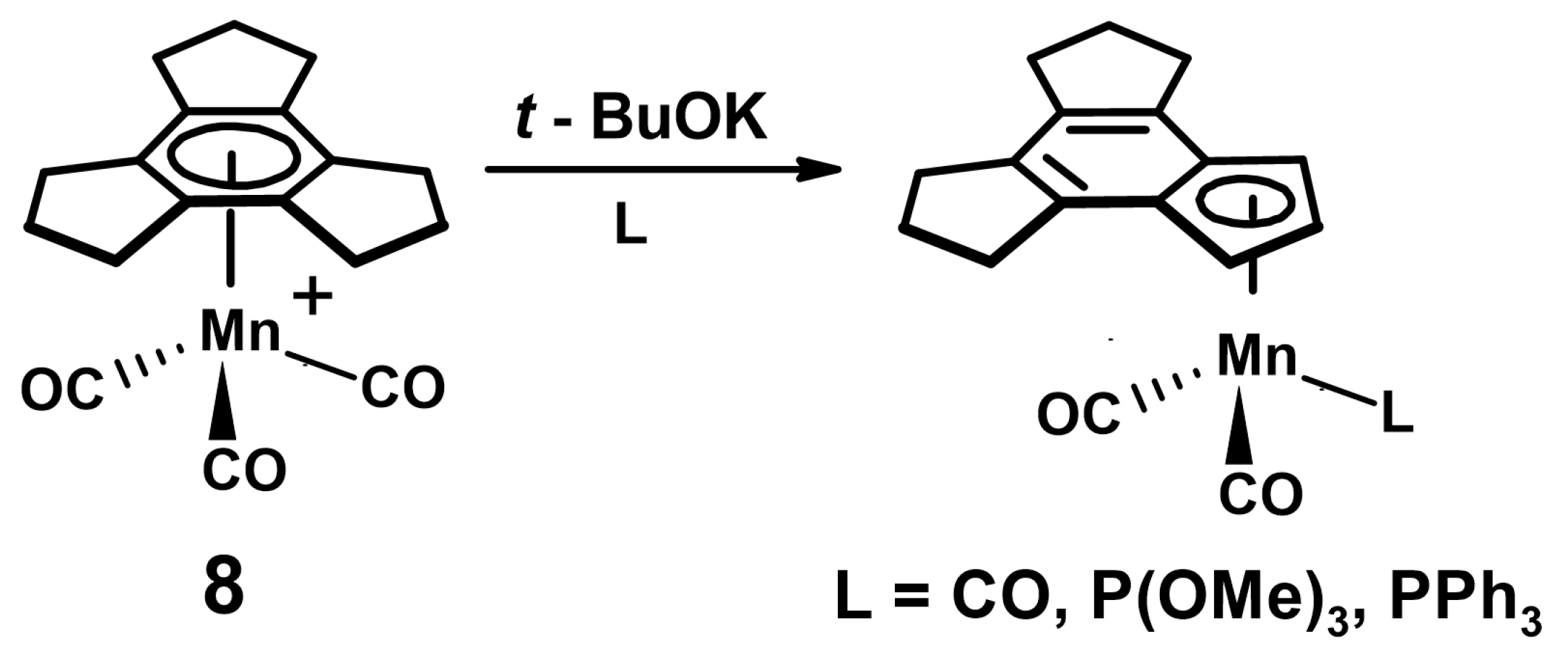
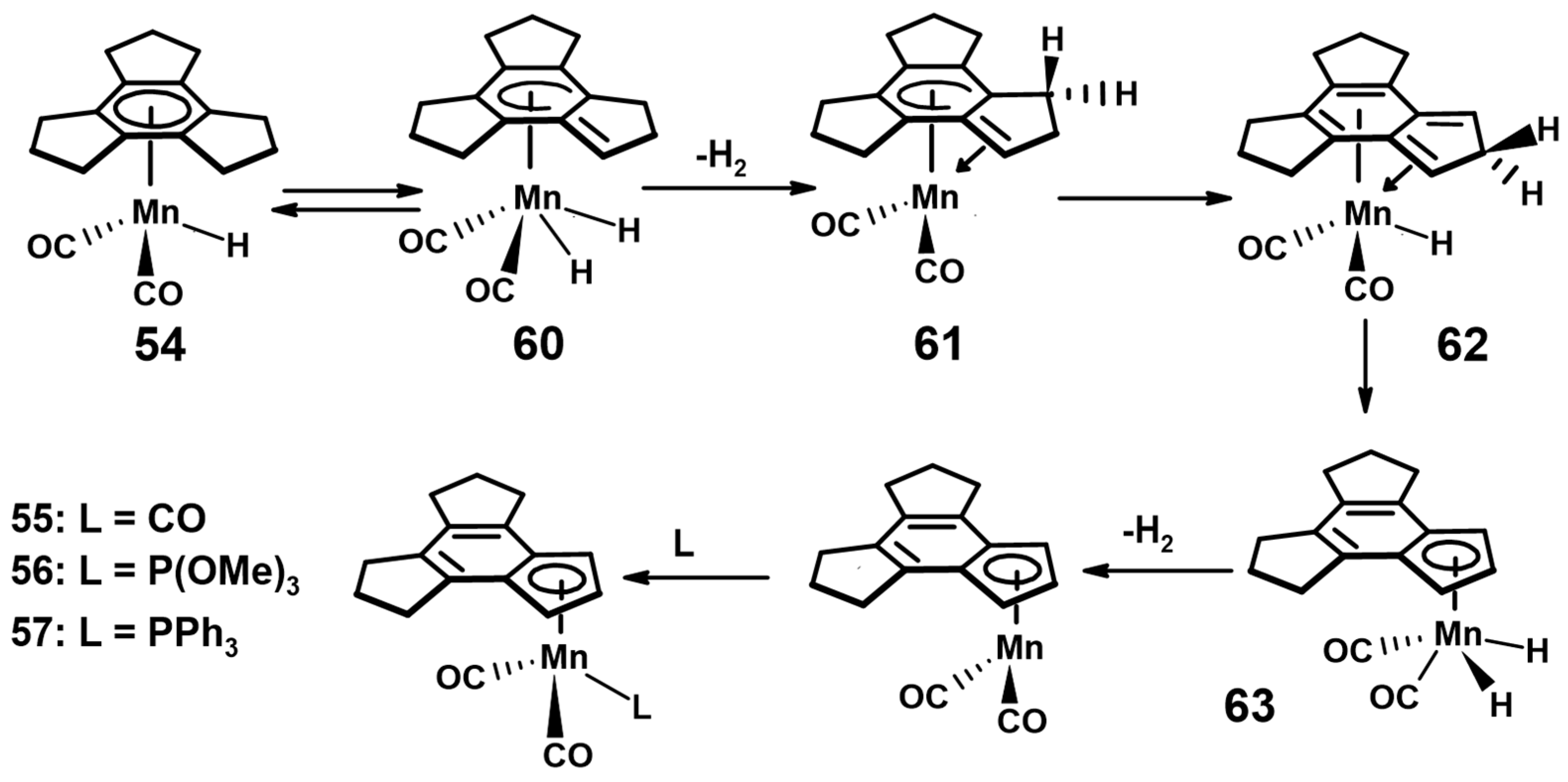
- Reginato, N.; McGlinchey, M.J. Unexpected reaction of (trindane)Mn(CO)3+ BF4− with potassium tert-butoxide: Three C-H insertions and a haptotropic shift. Organometallics 2001, 20, 4147–4149. [Google Scholar] [CrossRef]
- Bernhardt, R.J.; Eyman, D.P. Synthesis and characterization of (arene)manganese dicarbonyl halides and hydride. Organometallics 1984, 3, 1445–1446. [Google Scholar] [CrossRef]
- Schlom, P.J.; Morken, A.M.; Eyman, D.P.; Baenziger, N.C.; Schauer, S.J. Reactivity and structure of (η6-C6(CH3)6Mn(CO)2H: Stable alkylrhenium analogues. Organometallics 1993, 12, 3461–3467. [Google Scholar] [CrossRef]
- Bernhardt, R.J.; Wilmoth, M.A.; Weers, J.J.; LaBrush, D.M.; Eyman, D.P.; Huffman, J.C. Derivatives of the (arene)M(CO)2 fragment. The molecular structure of (η6-C6Me6)Mn(CO)2Cl. Organometallics 1986, 5, 883–888. [Google Scholar] [CrossRef]
- Walker, P.J.C.; Mawby, R.J. Patterns of nucleophilic attack on tricarbonyl π-arene complexes of manganese(I). Inorg. Chim. Acta 1973, 7, 621–625. [Google Scholar] [CrossRef]
