
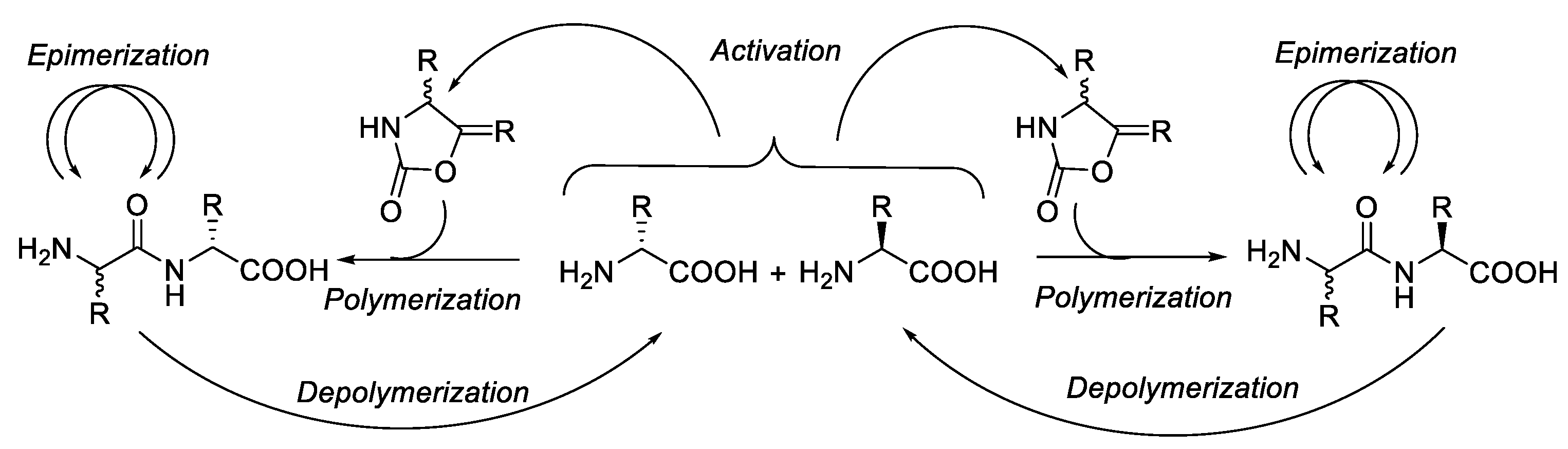
Epimerisation in Peptide Synthesis
Abstract
:
1. Introduction
2. Epimerisation-Inducing Factors
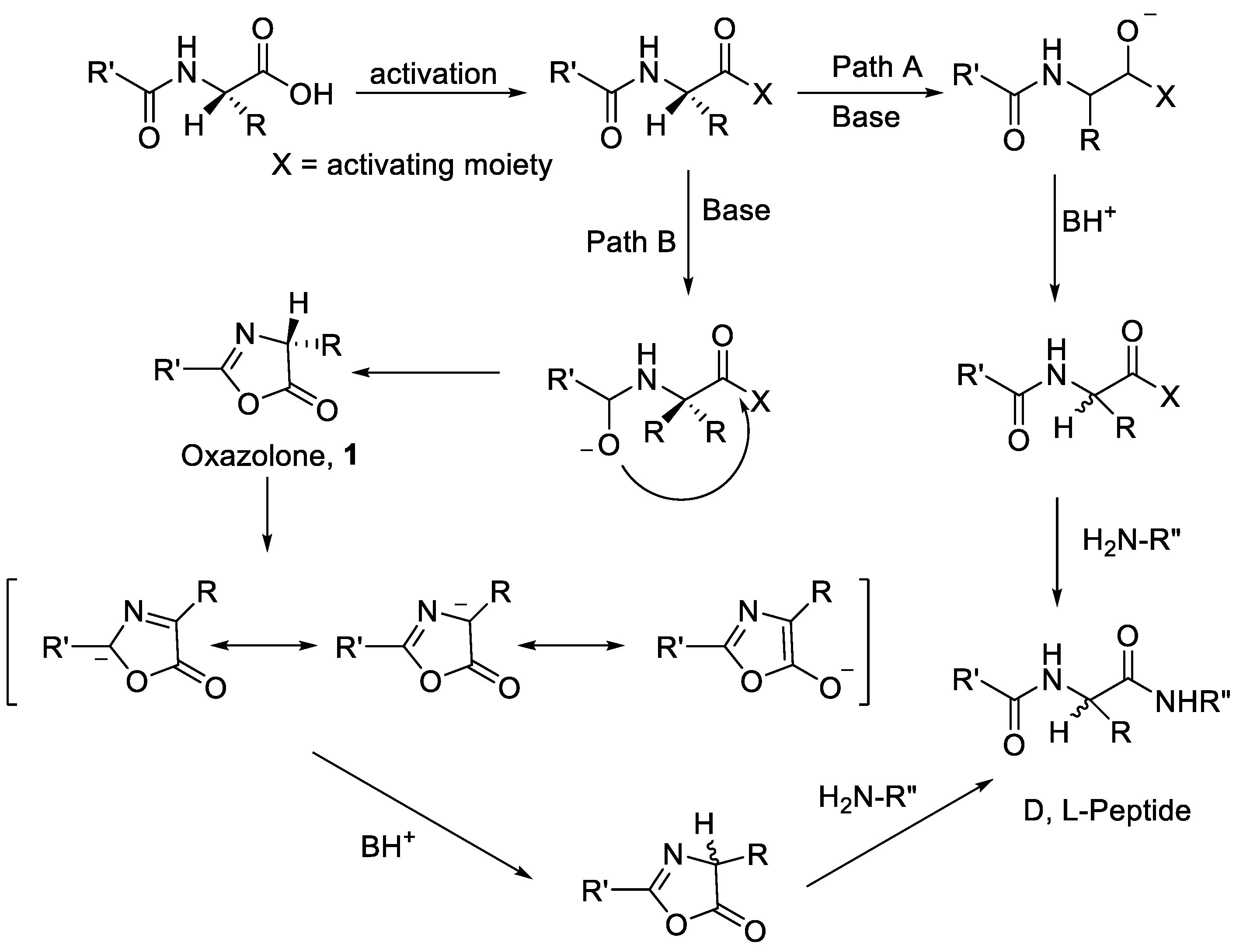
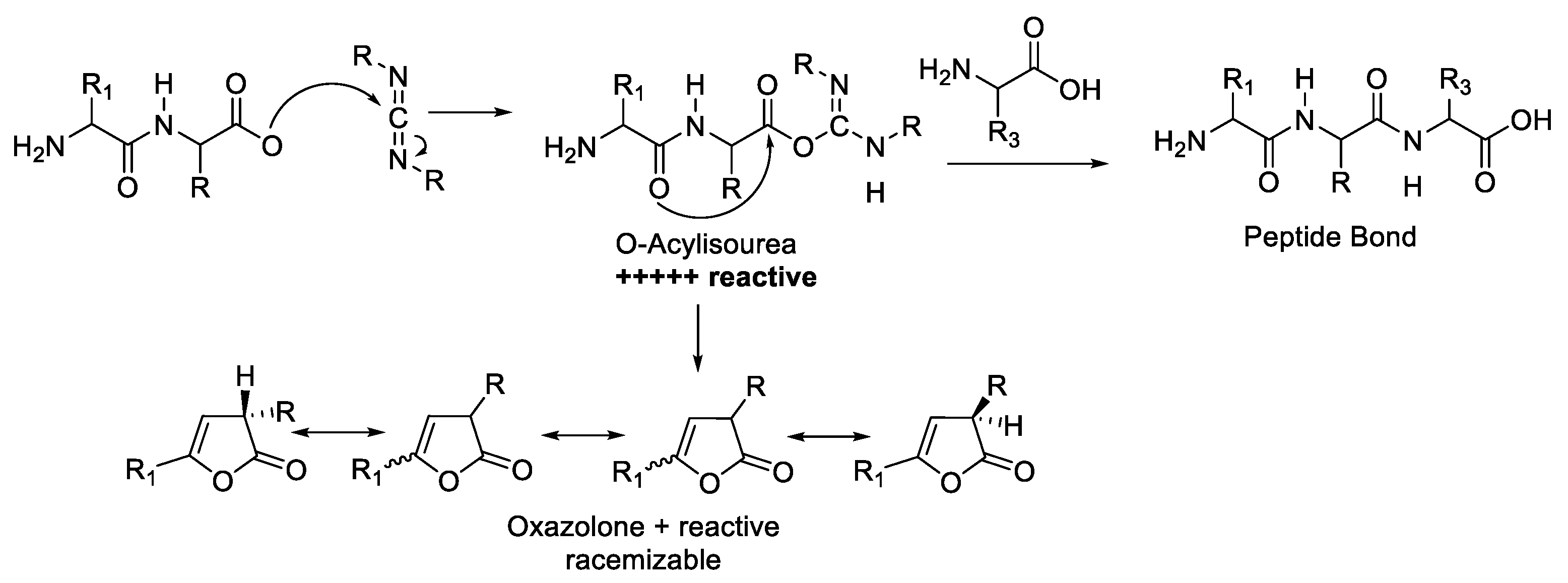
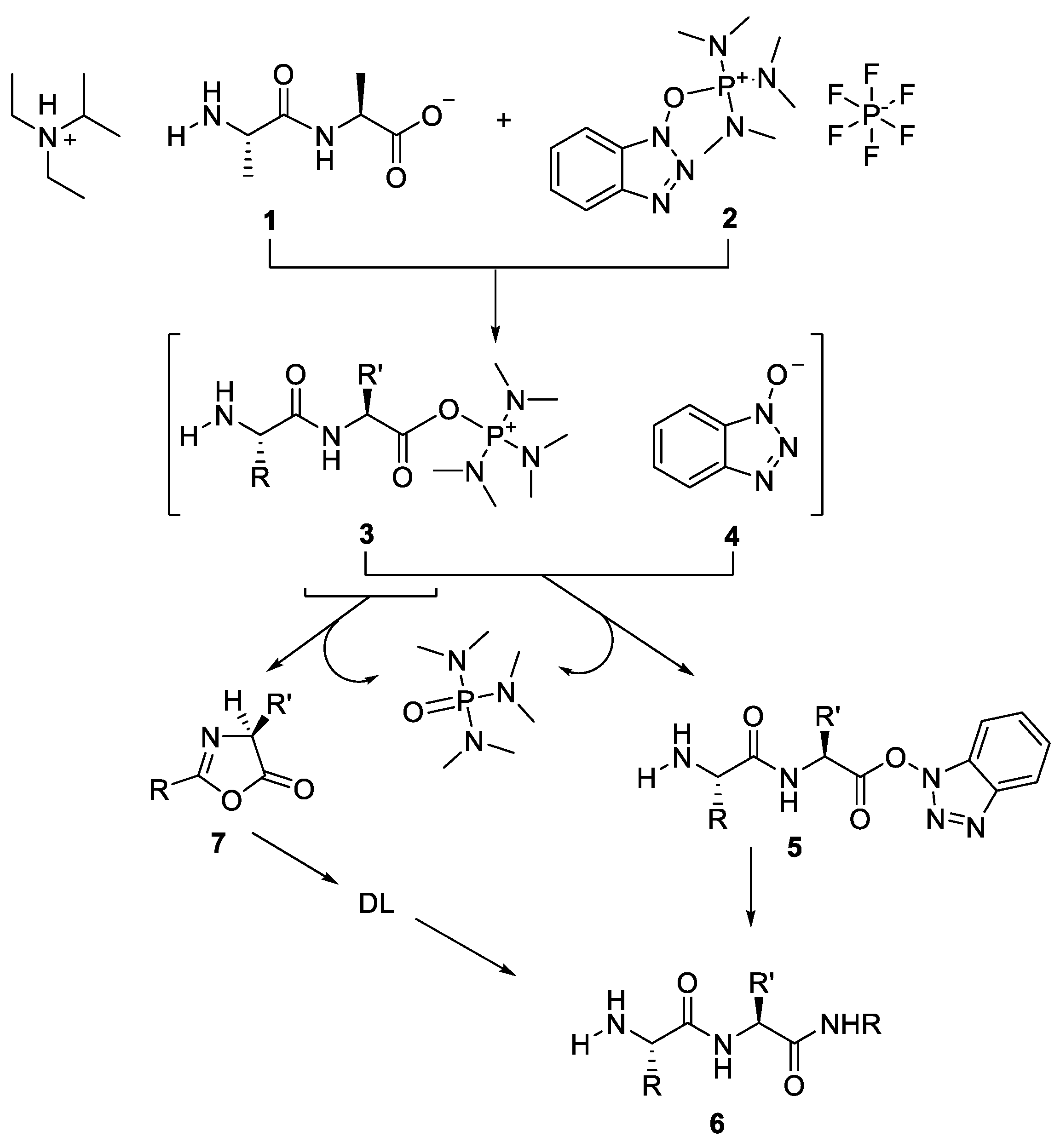
2.1. Activation of Carboxylic Groups to Active Esters
2.2. Amino Acids
2.3. Bases
2.3.1. Bases Used in Amino Acid Activation
2.3.2. Bases Used in Fmoc Deprotection
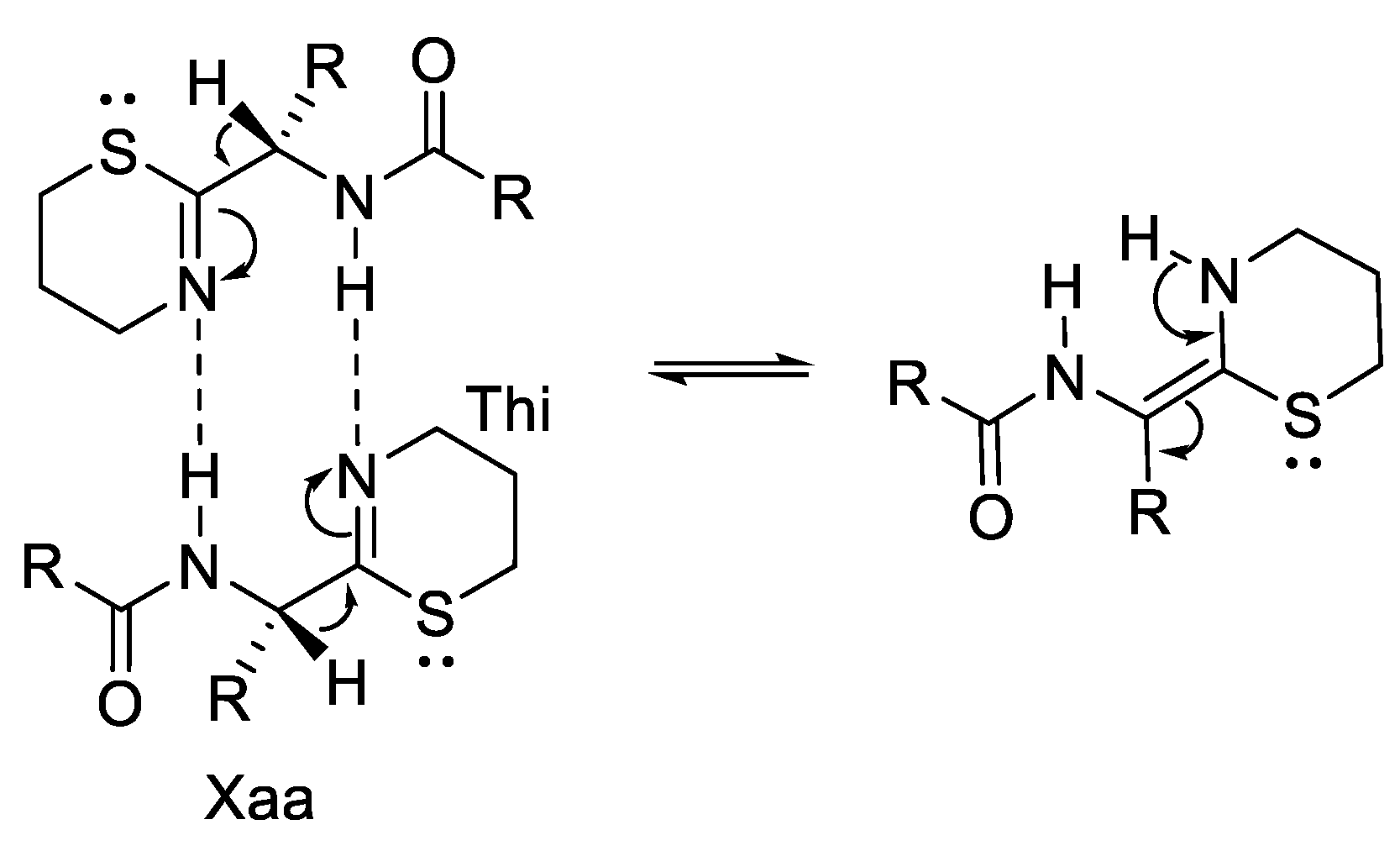
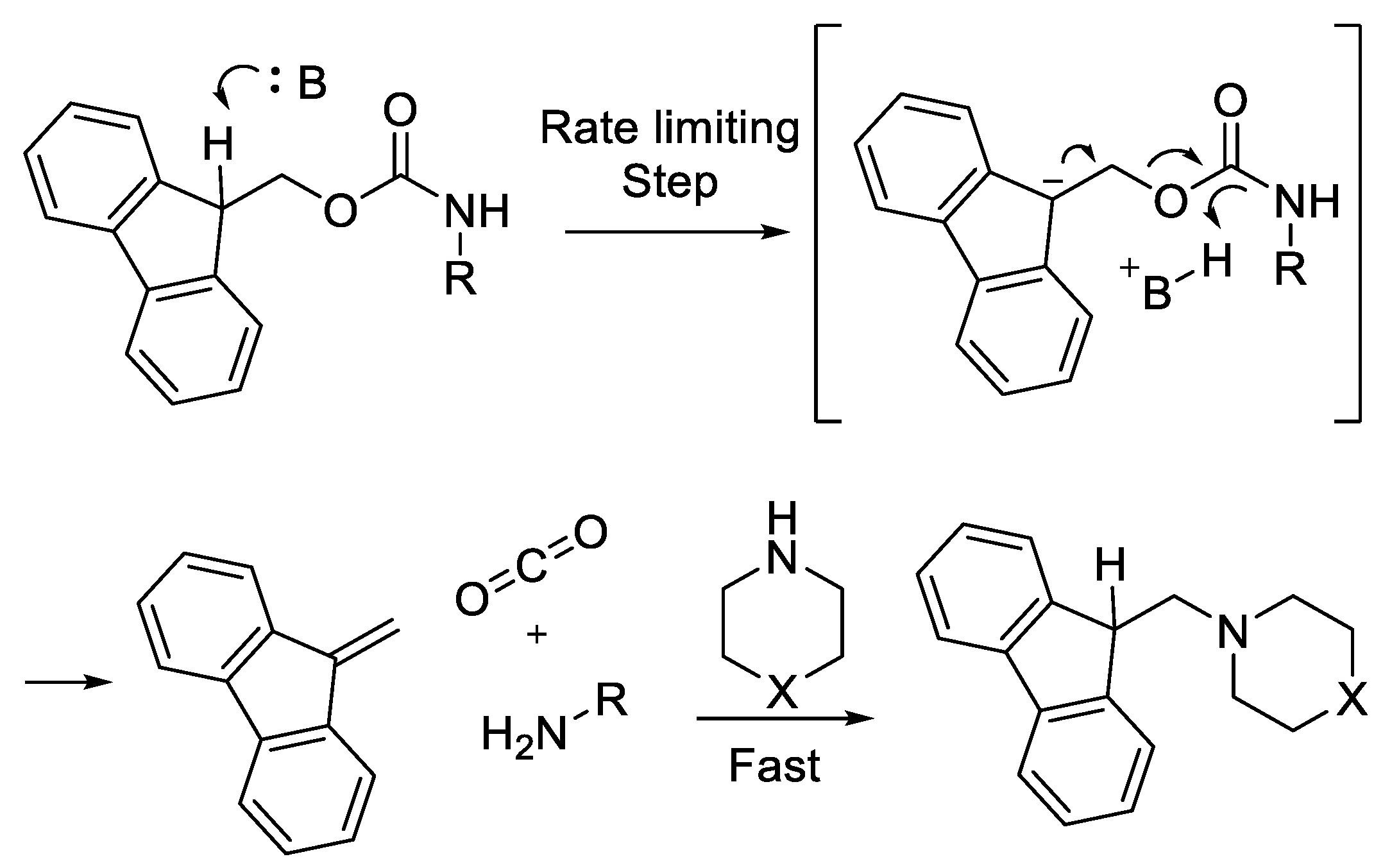
2.4. Steric Factor and Ring Orientation
2.5. Solvent
2.6. Temperature
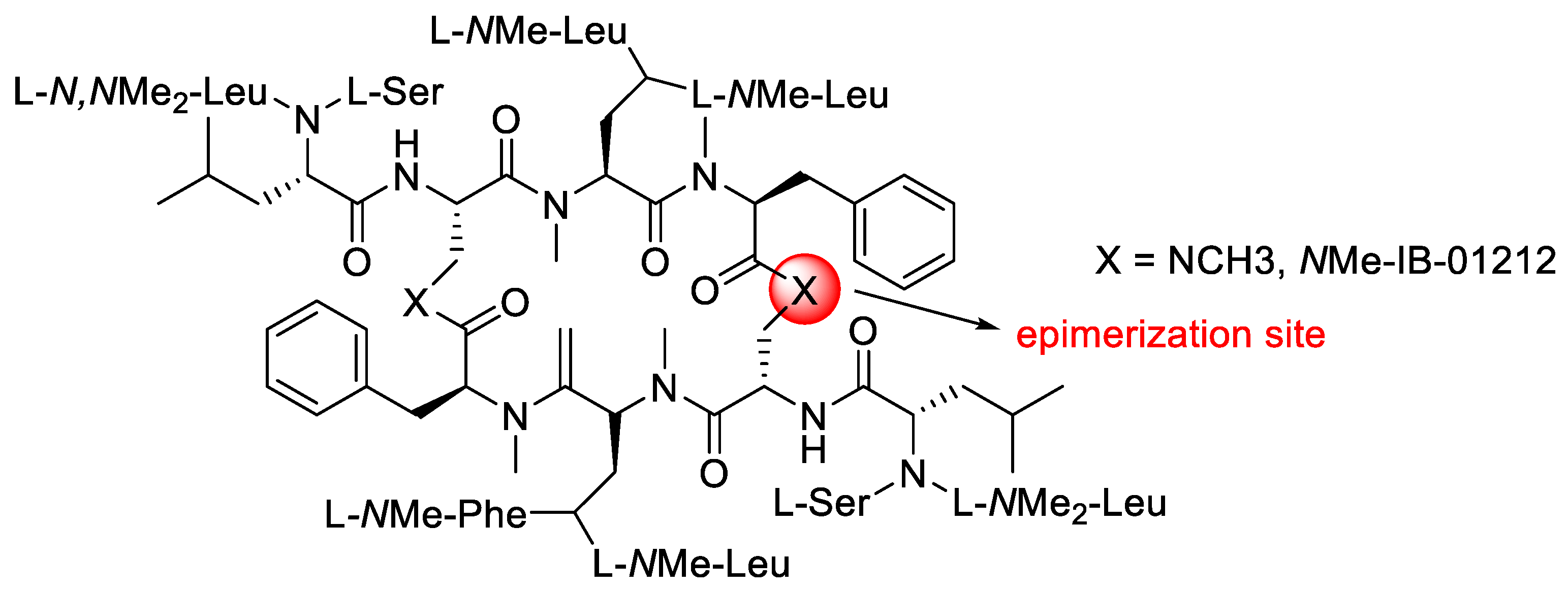
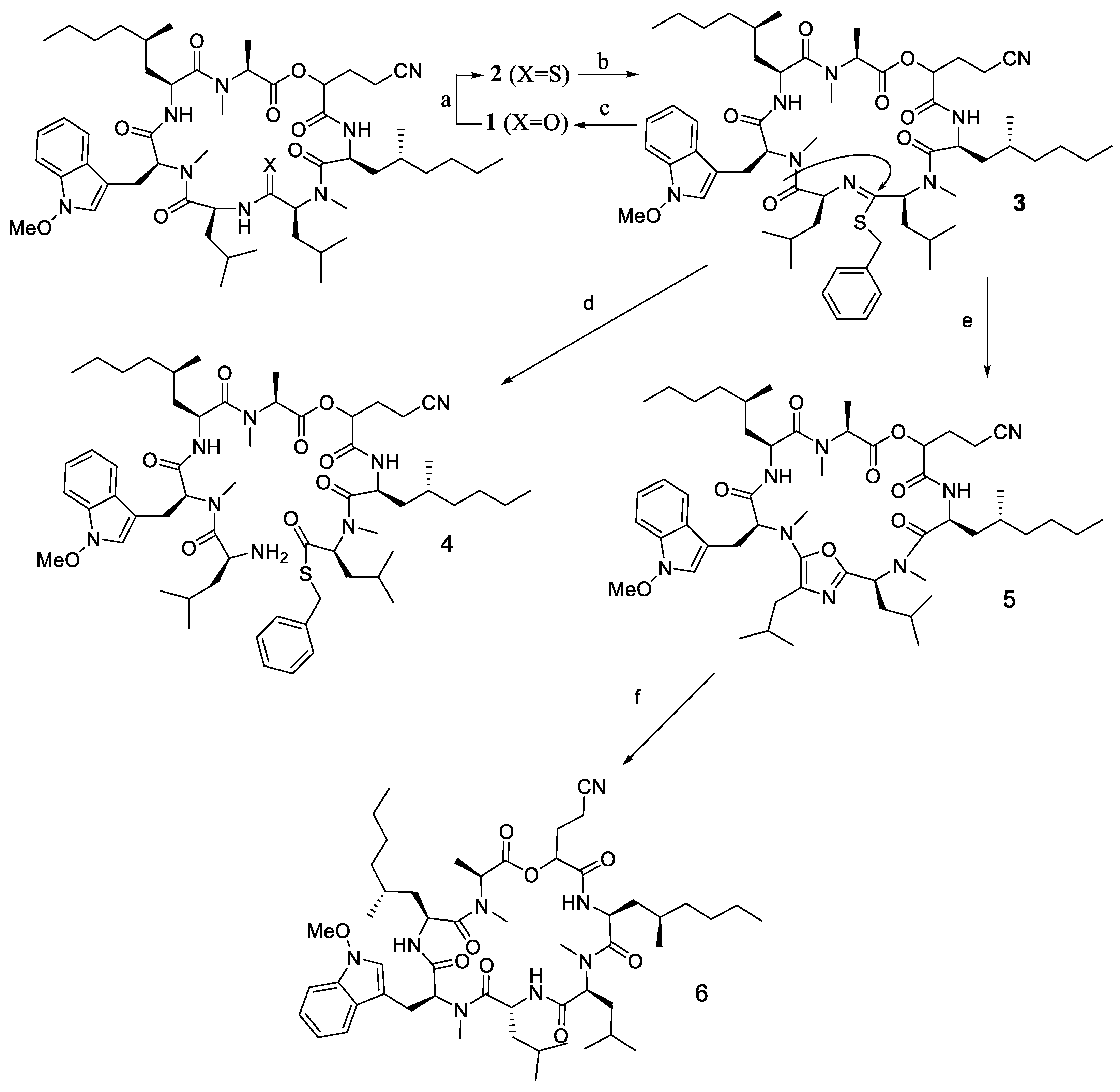
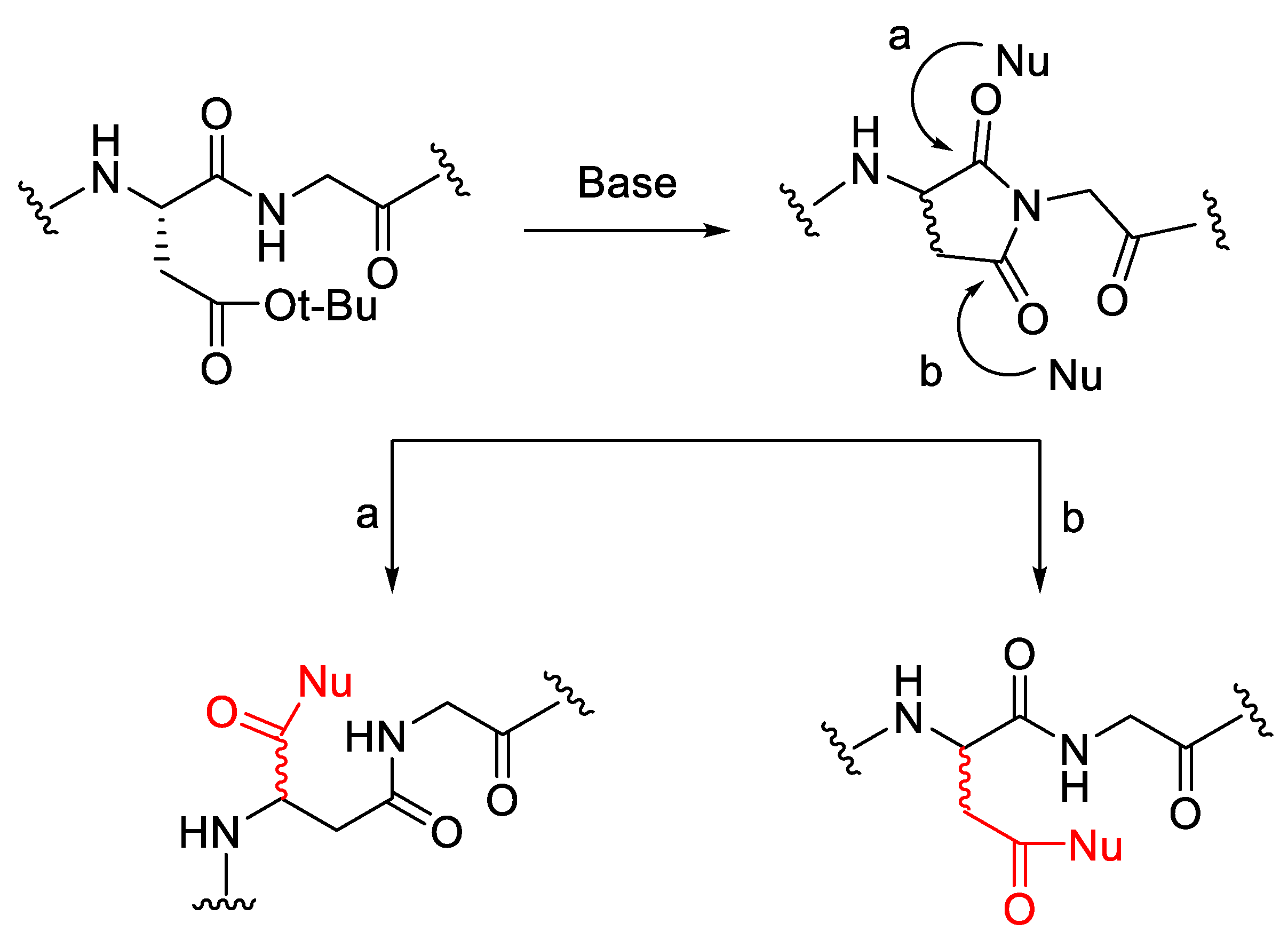
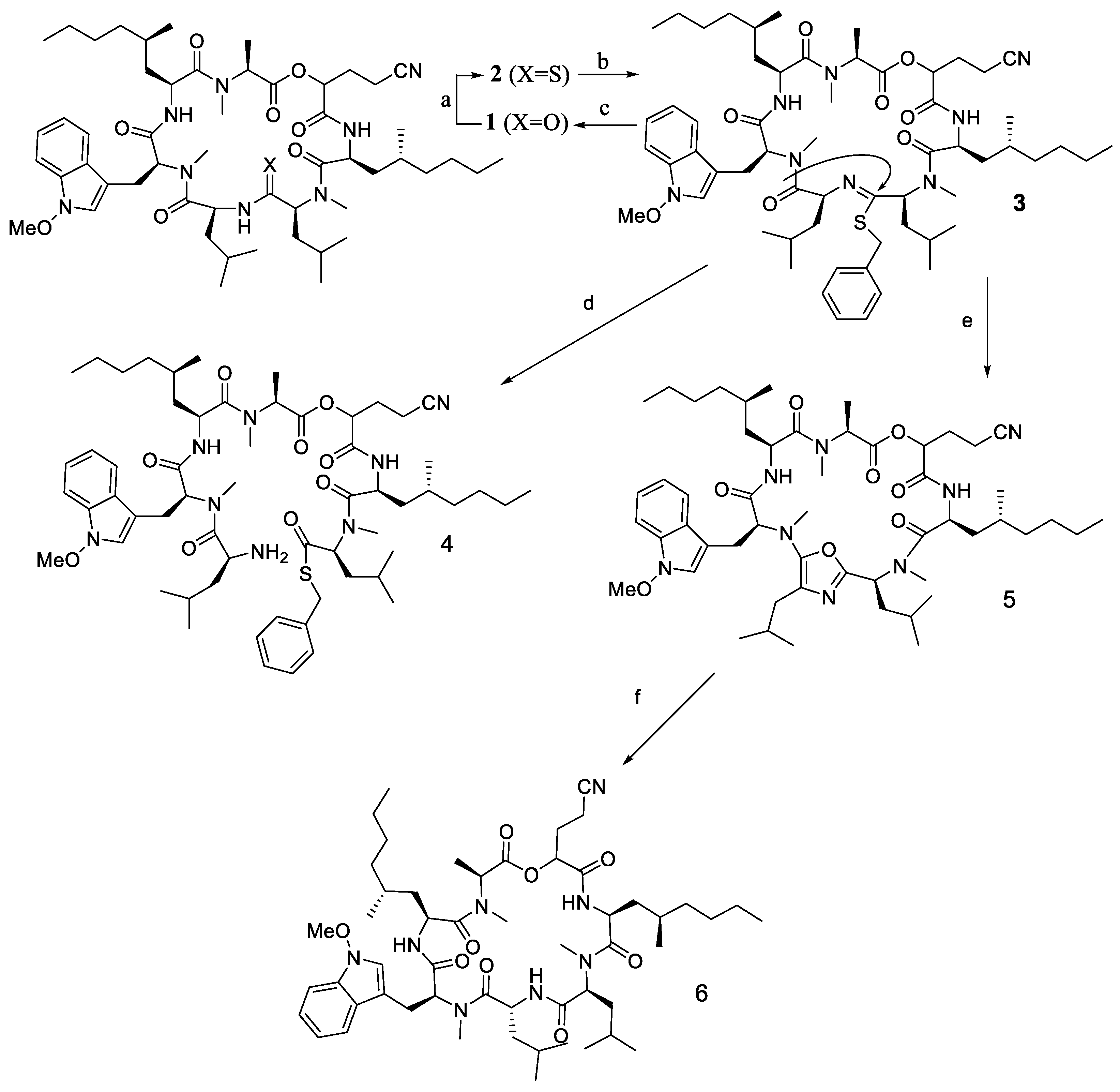
2.7. Epimerisation in a Peptide Cyclisation
3. Suppressing Epimerisation
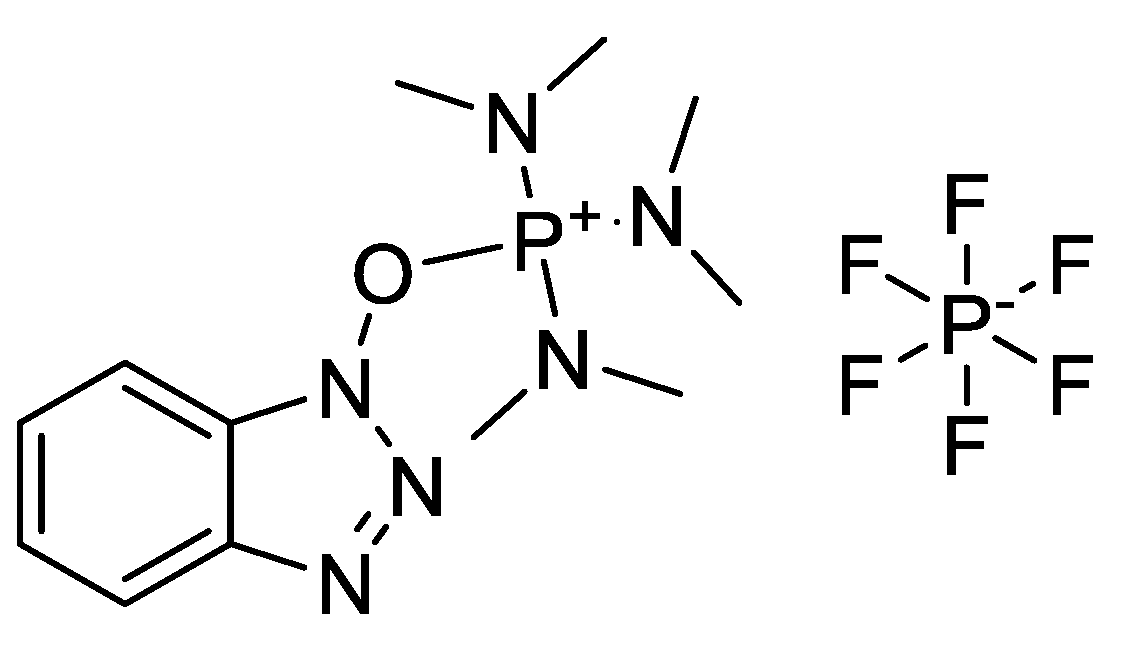
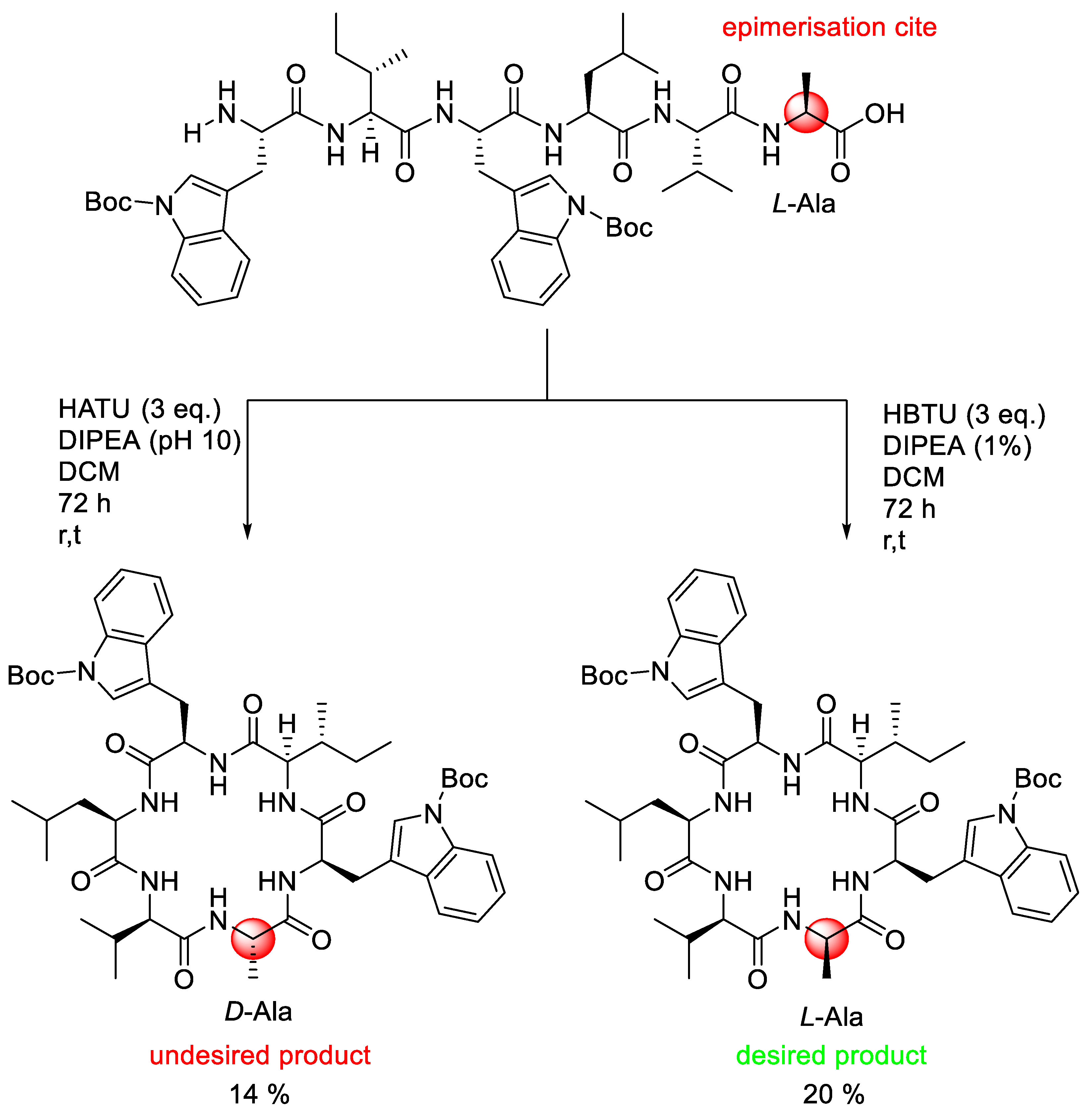
3.1. Coupling Agent Selection
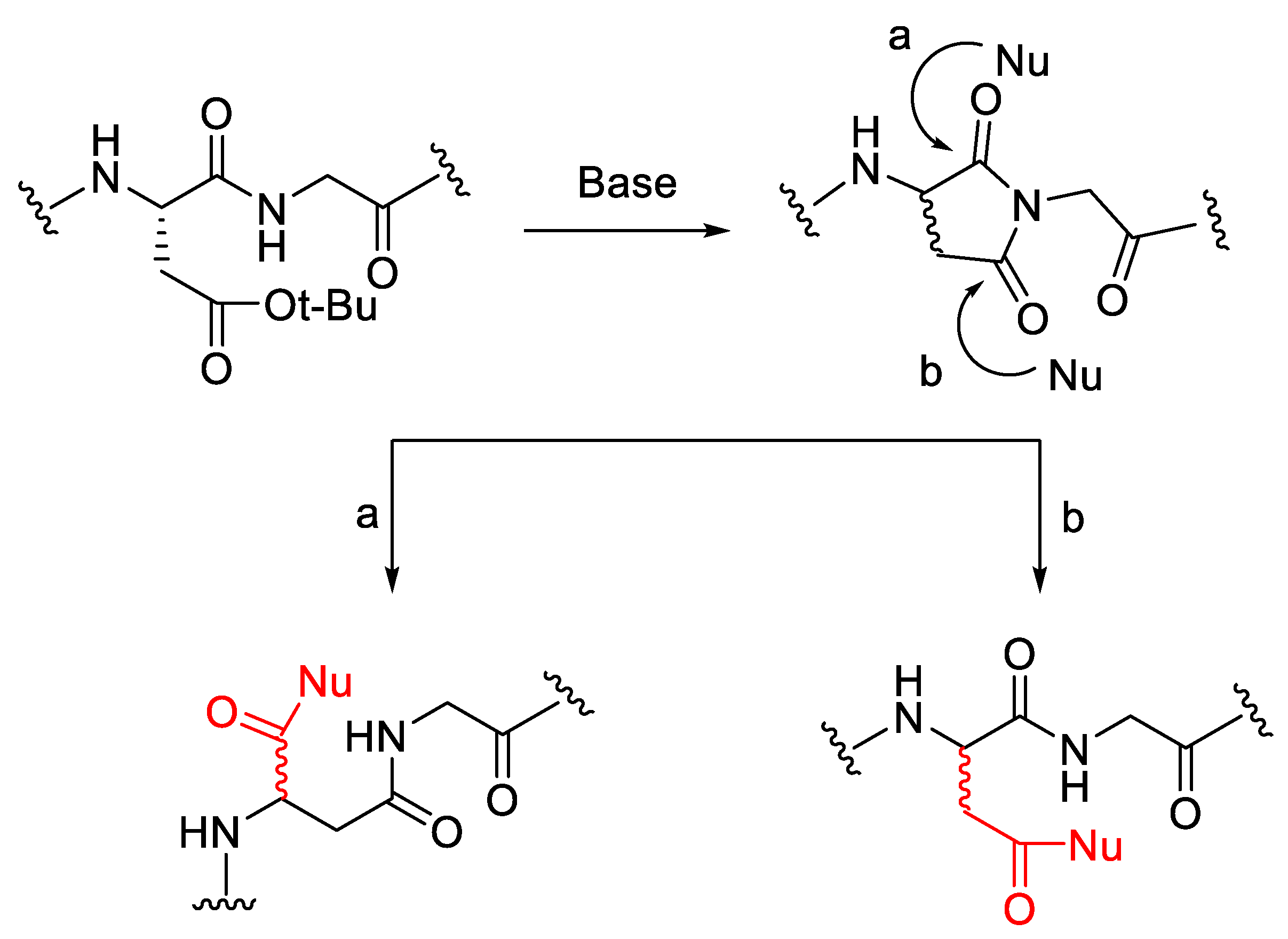
3.2. Side Chain Protection Group
3.3. Coupling Strategy
3.3.1. Pseudoproline
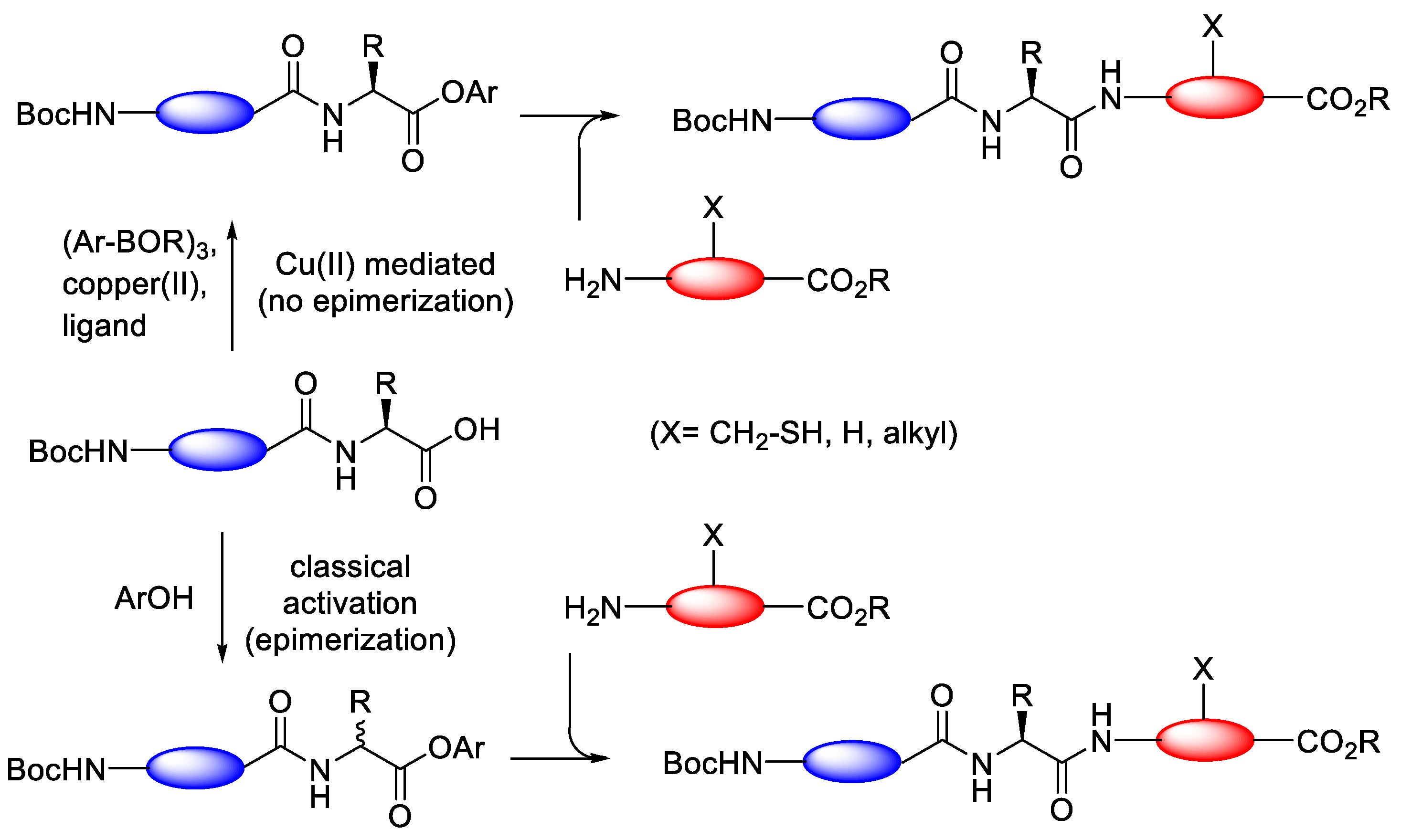
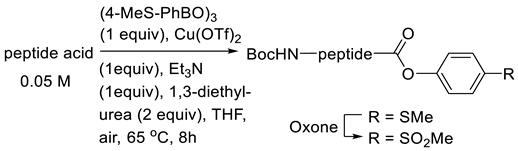
3.3.2. Copper(II)-Mediated Chan–Lam-Type Coupling
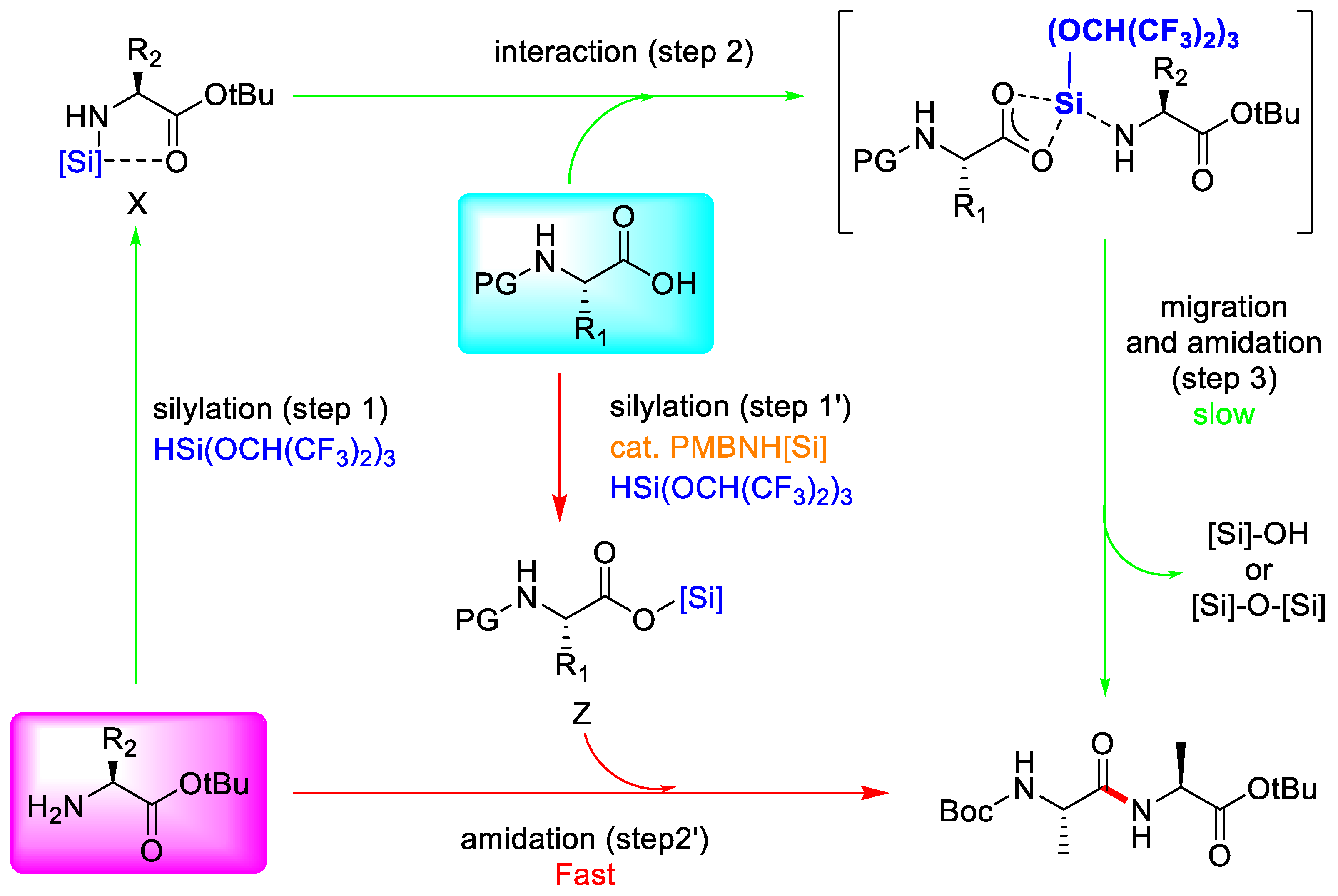
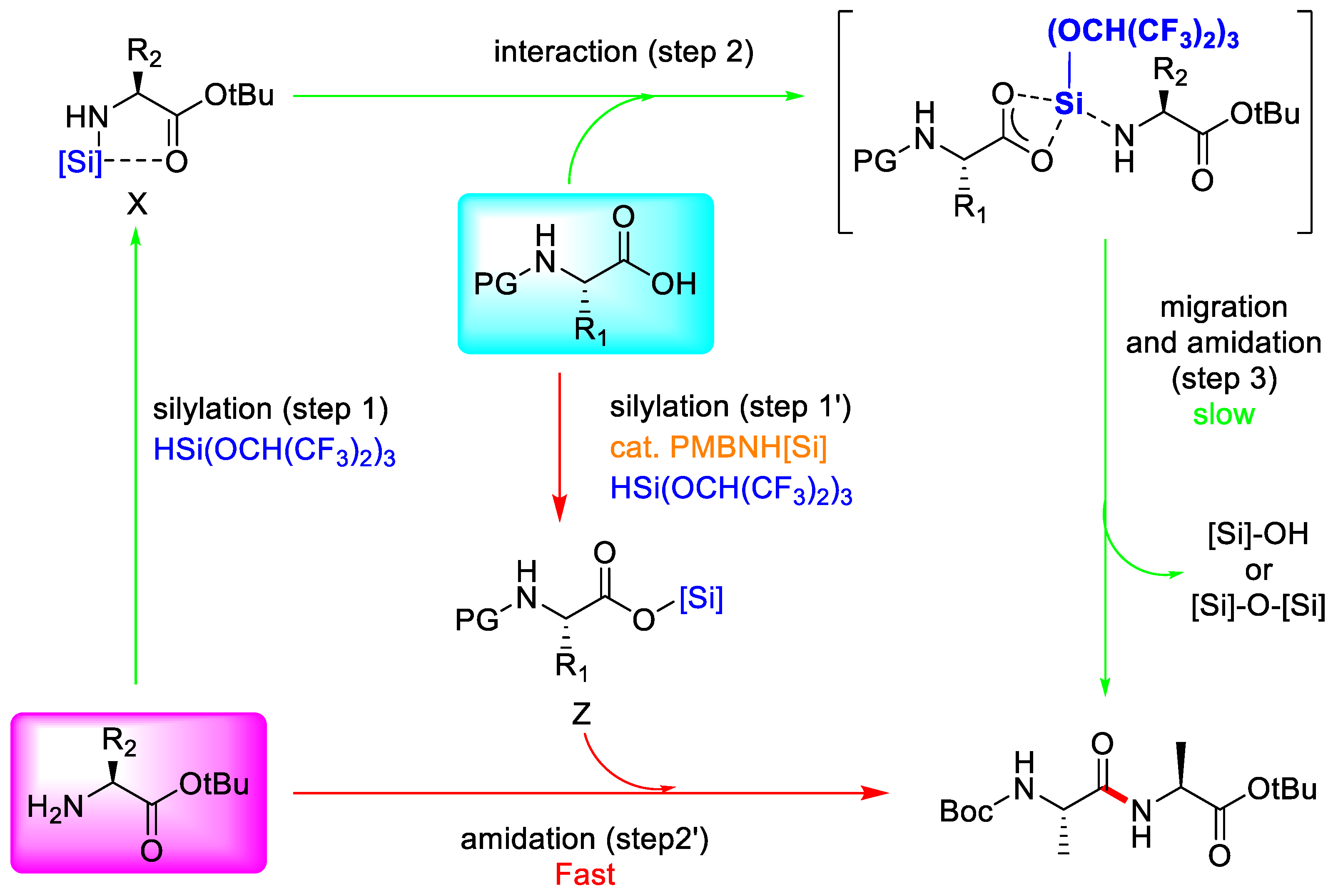
3.3.3. Hydrosilane-Mediated Type Coupling
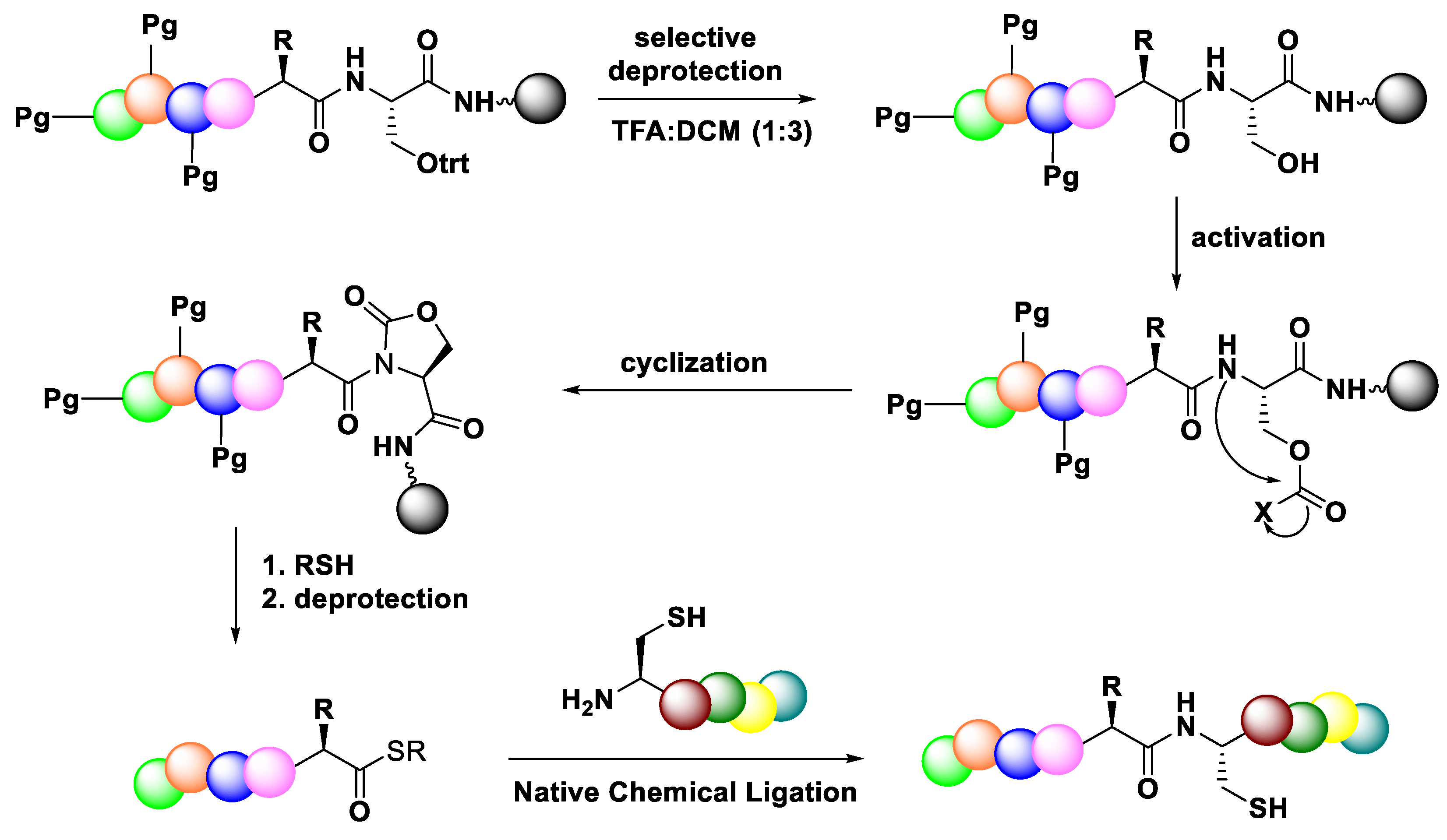
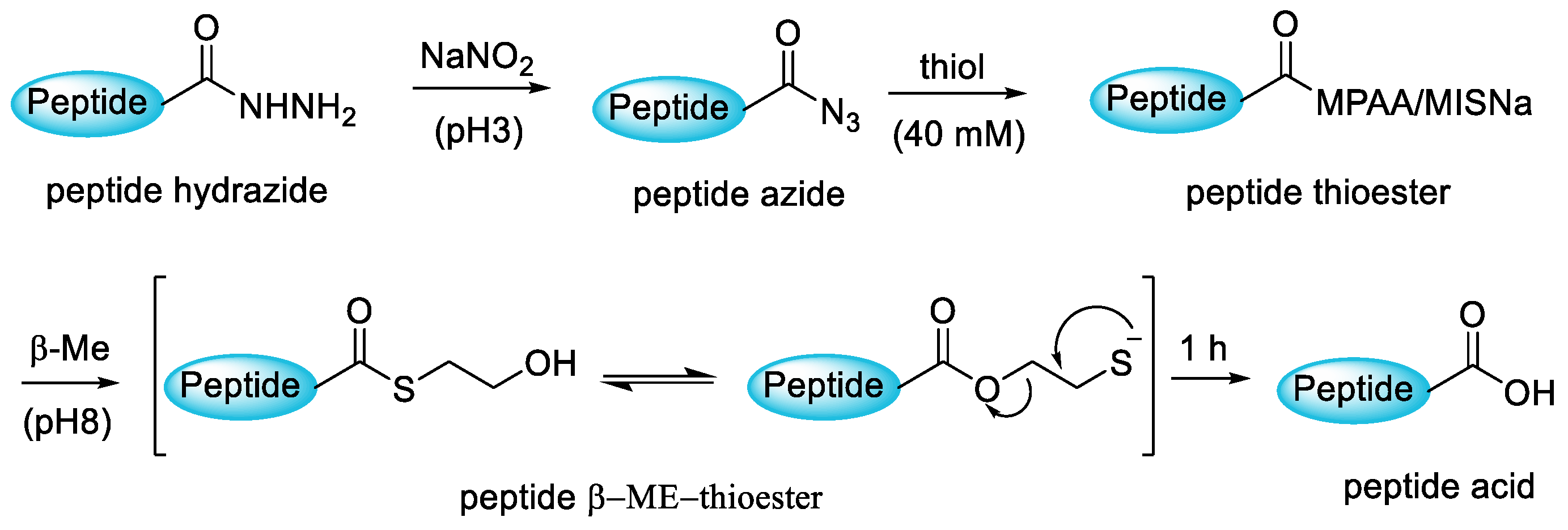
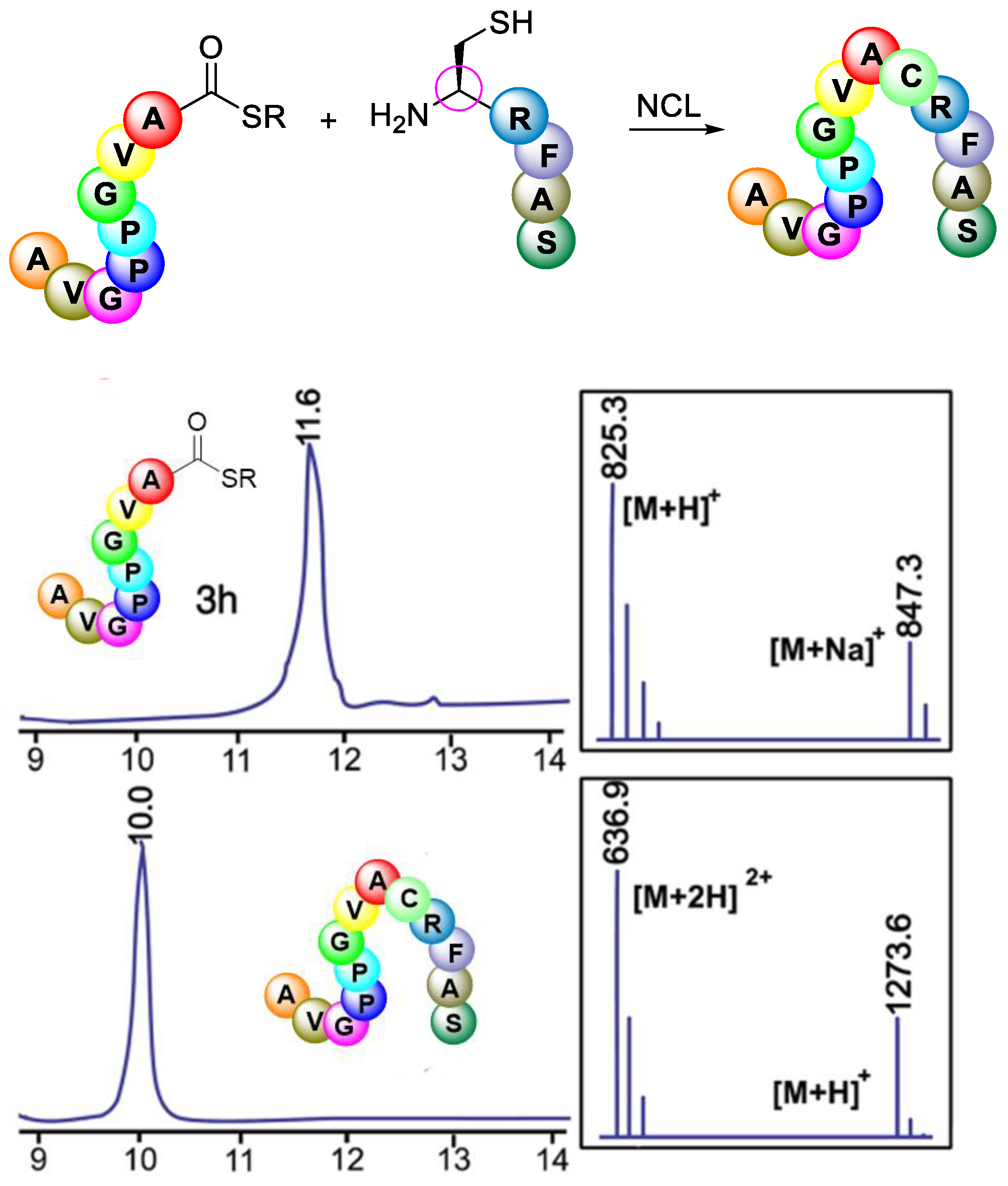
3.3.4. Native Chemical Ligation (NCL)
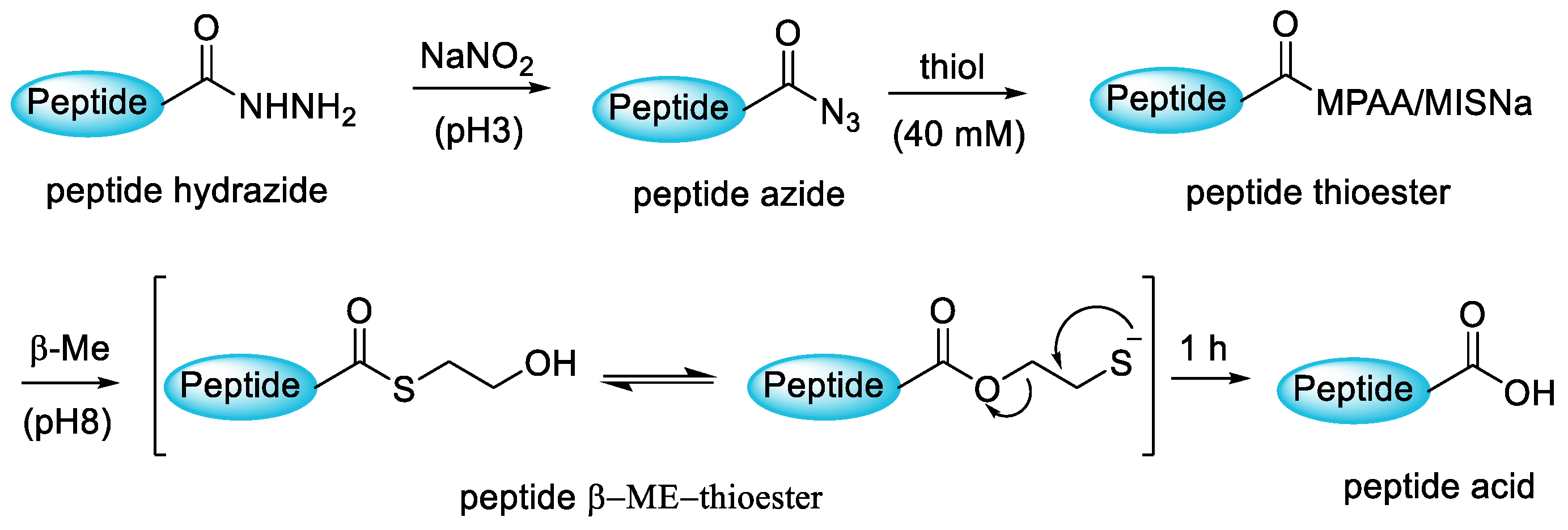
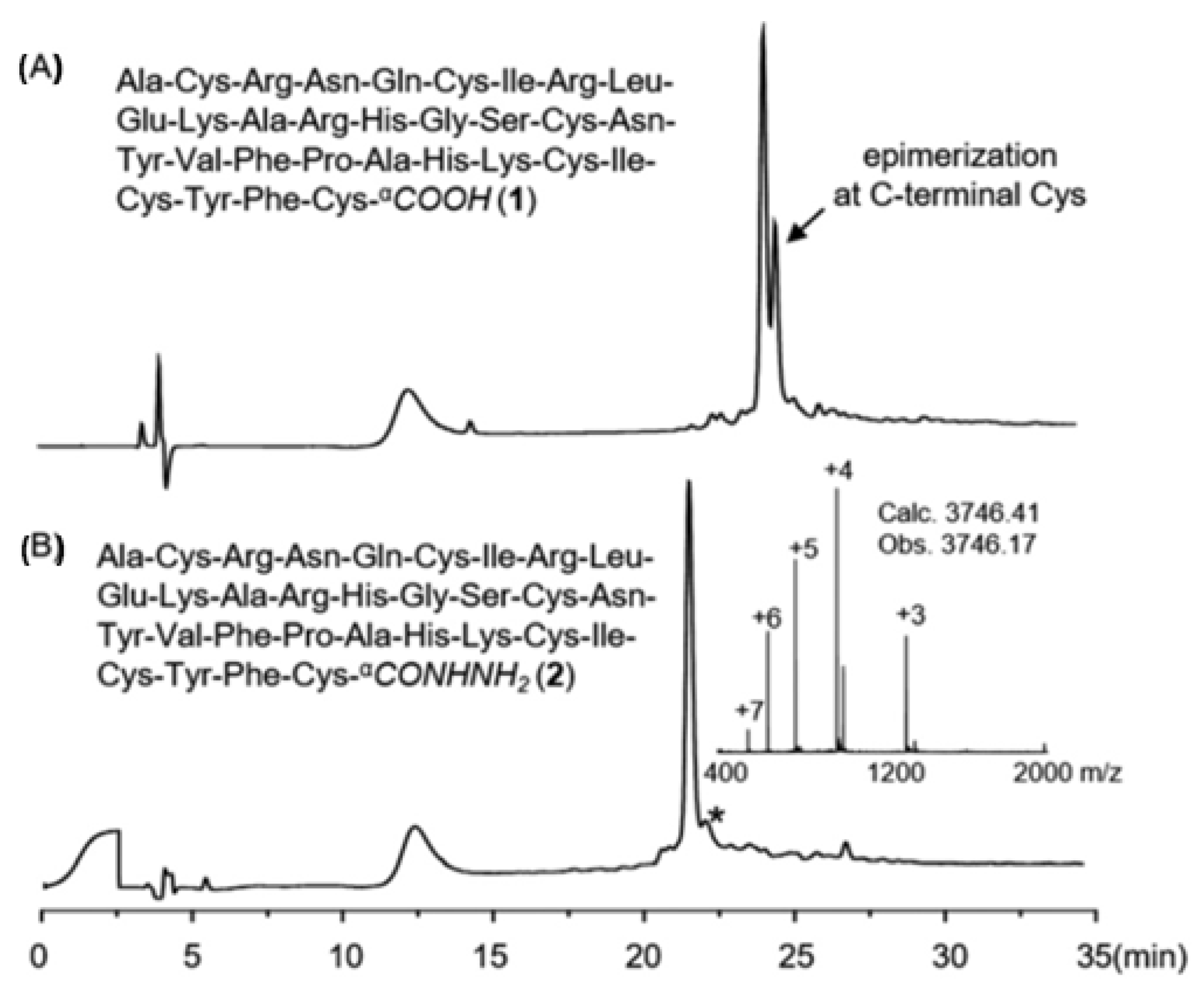
3.4. Hydrolisis Strategy of Peptide from Resin
3.5. Ball Milling-Assisted
3.6. Microwave- and Flow Synthesis-Assisted
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arbour, C.A.; Kondasinghe, T.D.; Saraha, H.Y.; Vorlicek, T.L.; Stockdill, J.L. Epimerization-free access to C-terminal cysteine peptide acids, carboxamides, secondary amides, and esters via complimentary strategies. Chem. Sci. 2018, 9, 350–355. [Google Scholar] [CrossRef] [PubMed]
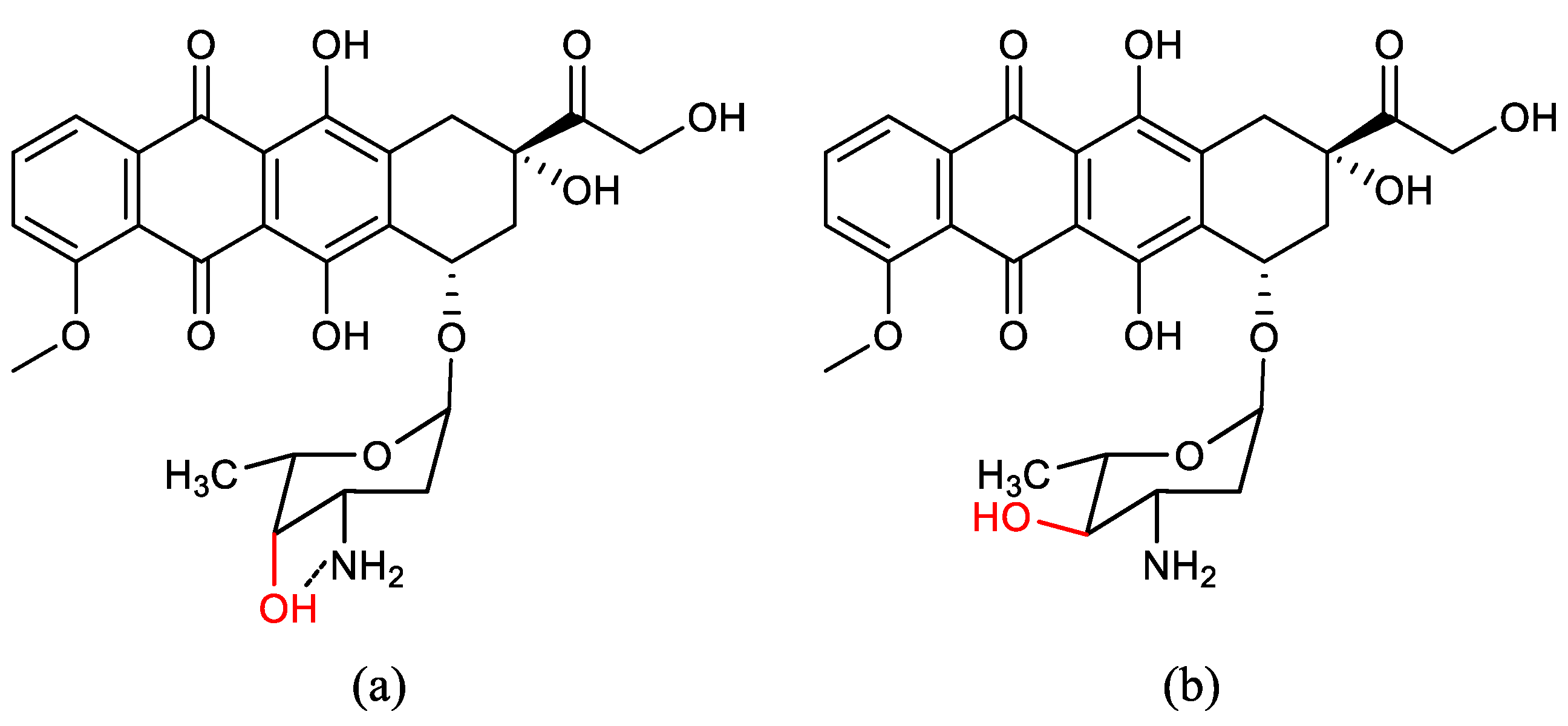
- Paul Launchbury, A.; Habboubi, N. Epirubicin and doxorubicin: A comparison of their characteristics, therapeutic activity and toxicity. Cancer Treat. Rev. 1993, 19, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.F.; Payne, C.D.; Rosengren, K.J.; Mylne, J.S. An Orbitide from Ratibida columnifera Seed Containing 16 Amino Acid Residues. J. Nat. Prod. 2019, 82, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Heck, S.D.; Siok, C.J.; Krapcho, K.J.; Kelbaugh, P.R.; Thadeio, P.F.; Welch, M.J.; Williams, R.D.; Ganong, A.H.; Kelly, M.E.; Lanzetti, A.J.; et al. Functional consequences of posttranslational isomerization of Ser46 in a calcium channel toxin. Science 1994, 266, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.D.; Shen, J.; Burnett, P.G.; Yang, J.; Sammynaiken, R.; Reaney, M.J.T. Methionine epimerisation in cyclic peptides. RSC Adv. 2021, 11, 20859–20864. [Google Scholar] [CrossRef]
- Heck, S.D.; Faraci, W.S.; Kelbaugh, P.R.; Saccomano, N.A.; Thadeio, P.F.; Volkmann, R.A. Posttranslational amino acid epimerisation: Enzyme-catalyzed isomerization of amino acid residues in peptide chains. Proc. Natl. Acad. Sci. USA 1996, 93, 4036–4039. [Google Scholar] [CrossRef] [PubMed]
