2.1. Interpretation of VT-NMR and Characterization of the Studied Compound 1
Compound
1 was synthesized recently by an alternative synthetic approach [
28,
29,
30]. Its structure has been confirmed by NMR and X-ray analyses. However, this compound shows flexibility, whose rate falls in the NMR time scale. We studied the amide and enamine rotational barriers at both the CC–N and C(O)–N bonds (
Figure 1). Before investigating the rotational barriers using
1H VT NMR, the four N-Me signals were assigned in order to understand which rotations occur. If we number the methyl groups by increasing chemical shift, then the methyl signals 1 and 4 are exchanging, whereas signals 2 and 3 are exchanging as well, albeit slightly more slowly (
Figures S1 and S6). The assignment of the methyl signals in the proton spectrum is supported by the following observations: the NOESY spectrum shows proximity between methyl 3 and CH (
Figure S2); the
1H,
15N-HMBC spectrum shows the proton–nitrogen correlations between the methine proton and those of enamine methyl groups (
Figure S5). A coupling constant of 0.4 Hz between protons from
N-methyl 2 and the methine proton is observed. The signals of amide methyls 1 and 4 were determined using the anisotropy effect of the carbonyl group. The assignments of the methyl signals in the carbon spectrum are based on the HSQC correlations (
Figure S4).
2.2. Dynamic NMR Studies
We measured the VT spectra of compound
1 in two aprotic solvents: in CDCl
3 in steps of 5 K between 283 and 328 K and in TCE-d2 in steps of 10 K between 263 and 393 K. The
N-methyl signals of the amide group (denoted as 1 and 4 in
Figure 1) are more sensitive to the increase in the temperature and in CDCl
3 become broadened at 313 K, while the
N-methyl signals of the enamine group (denoted as 2 and 3 in
Figure 1) remain in a slow range of exchange until 323 K (
Figure S8). In TCE-d2, the signals of both exchanging methyl pairs shift from a slow range of exchange to a fast range of exchange (
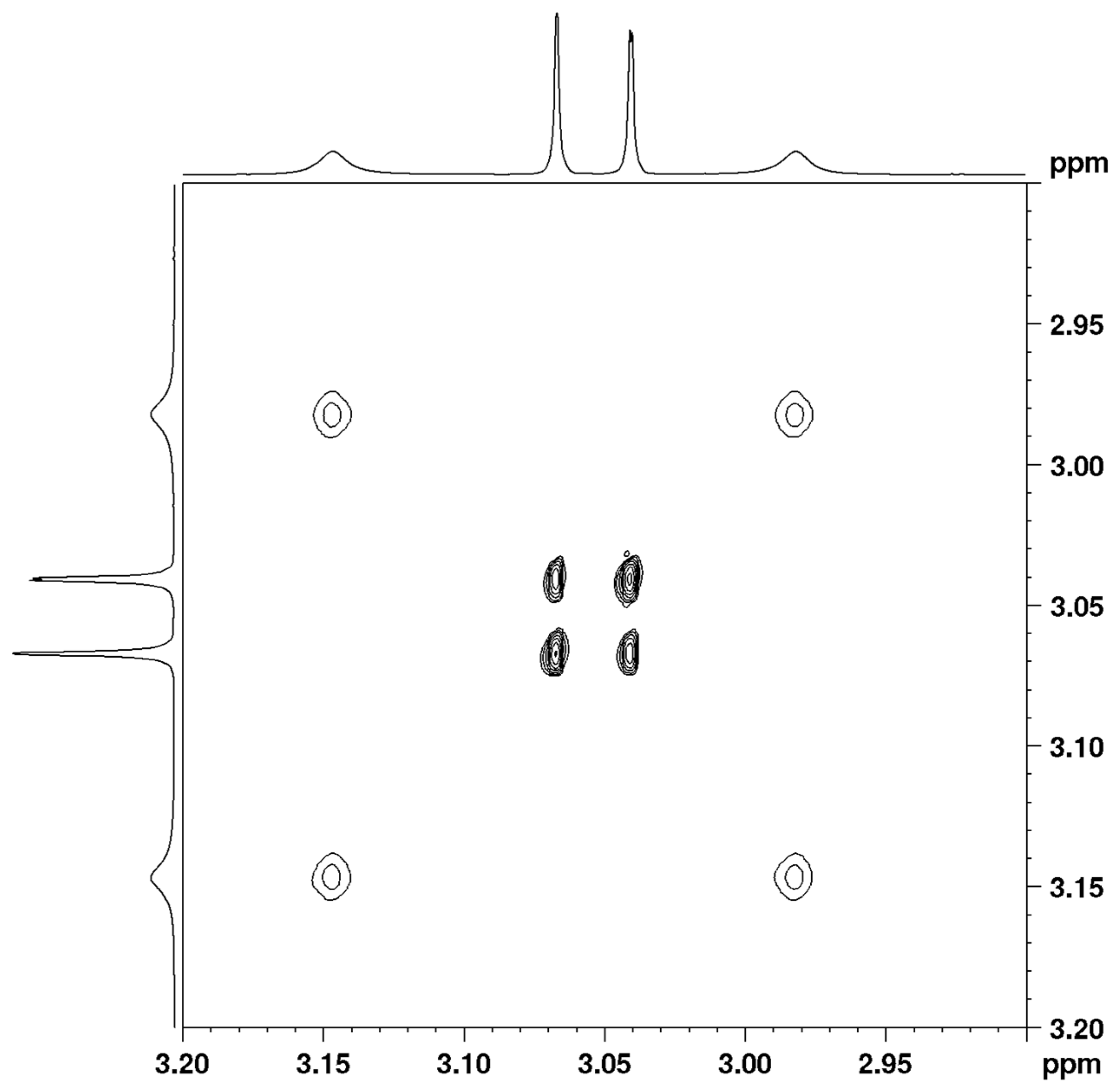
Figure 2 and
Figure S11). The methyl signals of the amide group merge into a lower-field signal (3.10 ppm) at 393 K, while the methyl signals of the enamine group merge into a single stronger-field signal (3.08 ppm) at 393 K.
The VT spectra of compound 1 in CDCl3 are in the slow range of exchange and therefore they are suitable for study using 2D EXSY, 1D EXSY, or magnetization–transfer experiments. We prefer to adopt 2D EXSY experiments as a well-established approach and an easy way to derive rate constants from 2D integrals.
The VT spectra of compound
1 in TCE-d2 were also studied using 2D EXSY spectra in a slow range of exchange (
Figure 3 and
Figure S12) and using complete line shape analysis (CLSA) in the entire temperature range (
Figure S30).
The 2D EXSY results in both solvents are almost equal and show no solvent dependence of the rotational barriers (
Table 1 and