- Reginato, N.; Harrington, L.E.; Ortin, Y.; McGlinchey, M.J. Unexpectedly different reactions of [(arene)Mn(CO)3]+ cations (arene = trindane, indane, tetralin or dibenzosuberane) with potassium t-butoxide—C-H insertions, haptotropic shifts, dimerization, or elimination. Can. J. Chem. 2009, 87, 232–246. [Google Scholar] [CrossRef]
- King, R.B. Organometallic chemistry of the transition metals XXIII. Some indenyl and fluorenyl derivatives of manganese carbonyl. J. Organomet. Chem. 1970, 23, 527–529. [Google Scholar] [CrossRef]
- Crabtree, R.H.; Parnell, C.P. Haptotropic rearrangements and C-H activation in indane, indenyl, and naphthalene complexes of iridium. Organometallics 1984, 3, 1727–1731. [Google Scholar] [CrossRef]
- Trifonova, O.I.; Ochertyanova, E.A.; Akhmedov, N.G.; Roznyatovsky, V.A.; Laikov, D.N.; Ustynyuk, N.A.; Ustynyuk, Y.A. Ricochet inter-ring haptotropic rearrangement of σ-methyl-(η5-indenyl)chromium tricarbonyls. Experimental, kinetic and theoretical DFT study. Inorg. Chim. Acta 1998, 280, 328–338. [Google Scholar] [CrossRef]
- Sakurai, H.T.; Daiko, T.; Hirao, T.A. Synthesis of sumanene, a fullerene fragment. Science 2003, 301, 1878. [Google Scholar] [CrossRef] [PubMed]