- Plumbridge, J.; Vimr, E. Convergent pathways for utilization of the amino sugars N-acetylglucosamine, N-acetylmannosamine, and N-acetylneuraminic acid by Escherichia coli. J. Bacteriol. 1999, 181, 47–54. [Google Scholar] [CrossRef] [PubMed]
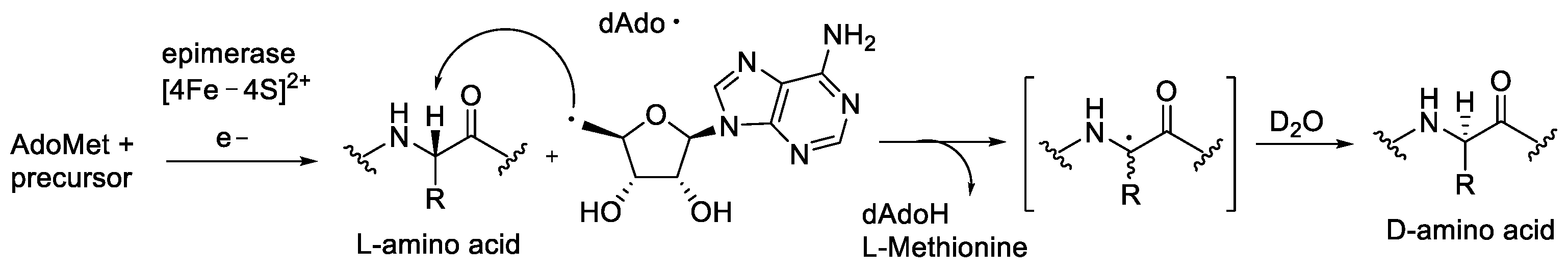
- Vagstad, A.L.; Kuranaga, T.; Püntener, S.; Pattabiraman, V.R.; Bode, J.W.; Piel, J. Introduction of d-Amino Acids in Minimalistic Peptide Substrates by an S-Adenosyl-l-Methionine Radical Epimerase. Angew. Chem—Int. Ed. 2019, 58, 2246–2250. [Google Scholar] [CrossRef]
- Ali, I.; Gupta, V.K.; Aboul-Enein, H.Y.; Singh, P.; Sharma, B. Role of racemisation in optically active drugs development. Chirality Pharmacol. Biol. Chem. Conseq. Mol. Asymmetry 2007, 19, 453–463. [Google Scholar]
- D’Hondt, M.; Bracke, N.; Taevernier, L.; Gevaert, B.; Verbeke, F.; Wynendaele, E.; De Spiegeleer, B. Related impurities in peptide medicines. J. Pharm. Biomed. Anal. 2014, 101, 2–30. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability challenges in peptide synthesis and purification: From R&D to production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [PubMed]
- Sturabotti, E.; Vetica, F.; Toscano, G.; Calcaterra, A.; Martinelli, A.; Migneco, L.M.; Leonelli, F. N-Acetyl-l-phenylalanine Racemization during TBTU Amidation: An In-Depth Study for the Synthesis of Anti-Inflammatory 2-(N-Acetyl)-l-phenylalanylamido-2-deoxy-d-glucose (NAPA). Molecules 2023, 28, 581. [Google Scholar] [CrossRef] [PubMed]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, P.; Papas, S.; Kostidis, S.; Tsikaris, V. α- and β-aspartyl peptide ester formation via aspartimide ring opening. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2005, 11, 658–664. [Google Scholar]
- Jaradat, D.S.M. Thirteen decades of peptide synthesis: Key developments in solid phase peptide synthesis and amide bond formation utilized in peptide ligation. Amino Acids 2018, 50, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Kvsrg, P.; Bharathi, K.; Haseena Banu, B. Applications of peptide coupling reagents–an update. Int. J. Pharm. Sci. Rev. Res. 2011, 8, 108–119. [Google Scholar]
- Bodanszky, M. Principles of Peptide Synthesis; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 16. [Google Scholar]
- Lloyd-Williams, P.; Albericio, F.; Giralt, E. Chemical Approaches to the Synthesis of Peptides and Proteins; CRC Press: Boca Raton, FL, USA, 1997. [Google Scholar]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. [Google Scholar] [CrossRef]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester coupling reactions—An enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 2015, 32, 605–632. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Magano, J.; Weisenburger, G.A. Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. [Google Scholar] [CrossRef]
- Carpino, L.A.; El-Faham, A. The diisopropylcarbodiimide/1-hydroxy-7-azabenzotriazole system: Segment coupling and stepwise peptide assembly. Tetrahedron 1999, 55, 6813–6830. [Google Scholar] [CrossRef]