Table 2). The unexpected result is that the enamine rotational barrier is 18.6 kcal/mol, a much higher value than the amide rotational barrier of 16.4 kcal/mol. The analogue of compound
1 in which the X is CH (compound
2) was studied in the mid-1970s by
13C NMR spectra, and the rotational barriers of the amide and enamine groups were estimated using the coalescence method [
31]. The experiment was performed in acetone-d6 and the enamine rotational barrier (11.7 kcal/mol) was lower than the amide rotational barrier (12.4 kcal/mol). The small structural change in the molecule causes a big change in the flexibility. This fact led us to study compound
1 in depth and find an answer to the question of why the two compounds (
1 and
2) have such different rotational flexibility.
The experiment in TCE-d2 was designed with the idea of studying compound
1 in the most rigorous way possible. The sample was prepared by adding one drop of anisole [
32,
33] (a compound with a singlet signal at 3.81 ppm), which is a suitable compound for the application of the reference deconvolution procedure [
34,
35]. The relaxation times T
1 and T
2 of the reference signal at different temperatures were measured (
Table S5) and their temperature dependence was determined (
Figures S14 and S15). From this dependence, the values of T
2 were calculated, which were used in the reference deconvolution procedure in order to remove both line distortion and the signals that we were interested in to be of natural half-width. The newly developed signal-to-noise ratio enhancement program [
36] was used to process the FIDs, instead of the previously used LP [
32] and MEM [
37]. Slow exchange chemical shifts were extrapolated to high temperatures. Thus, all that remained was to fit the rate constants in the procedure of CLSA. The CLSA results are very close to the 2D EXSY results and are complementary (
Table 1 and
Table 2). In general, 2D EXSY is applicable for slow exchange and the line shape analysis is most sensitive at intermediate exchange.
Figure 4 presents the Eyring plot of the rate constants obtained by different methods. These plots demonstrate very good linear dependence for all sets of kinetic data. The correlation coefficients of all plots are very good and are presented in
Tables S2, S4, S7, and S8.
2.4. Origin of Rotational Barriers
The studied compound
1 is a push–pull molecular system, for which several resonance structures can be written (
Figure 6). Even from these structures, it can be concluded that there is a large delocalization over the heavy atom framework and the C–N enamine single bond has a partial double bond character.
The computed geometrical parameters of
1 are in excellent agreement with the X-ray crystal structure [
30] (
Table 5). The heavy atom frameworks of
1 and
2 are planar in GS. When comparing the structural parameters of
1 and
2, only the bond lengths of X4 with neighboring C2 and C5 differ significantly, which is to be expected.