- Amaya, T.; Hirao, T.A. Chemistry of sumanene. Chem. Rec. 2015, 15, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Alvi, S.; Ali, R. Synthetic approaches to bowl-shaped π-conjugated sumanene and its congeners. Beilstein J. Org. Chem. 2020, 16, 2212–2259. [Google Scholar] [CrossRef] [PubMed]
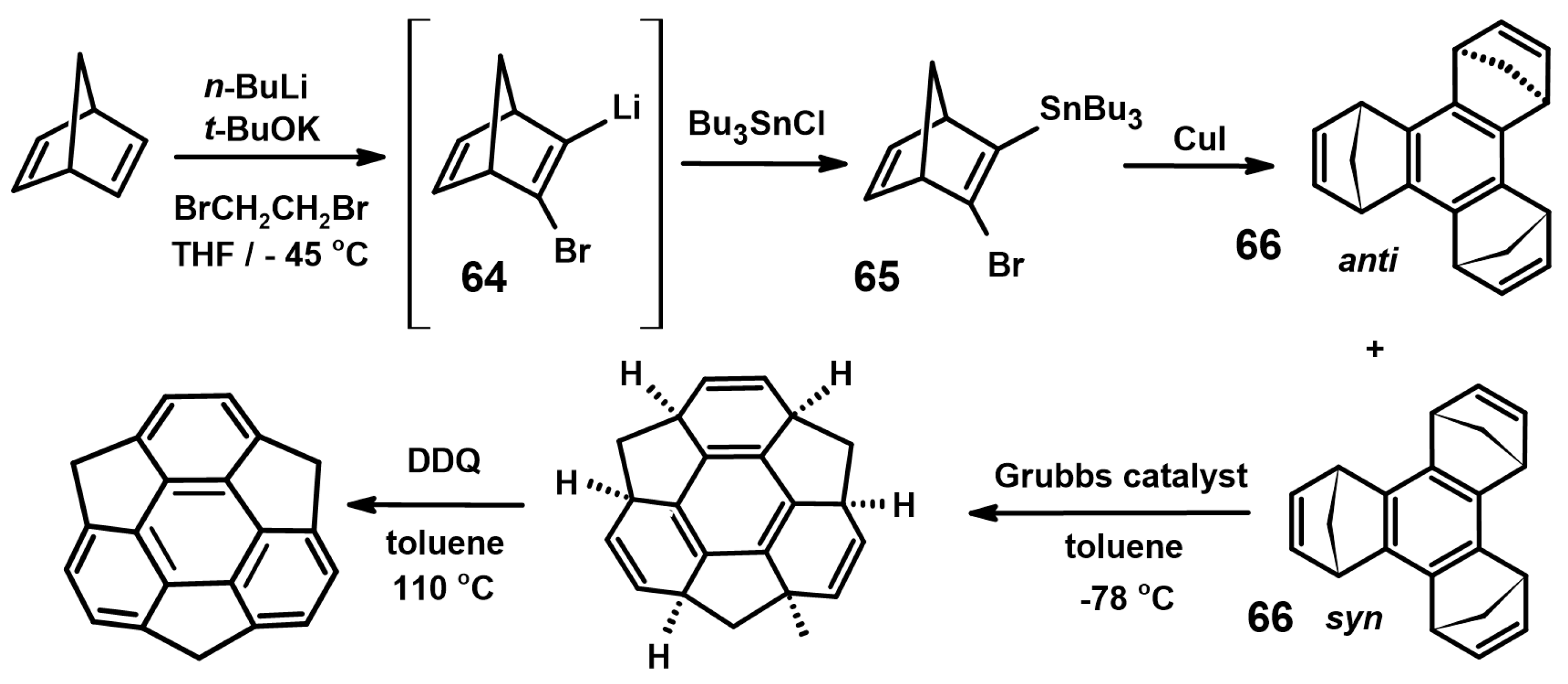
- Ansems, R.B.M.; Scott, L.T. Circumtrindene: A geodesic dome of molecular dimensions. Rational synthesis of 60% of C60. J. Am. Chem. Soc. 2000, 122, 2719–2724. [Google Scholar] [CrossRef]
- Forkey, D.M.; Attar, S.; Noll, B.C.; Koerner, R.; Olmstead, M.; Balch, A.L. Crystallographic characterization of the molecular structure and solid state packing of the fullerene-shaped hydrocarbon C36H12. J. Am. Chem. Soc. 1997, 119, 5766–5767. [Google Scholar] [CrossRef]
- Scott, L.T.; Boorum, M.M.; McMahon, B.J.; Hagen, S.; Mack, J.; Blank, J.; Wegner, H.; de Meijere, A. A rational synthesis of C60. Science 2002, 295, 1500–1503. [Google Scholar] [CrossRef] [PubMed]
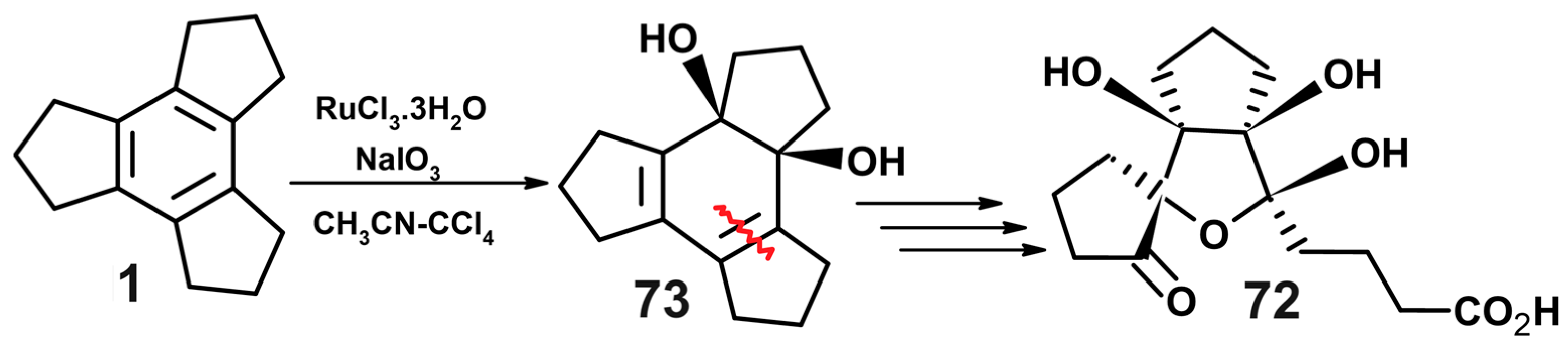
- Ranganathan, S.; Muraleedharan, K.M.; Bharadwaj, P.; Madhusudanan, K.P. One step trans formation of tricyclopentabenzene (trindane) [C15H18] to 4-[1R,2S,4R,5S)-1,2,5-tri hydroxy-3-oxabicyclo[3,3,0]octane-4-spiro-1’-(2’-oxocyclopentan)-2-yl]butanoic acid [C15H22O7]. J. Chem. Soc. Chem. Commun. 1998, 3, 2239–2240. [Google Scholar] [CrossRef]