- Lei, Q.P.; Lamb, D.H.; Shannon, A.G.; Cai, X.; Heller, R.K.; Huang, M.; Zablackis, E.; Ryall, R.; Cash, P. Quantification of residual EDU (N-ethyl-N′-(dimethylaminopropyl) carbodiimide (EDC) hydrolyzed urea derivative) and other residual by LC-MS/MS. J. Chromatogr. Anal. Technol. Biomed. Life Sci. 2004, 813, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Spatola, A.F.; Crozet, Y.; DeWit, D.; Yanagisawa, M. Rediscovering an endothelin antagonist (BQ-123): A self-deconvoluting cyclic pentapeptide library. J. Med. Chem. 1996, 39, 3842–3846. [Google Scholar] [CrossRef] [PubMed]
- Spatola, A.F.; Darlak, K.; Romanovskis, P. An approach to cyclic peptide libraries: Reducing epimerization in medium sized rings during solid phase synthesis. Tetrahedron Lett. 1996, 37, 591–594. [Google Scholar] [CrossRef]
- Morinaka, B.I.; Vagstad, A.L.; Helf, M.J.; Gugger, M.; Kegler, C.; Freeman, M.F.; Bode, H.B.; Piel, J. Radical S-Adenosyl Methionine Epimerases: Regioselective Introduction of Diverse D-Amino Acid Patterns into Peptide Natural Products. Angew. Chem. Int. Ed. 2014, 53, 8503–8507. [Google Scholar] [CrossRef] [PubMed]
- Yeboue, Y.; Jean, M.; Subra, G.; Martinez, J.; Lamaty, F.; Métro, T.X. Epimerisation-Free C-Term Activation of Peptide Fragments by Ball Milling. Org. Lett. 2021, 23, 631–635. [Google Scholar] [CrossRef]
- Yang, Y. Peptide racemisation. In Side Reactions in Peptide Synthesis; Academic Press: Cambridge, MA, USA, 2016; pp. 257–292. [Google Scholar]
- Marcucci, E.; Tulla-Puche, J.; Albericio, F. Solid-phase synthesis of N Me-IB-01212, a highly N-methylated cyclic peptide. Org. Lett. 2012, 14, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Nabika, R.; Suyama, T.L.; Hau, A.M.; Misu, R.; Ohno, H.; Ishmael, J.E.; McPhail, K.L.; Oishi, S.; Fujii, N. Synthesis and biological evaluation of the [d-MeAla11]-epimer of coibamide A. Bioorg. Med. Chem. Lett. 2015, 25, 302–306. [Google Scholar] [CrossRef]
- Liang, C.; Behnam, M.A.M.; Sundermann, T.R.; Klein, C.D. Phenylglycine racemisation in Fmoc-based solid-phase peptide synthesis: Stereochemical stability is achieved by choice of reaction conditions. Tetrahedron Lett. 2017, 58, 2325–2329. [Google Scholar] [CrossRef]
- Al Toma, R.S.; Brieke, C.; Cryle, M.J.; Süssmuth, R.D. Structural aspects of phenylglycines, their biosynthesis and occurrence in peptide natural products. Nat. Prod. Rep. 2015, 32, 1207–1235. [Google Scholar] [CrossRef]
- Buskas, T.; Ingale, S.; Boons, G.-J. Glycopeptides as versatile tools for glycobiology. Glycobiology 2006, 16, 113R–136R. [Google Scholar] [CrossRef]
- Batsalova, T.; Dzhambazov, B.; Klaczkowska, D.; Holmdahl, R. Mice producing less reactive oxygen species are relatively resistant to collagen glycopeptide vaccination against arthritis. J. Immunol. 2010, 185, 2701–2709. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Warren, J.D.; Danishefsky, S.J. Synthetic carbohydrate-based anticancer vaccines: The Memorial Sloan-Kettering experience. Expert Rev. Vaccines 2009, 8, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Buskas, T.; Thompson, P.; Boons, G.-J. Immunotherapy for cancer: Synthetic carbohydrate-based vaccines. Chem. Commun. 2009, 36, 5335–5349. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Röder, A.; Johannes, M. Synthesis of a MUC1-glycopeptide–BSA conjugate vaccine bearing the 3′-deoxy-3′-fluoro-Thomsen–Friedenreich antigen. Chem. Commun. 2011, 47, 9903–9905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Muthana, S.M.; Farnsworth, D.; Ludek, O.; Adams, K.; Barchi, J.J.; Gildersleeve, J.C. Enhanced epimerisation of glycosylated amino acids during solid-phase peptide synthesis. J. Am. Chem. Soc. 2012, 134, 6316–6325. [Google Scholar] [CrossRef] [PubMed]