The C2–N1 amide bond was studied intensively in the past, and the heavy atom framework of the studied amides in the GS was found to be essentially planar and close to C
S symmetry. The transition state geometries of the studied amides can be optimized in C
S symmetry and yield two transition states:
syn and
anti. The most significant structural changes in the process of the rotation of amides towards the transition states are that the nitrogen is pyramidalized and the C–N bond lengthens by 0.07–0.08 Å. However, the C=O bond length shortens by 0.01–0.02 Å only. This indicates that the carbonyl group is relatively unaffected by this rotation. The origin of the C–N amide rotational barrier and its relation to amide resonance has also received much attention in the last few years [
11,
12,
13,
14,
15,
16] and therefore we will discuss here mainly the enamine rotational barrier, which has only been discussed once in the literature [
43]. The X4–C5 bond (
Figure 3) of both molecules does not change appreciably from GS to TSs. However, the change in bond length in the GS and TS geometries is significant for the C5–N6 bond. In
1, it increases in
syn-TS with 0.091 Å and in
anti-TS with 0.083 Å. In
2, it increases in
syn-TS with 0.087 Å and in
anti-TS with 0.078 Å. In the GS, the C5–N6 bond adopts a partial double bond character (as indicated by the Wiberg Bond Index values of 1.298 and 1.237 for
1 and
2, respectively), but in the rotated TS, it becomes a single bond (
Table 6). Moreover, the enamine nitrogen atom adopts a planar geometry in the GS, but it is pyramidalized in the TS. This is further supported by the calculated and observed C–N–H and H–N–H bond angles around the enamine nitrogen atom, which correspond to the sp
2 hybridized nitrogen atom in the GS (
Table 6). However, in the TS, the hybridization changes to sp
3, as indicated by the respective bond angles (
Figure 5). The above data indicate that the actual ground state geometry can be envisaged as a resonance hybrid of three structures (
Figure 6). The left and middle structures represent the resonance of the amide bond, while the middle and right structures represent the resonance of the enamine bond. The left structure contains a double bond between C2 and N1 and this partial double bond is responsible for the observed barrier of the amide group. The right structure contains a double bond between C5 and N6 and this partial double bond is responsible for the observed barrier of the enamine group.
The insight obtained about the nature of the C5–N6 bond (
Figure 3) from the NBO analysis agrees well with those acquired from the geometrical parameters. Our discussion of the NBO analysis of
1 and
2 repeats most of the conclusions made earlier on the origin of the enamine rotational barrier [
43]. In GS, this bond has a partial double bond character, while, in TSs, it is a single bond and the nitrogen atom changes from sp
2 in GS to a sp
3 hybridized atom in TSs. The lone pair of electrons on N6 has reduced occupancy in the GS than in the TSs (
Table 6) because, in the GS, a part of the electron density of the lone pair is delocalized into the antibonding orbital of the adjacent C–N double bond. Such delocalization is not possible in the TS as the participating orbitals are orthogonal to each other. The orbital housing the lone pair in the GS is an in-plane p orbital, but it attains some s character in the TS, thereby making the nitrogen atom more electronegative in the TS.
Natural charges computed at the same level of theory predict a higher negative charge for the N6 atom in the TSs (
Table 7), which correlates well with the increased occupancy of N6 in the TS (
Table 6). However, the charge on the C5 atom does not change appreciably in both the GS and TSs. On the other hand, the N4 atom, which is double bonded to C5, carries a higher negative charge in the GS.
The stabilization energies resulting from the delocalization of the N6 lone pair of electrons into the antibonding orbital of the neighboring C5–N4 double bond decrease from
1 to
2 (
Table 8). Along with them, the rotational barrier also decreases from
1 to
2. This stabilizing interaction is responsible for the partial double bond character of the C5–N6 single bond in the GS (right structure of
Figure 6) and therefore the barrier height.
The calculated main steric exchange energies (
Table 9) for the GS and TS structures of
1 and
2 did not show significant differences that could explain the significant difference in the enamine barrier of
1 and
2.
All these data are consistent with our resonance-based explanation of the origin of the rotational barrier about the C5–N6 bond. This barrier has a purely electronic origin, which recently was demonstrated by the relationship between the C–N bond order difference and calculated energy barrier [
20]. The proportion of experimentally determined enamine to amide barriers in
1 (18.6/16.4 = 1.13) is in very good agreement with the proportion of Wiberg bond orders (
Table 10) of enamine to amide C–N bonds in GS of
1 (1.298/1.169 = 1.11).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}