- Ranganathan, S.; Muraleedharan, K.M.; Rao, C.C.; Vairamani, M.; Karle, I.L. One-step transformation of tricyclopentabenzene (trindane, C15H18) to bicyclo(10.3.0)pentadec-1(12)ene-2,6,7,11-tetrone (C15H18O4) and its aldol product 12-hydroxy-16-oxatetracyclo-(10.3.1.0.1,5O7,11))hexadec-7(11)ene-2,6-dione (C15H18O4). Org. Lett. 2001, 3, 2447–2449. [Google Scholar] [CrossRef]
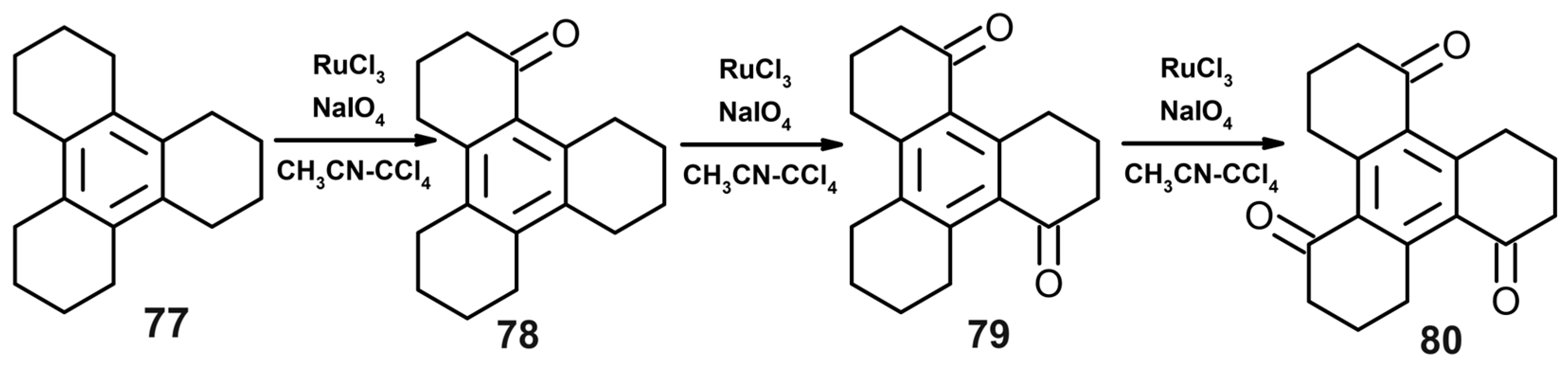
- Bhat, G.J.; Deota, P.T.; Upadhyay, D.; Jha, P.K. Site-selective unidirectional benzylic sp3 C-H oxidation of dodecahydrotriphenylene with RuCl3-NaIO4: Formation of benzylic ketones. RSC Adv. 2021, 55, 34498–34502. [Google Scholar] [CrossRef]
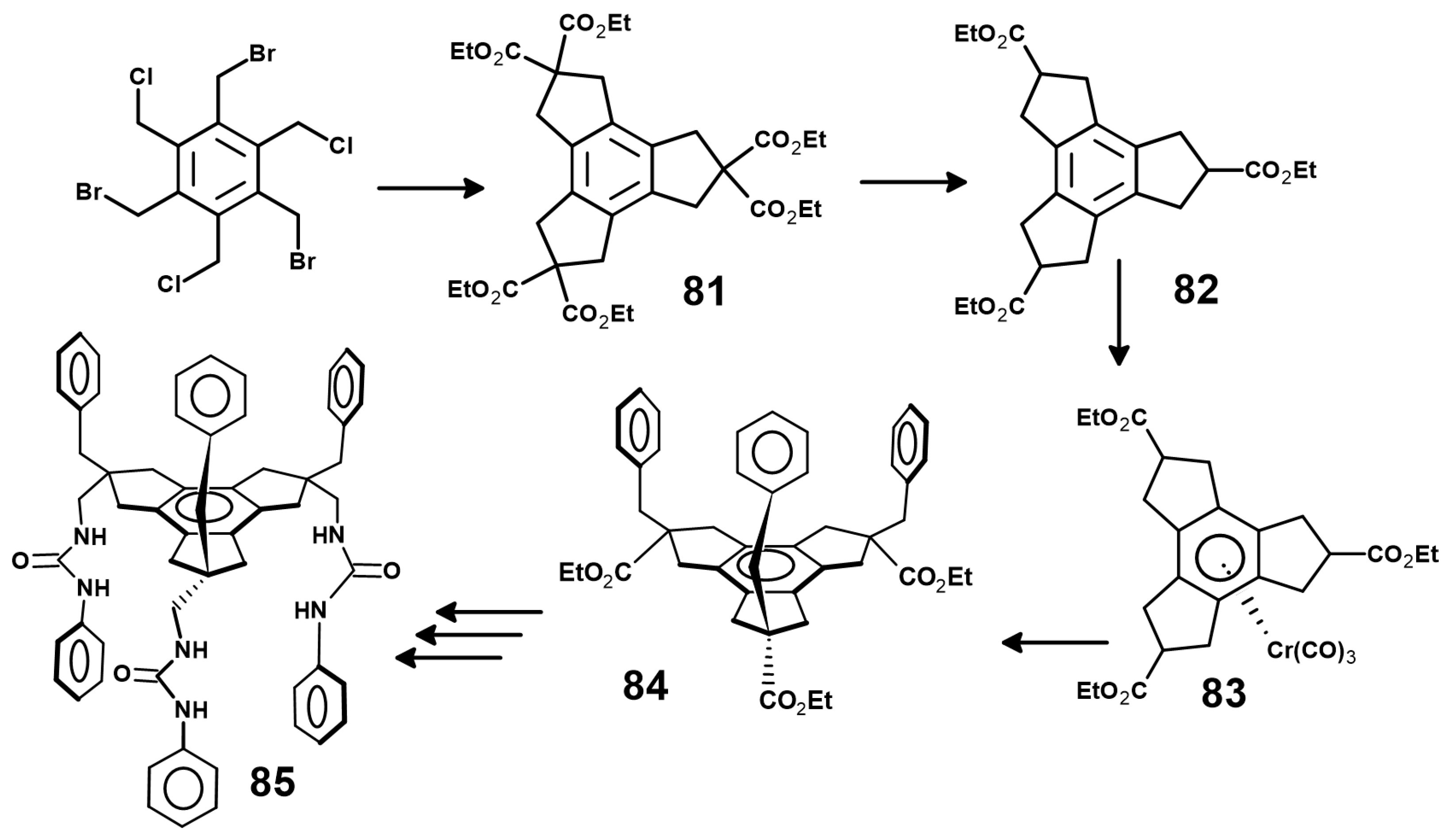
- Choi, H.-J.; Park, Y.S.; Yun, S.H.; Kim, H.-S.; Cho, C.S.; Ko, K.; Ahn, K.H. Novel C3v-symmetric tripodal scaffold, triethyl cis,cis,cis-2,5,8-tribenzyltrindane-2,5,8-tricarboxylate, for the construction of artificial receptors. Org. Lett. 2002, 4, 795. [Google Scholar] [CrossRef]
- Oded, Y.N.; Agranat, I. A simple synthesis of truxene, a building block for optoelectronics and fullerene fragments. Tetrahedron Lett. 2014, 55, 636–638. [Google Scholar] [CrossRef]