- Di Fenza, A.; Tancredi, M.; Galoppini, C.; Rovero, P. Racemisation studies of Fmoc-Ser (tBu)-OH during stepwise continuous-flow solid-phase peptide synthesis. Tetrahedron Lett. 1998, 39, 8529–8532. [Google Scholar] [CrossRef]
- Bolchi, C.; Pallavicini, M.; Bernini, S.K.; Chiodini, G.; Corsini, A.; Ferri, N.; Fumagalli, L.; Straniero, V.; Valoti, E. Thiazole-and imidazole-containing peptidomimetic inhibitors of protein farnesyltransferase. Bioorg. Med. Chem. Lett. 2011, 21, 5408–5412. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Safina, B.S.; Zak, M.; Lee, S.H.; Nevalainen, M.; Bella, M.; Estrada, A.A.; Funke, C.; Zécri, F.J.; Bulat, S. Total synthesis of thiostrepton. Retrosynthetic analysis and construction of key building blocks. J. Am. Chem. Soc. 2005, 127, 11159–11175. [Google Scholar] [CrossRef]
- Tumminakatti, S.; Reddy, D.N.; Lakshmi, A.N.; Prabhakaran, E.N. Synthesis of 5,6-dihydro-4H-1,3-thiazine containing peptide mimics from N-(3-hydroxypropyl)thioamides and epimerisation studies. Tetrahedron Lett. 2014, 55, 4731–4735. [Google Scholar] [CrossRef]
- Sewald, N.; Jakubke, H.D. Peptides: Chemistry and Biology; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Steinauer, R.; Chen, F.M.; Leo Benoiton, N. Studies on racemization associated with the use of benzotriazol-1-yl-tris (dimethylamino) phosphonium hexafluorophosphate (BOP). Int. J. Pept. Protein Res. 1989, 34, 295–298. [Google Scholar] [CrossRef]
- Castro, B.; Dormoy, J.R.; Dourtoglou, B.; Geneviève, E.V.I.N.; Selve, C.; Ziegler, J.C. Peptide Coupling Reagents VI1. A Novel, Cheaper Preparation of Benzotriazolyloxytris [dimethylamino] phosphonium Hexafluorophosphate (BOP Reagent). Synthesis 1976, 1976, 751–752. [Google Scholar] [CrossRef]
- Li, F.; Guo, X.X.; Zeng, G.Z.; Qin, W.W.; Zhang, B.; Tan, N.H. Design and synthesis of plant cyclopeptide Astin C analogues and investigation of their immunosuppressive activity. Bioorg. Med. Chem. Lett. 2018, 28, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Ralhan, K.; KrishnaKumar, V.G.; Gupta, S. Piperazine and DBU: A safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. RSC Adv. 2015, 5, 104417–104425. [Google Scholar] [CrossRef]
- Ruczyński, J.; Lewandowska, B.; Mucha, P.; Rekowski, P. Problem of aspartimide formation in Fmoc-based solid-phase peptide synthesis using Dmab group to protect side chain of aspartic acid. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2008, 14, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Danger, G.; Plasson, R.; Pascal, R. An experimental investigation of the evolution of chirality in a potential dynamic peptide system: N-terminal epimerisation and degradation into diketopiperazine. Astrobiology 2010, 10, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.G.; Reddy, G.V. Effect of the side chain on the racemisation of amino acids in aqueous solution. J. Org. Chem. 1989, 54, 4529–4535. [Google Scholar] [CrossRef]