- Goubard, F.; Dumur, F. Truxene: A promising scaffold for future materials. RSC Advan. 2015, 5, 3521–3551. [Google Scholar] [CrossRef]
- Wagay, S.A.; Rather, I.A.; Ali, R. Functionalized truxene scaffold: A promising advanced organic material for digital era. Chem. Select. 2019, 4, 12272–12288. [Google Scholar] [CrossRef]
- Destrade, C.; Gasparoux, H.; Babeau, A.; Tinh, N.H.; Malthete, J. Truxene derivatives: A new family of disc-like liquid crystal with inverted nematic-columnar sequence. Mol. Cryst. Liq. Cryst. 1981, 67, 37–47. [Google Scholar] [CrossRef]
- Singh, H.; Tomer, V.K.; Jena, N.; Bala, I.; Sharma, N.; Nepak, D.; De Sarkar, A.; Kailasam, K.; Pal, S.K. A porous crystalline truxene-based covalent organic framework and its application in humidity sensing. J. Mater. Chem. A 2017, 5, 21820–21827. [Google Scholar] [CrossRef]
- Guadaupe, J.; Ray, A.M.; Maya, E.M.; Gómez-Lor, B.; Iglesias, M. Truxene-based porous polymers: From synthesis to catalytic activity. Polym. Chem. 2018, 9, 4585–4595. [Google Scholar] [CrossRef]
- Lv, Q.; Feng, S.; Jing, L.; Zhang, Q.; Qi, M.; Wang, J.; Bai, H.; Fu, R. Features of a truxene-based stationary phase in capillary gas chromatography for separation of some challenging