- Shim, Y.Y.; Reaney, M.J. Kinetic interactions between cyclolinopeptides and immobilized human serum albumin by surface plasmon resonance. J. Agric. Food Chem. 2015, 63, 1099–1106. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Manikandan, V.; Lee, N.Y. Reduced graphene oxide: Biofabrication and environmental applications. Chemosphere 2023, 311, 136934. [Google Scholar] [CrossRef]
- Bu, X.; Wu, X.; Xie, G.; Guo, Z. Synthesis of tyrocidine A and its analogues by spontaneous cyclization in aqueous solution. Org. Lett. 2002, 4, 2893–2895. [Google Scholar] [CrossRef]
- Grotenbreg, G.M.; Timmer, M.S.; Llamas-Saiz, A.L.; Verdoes, M.; van der Marel, G.A.; van Raaij, M.J.; Overkleeft, H.S.; Overhand, M. An unusual reverse turn structure adopted by a furanoid sugar amino acid incorporated in gramicidin S. J. Am. Chem. Soc. 2004, 126, 3444–3446. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Roller, P.P. Cyclization strategies in peptide derived drug design. Curr. Top. Med. Chem. 2002, 2, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Sikandar, A.; Franz, L.; Adam, S.; Santos-Aberturas, J.; Horbal, L.; Luzhetskyy, A.; Truman, A.W.; Kalinina, O.V.; Koehnke, J. The bottromycin epimerase BotH defines a group of atypical α/β-hydrolase-fold enzymes. Nat. Chem. Biol. 2020, 16, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Oberhauser, B.; Baumann, K.; Grohmann, B.; Sperner, H. Site-selective epimerisation of a fungal cyclodepsipeptide via a 5-aminooxazole intermediate. Synlett 1999, 1999, 893–896. [Google Scholar] [CrossRef]
- Muhajir, M.I.; Hardianto, A.; Al-Anshori, J.; Sumiarsa, D.; Mayanti, T.; Nurlelasari Harneti, D.; Hidayat, A.T.; Supratman, U.; Maharani, R. Total Synthesis of Nocardiotide A by Using a Combination of Solid- and Solution-Phase Methods. ChemistrySelect 2021, 6, 12941–12946. [Google Scholar] [CrossRef]
- Todorovic, M.; Perrin, D.M. Recent developments in catalytic amide bond formation. Pept. Sci. 2020, 112, e24210. [Google Scholar] [CrossRef]
- Hu, L.; Xu, S.; Zhao, Z.; Yang, Y.; Peng, Z.; Yang, M.; Wang, C.; Zhao, J. Ynamides as racemisation-free coupling reagents for amide and peptide synthesis. J. Am. Chem. Soc. 2016, 138, 13135–13138. [Google Scholar] [CrossRef]
- Xu, X.; Lai, R. The chemistry and biological activities of peptides from amphibian skin secretions. Chem. Rev. 2015, 115, 1760–1846. [Google Scholar] [CrossRef]
- Koshizuka, M.; Makino, K.; Shimada, N. Diboronic Acid Anhydride-Catalyzed Direct Peptide Bond Formation Enabled by Hydroxy-Directed Dehydrative Condensation. Org. Lett. 2020, 22, 8658–8664. [Google Scholar] [CrossRef]
- Shimada, N.; Hirata, M.; Koshizuka, M.; Ohse, N.; Kaito, R.; Makino, K. Diboronic acid anhydrides as effective catalysts for the hydroxy-directed dehydrative amidation of carboxylic acids. Org. Lett. 2019, 21, 4303–4308. [Google Scholar] [CrossRef]
- Muramatsu, W.; Hattori, T.; Yamamoto, H. Game change from reagent-to substrate-controlled peptide synthesis. Bull. Chem. Soc. Jpn. 2020, 93, 759–767. [Google Scholar] [CrossRef]
- Parkin, G. Synthetic analogues relevant to the structure and function of zinc enzymes. Chem. Rev. 2004, 104, 699–768. [Google Scholar] [CrossRef] [PubMed]
- Vera-Aviles, M.; Vantana, E.; Kardinasari, E.; Koh, N.L.; Latunde-Dada, G.O. Protective role of histidine supplementation against oxidative stress damage in the management of anemia of chronic kidney disease. Pharmaceuticals 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
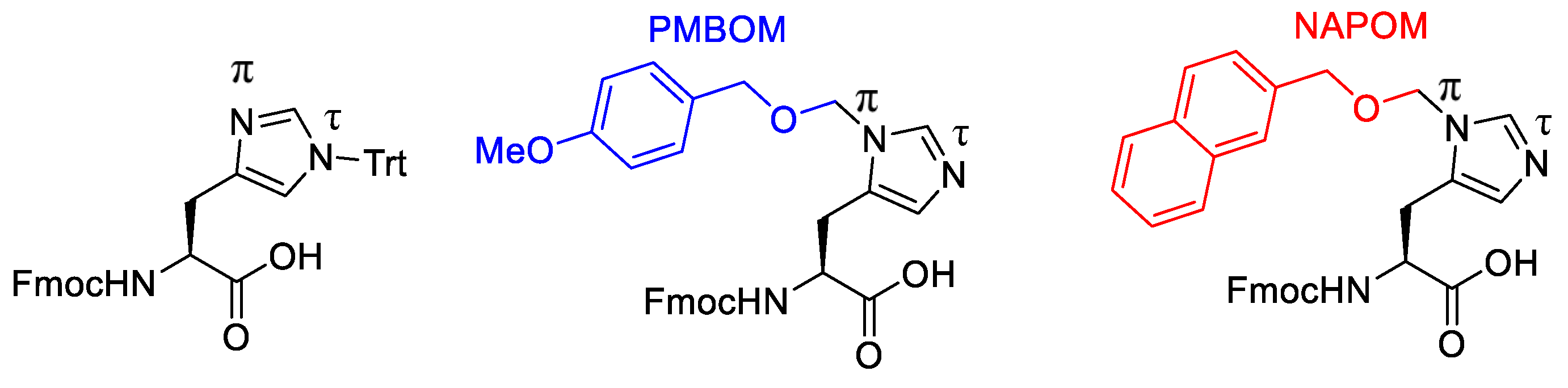
- Torikai, K.; Yanagimoto, R.; Watanabe, L.A. N(Ï)-2-Naphthylmethoxymethyl-Protected Histidines: Scalable, Racemisation-Free Building Blocks for Peptide Synthesis. Org. Process Res. Dev. 2020, 24, 448–453. [Google Scholar] [CrossRef]
- Eritja, R.; Ziehler-Martin, J.P.; Walker, P.A.; Lee, T.D.; Legesse, K.; Albericio, F.; Kaplan, B.E. On the use of st-butylsulphenyl group for protection of cysteine in solid-phase peptide synthesis using fmoc-amino acids. Tetrahedron 1987, 43, 2675–2680. [Google Scholar] [CrossRef]
- Tsuda, S.; Masuda, S.; Yoshiya, T. Epimerisation-Free Preparation of C-Terminal Cys Peptide Acid by Fmoc SPPS Using Pseudoproline-Type Protecting Group. J. Org. Chem. 2020, 85, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.X.; Lam, P.Y.S. Copper-promoted carbon-heteroatom bond cross-coupling with boronic acids and derivatives. Synthesis 2011, 2011, 829–856. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, G.; Zhang, M.; Cheng, J. Cu (OTf) 2-mediated Chan-Lam reaction of carboxylic acids to access phenolic esters. J. Org. Chem. 2010, 75, 7472–7474. [Google Scholar] [CrossRef]
- King, A.E.; Huffman, L.M.; Casitas, A.; Costas, M.; Ribas, X.; Stahl, S.S. Copper-catalyzed aerobic oxidative functionalization of an arene C–H bond: Evidence for an aryl-copper (III) intermediate. J. Am. Chem. Soc. 2010, 132, 12068–12073. [Google Scholar] [CrossRef]
- Popovic, S.; Bieräugel, H.; Detz, R.J.; Kluwer, A.M.; Koole, J.A.A.; Streefkerk, D.E.; Hiemstra, H.; Van Maarseveen, J.H. Epimerisation-free C-terminal peptide activation. Chem.—Eur. J. 2013, 19, 16934–16937. [Google Scholar] [CrossRef]
- Miyazawa, T.; Donkai, T.; Yamada, T.; Kuwata, S. Racemisation suppression by copper (II) chloride in peptide synthesis by the mixed anhydride and related methods. Chem. Lett. 1989, 18, 2125–2128. [Google Scholar] [CrossRef]
- Chalker, J.M.; Wood, C.S.C.; Davis, B.G. A convenient catalyst for aqueous and protein Suzuki−Miyaura cross-coupling. J. Am. Chem. Soc. 2009, 131, 16346–16347. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T.; Otomatsu, T.; Fukui, Y.; Yamada, T.; Kuwata, S. Racemisation-free and efficient peptide synthesis by the carbodiimide method using 1-hydroxybenzotriazole and copper (II) chloride simultaneously as additives. J. Chem. Soc. Chem. Commun. 1988, 419–420. [Google Scholar] [CrossRef]
- Ryadnov, M.G.; Klimenko, L.V.; Mitin, Y.V. Suppression of epimerisation by cupric (II) salts in peptide synthesis using free amino acids as amino components. J. Pept. Res. 1999, 53, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Gibson, F.S.; Rapoport, H. Carboxy Terminus Coupling Using 1, 1′-Carbonylbis (3-methylimidazolium triflate)(CBMIT) in the Presence of Cu (II) Salts. J. Org. Chem. 1995, 60, 2615–2617. [Google Scholar] [CrossRef]
- Pattabiraman, V.R.; Bode, J.W. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef] [PubMed]
- McKnelly, K.J.; Sokol, W.; Nowick, J.S. Anaphylaxis Induced by peptide coupling agents: Lessons learned from repeated exposure to HATU, HBTU, and HCTU. J. Org. Chem. 2019, 85, 1764–1768. [Google Scholar] [CrossRef] [PubMed]