isomers. J. Chromatogr. A 2016, 1454, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Liang, M.; Wang, Z.; Cheng, F.; Wang, C.; Sun, Z.; Xue, S. Synthesis of new truxene based organic sensitizers for iodine-free dye-sensitized solar cells. Tetrahedron 2013, 69, 10573–10580. [Google Scholar] [CrossRef]
- Lin, K.-H.; Prlj, A.; Corminboeuf, C. A rising star: Truxene as a promising hole transport material in perovskite solar cells. J. Phys. Chem. C 2017, 121, 21729–21739. [Google Scholar] [CrossRef]
- Sbrogiò, F.; Fabris, F.; De Lucchi, O.; Lucchini, V. 5,10,15-Trimethenetribenzo[a,f,k]trindene (truxenene). Synlett 1994, 1994, 761–762. [Google Scholar] [CrossRef]
- Dehmlow, E.V.; Kelle, T. Synthesis of new truxene derivatives: Possible precursors of fullerene partial structures. Synth. Commun. 1997, 27, 2021–2031. [Google Scholar] [CrossRef]
- Plater, M.J.; Praveen, M. A new synthesis of truxenone. Tetrahedron Lett. 1997, 38, 1081–1082. [Google Scholar] [CrossRef]
- Wu, M.; Liu, H.; Liu, H.; Lu, T.; Wang, S.; Niu, G.; Sui, L.; Bai, F.; Yang, B.; Wang, K.; et al. Pressure-induced restricting intermolecular vibration of a herringbone dimer for significantly enhanced multicolor emission in rotor-free truxene crystals. J. Phys. Chem. Lett. 2022, 13, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