- Dunetz, J.R.; Xiang, Y.; Baldwin, A.; Ringling, J. General and scalable amide bond formation with epimerisation-prone substrates using T3P and pyridine. Org. Lett. 2011, 13, 5048–5051. [Google Scholar] [CrossRef]
- Berger, A.; Wehrstedt, K.-D. Azodicarboxylates: Explosive properties and DSC measurements. J. Loss Prev. Process Ind. 2010, 23, 734–739. [Google Scholar] [CrossRef]
- Muramatsu, W.; Manthena, C.; Nakashima, E.; Yamamoto, H. Peptide Bond-Forming Reaction via Amino Acid Silyl Esters: New Catalytic Reactivity of an Aminosilane. ACS Catal. 2020, 10, 9594–9603. [Google Scholar] [CrossRef]
- Dawson, P.E.; Kent, S.B.H. Synthesis of native proteins by chemical ligation. Annu. Rev. Biochem. 2000, 69, 923–960. [Google Scholar] [CrossRef] [PubMed]
- Unverzagt, C.; Kajihara, Y. Chemical assembly of N-glycoproteins: A refined toolbox to address a ubiquitous posttranslational modification. Chem. Soc. Rev. 2013, 42, 4408–4420. [Google Scholar] [CrossRef] [PubMed]
- Elashal Hader, E.; Sim, Y.E.; Raj, M. Serine promoted synthesis of peptide thioester-precursor on solid support for native chemical ligation. Chem. Sci. 2016, 8, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Hackeng, T.M.; Griffin, J.H.; Dawson, P.E. Protein synthesis by native chemical ligation: Expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. USA 1999, 96, 10068–10073. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Zhang, H.; Huang, W.; Zhou, J.; Qian, H.; Chen, W. The application of an aryl hydrazine linker prevents ß-elimination side products in the SPPS of C-terminal cysteine peptides. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2010, 16, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Elashal Hader, E.; Cohen, R.D.; Raj, M. Fmoc solid-phase synthesis of C-terminal modified peptides by formation of a backbone cyclic urethane moiety. Chem. Commun. 2016, 52, 9699–9702. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Yan, B.J.; Zhu, H.Y.; Shi, W.W.; Xi, T.K.; Shi, J.; Fang, G.M. Robust synthesis of C-terminal cysteine-containing peptide acids through a peptide hydrazide-based strategy. Org. Biomol. Chem. 2019, 17, 5698–5702. [Google Scholar] [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2019 FDA TIDES (peptides and oligonucleotides) harvest. Pharmaceuticals 2020, 13, 40. [Google Scholar] [CrossRef]
- Olivos, H.J.; Alluri, P.G.; Reddy, M.M.; Salony, D.; Kodadek, T. Microwave-assisted solid-phase synthesis of peptoids. Org. Lett. 2002, 4, 4057–4059. [Google Scholar] [CrossRef]
- Erdelyi, M.; Gogoll, A. Rapid microwave-assisted solid phase peptide synthesis. Synthesis 2002, 2002, 1592–1596. [Google Scholar]
- Singh, S.K.; Collins, J.M. New developments in Microwave–Assisted solid phase peptide synthesis. Pept. Synth. Methods Protoc. 2020, 2103, 95–109. [Google Scholar]
- Mijalis, A.J.; Thomas, D.A.; Simon, M.D.; Adamo, A.; Beaumont, R.; Jensen, K.F.; Pentelute, B.L. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol. 2017, 13, 464–466. [Google Scholar] [CrossRef]
- Palasek Stacey, A.; Cox, Z.J.; Collins, J.M. Limiting racemisation and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2007, 13, 143–148. [Google Scholar]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ala (%) | Leu (%) | Phe (%) | Val (%) | Ile (%) | |
|---|---|---|---|---|---|
| DCC/DCM | 10 | 14 | 18 | 5 | 9 |
| EDC/DCM | 25 | 25 | 21 | 22 | 29 |