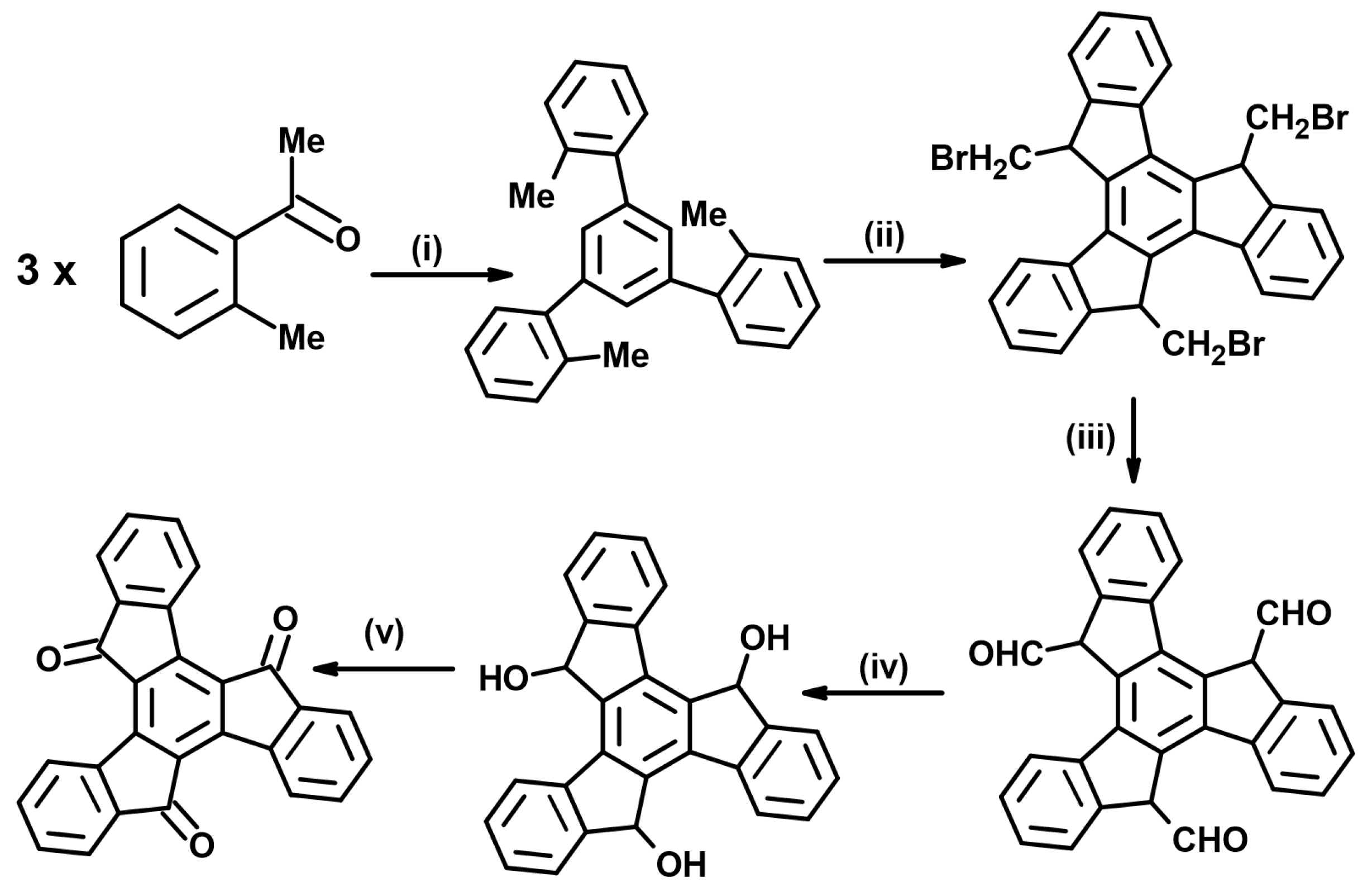
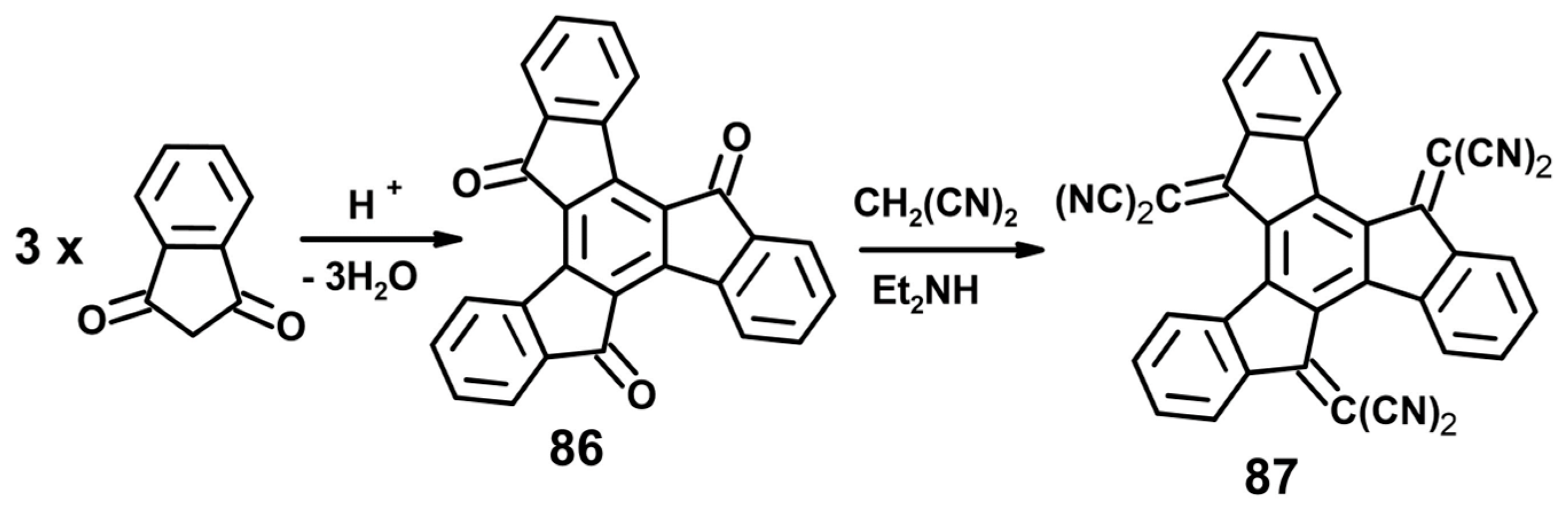
- Jacob, K.; Becker, J.Y.; Ellern, A.; Khordorkovsky, V. Synthesis of novel truxenequinone based electron acceptor. Tetrahedron Lett. 1999, 40, 8625–8628. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Voroshazi, E.; Holliday, S.; Cnops, K.; Rand, B.P.; McCulloch, A. Efficient truxenone-based acceptors for organic photovoltaics. J. Mater. Chem. A 2013, 1, 73–76. [Google Scholar] [CrossRef]
- Gómez-Esteban, S.; Benito-Hernandez, A.; Termine, R.; Hennrich, G.; López Navarrete, J.T.; Ruiz Delgado, M.C.; Golemme, A.; Gómez-Lor, B. High-mobility self-assembling truxenone-based n-type organic semiconductors. Chem. Eur. J. 2018, 24, 3576–3583. [Google Scholar] [CrossRef]
- Yang, X.; Hu, Y.; Dunlap, N.; Wang, X.; Huang, S.; Su, Z.; Sharma, S.; Jin, Y.; Huang, F.; Wang, X. A truxenone-based covalent organic framework as an all-solid-state lithium-ion battery cathode with high capacity. Angew. Chem. Int. Ed. 2020, 59, 20385–20389. [Google Scholar] [CrossRef]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lock, P.E.; Reginato, N.; Bruno-Colmenárez, J.; McGlinchey, M.J. Syntheses, Structures and Reactivity of Metal Complexes of Trindane, Trindene, Truxene, Decacyclene and Related Ring Systems: Manifestations of Three-Fold Symmetry. Molecules 2023, 28, 7796. https://doi.org/10.3390/molecules28237796
Lock PE, Reginato N, Bruno-Colmenárez J, McGlinchey MJ. Syntheses, Structures and Reactivity of Metal Complexes of Trindane, Trindene, Truxene, Decacyclene and Related Ring Systems: Manifestations of Three-Fold Symmetry. Molecules. 2023; 28(23):7796. https://doi.org/10.3390/molecules28237796
Chicago/Turabian StyleLock, Philippa E., Nada Reginato, Julia Bruno-Colmenárez, and Michael J. McGlinchey. 2023. "Syntheses, Structures and Reactivity of Metal Complexes of Trindane, Trindene, Truxene, Decacyclene and Related Ring Systems: Manifestations of Three-Fold Symmetry" Molecules 28, no. 23: 7796. https://doi.org/10.3390/molecules28237796
APA StyleLock, P. E., Reginato, N., Bruno-Colmenárez, J., & McGlinchey, M. J. (2023). Syntheses, Structures and Reactivity of Metal Complexes of Trindane, Trindene, Truxene, Decacyclene and Related Ring Systems: Manifestations of Three-Fold Symmetry. Molecules, 28(23), 7796. https://doi.org/10.3390/molecules28237796