| DCC-HOBt/DMF | 0.8 | 6.0 | |||
| EDC-HOBt/DMF | 2.0 | 9.0 |
| Reagent | Percentage |
|---|---|
| EDC/HOAt | 29.8 |
| EDC-HCl/HOAt | 24.1 |
| DIC/HOAt | 4.2 |
| No | Peptide | Mixture | Coupling Agent; Cyclisation Reagent | Mass Spectra FABMS; LCMS | D-Asp (%) Marfey’s Test (L-Asp) |
|---|---|---|---|---|---|
| 1 | C(Xxx-D-Leu-Val-D-Pro-Asp) | Xxx = Ala, Phe, Tyr, Trp | BOP/HOBt; BOP/HOBt | +; + | 30.0 |
| 2 | C(Xxx-D-Leu-Val-D-Pro-Asp) | Xxx = Phe, Tyr, Trp, Nal Yyy = Gly, Ala, Val, Leu | BOP/HOBt; BOP/HOBt | +; + | 26.5 |
| 3 | C(Xxx-D-Leu-Val-D-Pro-Asp) | Xxx = Ala, Phe, Tyr, Trp | Active ester; HATU/HOAt | +; + | 10.7 |
| Entry | Coupling Reagent | Base | Solvent | pH conv | Diastereomeric Ratio (D: L) |
|---|---|---|---|---|---|
| 1 2 3 4 | PyBOP/HOAt PyBOP/HOAt PyBOP/HOAt DIPCDI/HOAt | - DIEA Collidine DIEA | DMF-DCM DMF-DCM DMF DMF-DCM | 8 low 8 low 8 high 7 | 15:85 67:33 13:87 |
| Coupling Condition (Equiv) | Fmoc-Ser(R)-OH, Where R = | |||||
|---|---|---|---|---|---|---|
| Trt | Ac3GaINAca | Ac4GaIb1-3Ac2GaINAca | Ac3GlcNAca | Ac3GlcNAcb | ||
| AAs: 2.5; HATU/HOAt: 1.1/1.1; NMM 2.2; 0/3 h in DMF | Yield % (D/(D + L) % | 75.1 0.8 | 69.1 5.1 | 83.9 4.3 | 100 7.7 | 69.0 8.1 |
| AAs: 1.5; HATU/HOAt: 1.2/1.2; NMM 4.0; 0/8 h in DMF | Yield % (D/(D + L) % | 81.0 2.2 | 74.6 11.4 | 96.7 21.2 | 89.9 15.0 | 92.0 20.0 |
| AAs: 3.3; HATU/HOAt: 3.65/3.7; DIPEA 7.22; 0/12 h in DMF | Yield % (D/(D + L) % | 98.9 0.2 | 99.4 0.8 | 83.0 65.6 | 100 72.5 | 1.95 2.0 |
| AAs: 4.4; HATU/HOAt: 4.4/0; NMM: 8.8; 3/12 h in NMP | Yield % (D/(D + L) % | 63.4 37.6 | 99.4 69.8 | 83.0 65.6 | 100 72.5 | 1.9 52.0 |
| AAs: 5; HATU/HOAt: 6.25/6.25; NMM: 12.5; 0/6 h in NMP | Yield % (D/(D + L) % | 98.4 2.9 | 99.4 5.2 | 98.8 6.5 | 99.5 8.1 | 95.2 9.7 |
| Reaction | Time (h) | Yield (%) |
|---|---|---|
 | 12 h | 70 |
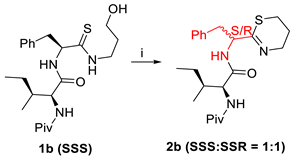 | 14 h | 51 |
| (i) MsCl (1.1 eq.), NEt3 (2 eq.), THF (0.06 M), 0 °C–rt. | ||
| Entry | Base | Solvent | pH Conv. | Diastereomeric Ratio (D:L) |
|---|---|---|---|---|
| 1 | DIEA | DMF-DCM | 8 low | 15:85 |
| 2 | Collidine | DMF-DCM | 8 low | 67:33 |
| 3 | DIEA | DMF | 8 high | 13:87 |
| Amine | Xxx (%) | |||
|---|---|---|---|---|
| Base | Equiv | Phe | Leu | Val |
| DIPEA | 1 | 2.4 | ||
| DIPEA | 3 | 11.4 | ||
| DIPEA | 2 | 3.7 | 2.5 | 1.7 |
| Bu3N | 2 | 4.1 | ||
| TEA | 2 | 5.3 | 2.4 | |
| NMP | 2 | 7.8 | ||
| NMM | 2 | 9.6 | 8.7 | 2.7 |
| NMM | 1 | 4.4 | ||
| NMM | 3 | 15.0 | ||
| Incubation Solution | Level of Epimerisation (D-Peptide/L-Peptide) × 100 |
|---|---|
| None | 1.39% |
| 20% piperidine | 1.56% |
| 5% piperazine + 2% DBU | 1.91% |
| 5% piperazine + 1% DBU + 1% FA | 1.41% |
| Incubation Solution | Relative Yield % | ||
|---|---|---|---|
| Target Peptide | D/L-Aspartimide | Piperidides or Piperazides | |
| No treatment | 97.6 | 2.4 | n.d. |
| 20% piperidine | 55.6 | 24.4 | 20.0 |
| 20% piperidine + 1% FA | 77.5 | 17.2 | 5.3 |
| 5% piperazine + 1% DBU | 9.2 | 13.7 | 77.1 |
| 5% piperazine + 1% FA | 82.8 | 7.2 | n.d. |
| 5% piperazine + 1% DBU + 1% FA | 86.0 | 14.0 | n.d. |
| No | Peptide Name | Sequence | Ring | % D to L |
|---|---|---|---|---|
| 1 | [1–8-NαC],[1-MetO2]-linusorb B1q | MetO2-Leu-Val-Phe-Pro-Leu-Phe-Ile | 8 | 61.16% |
| 2 | [1–9-NαC],[1-MetO2]-linusorb B2 | MetO2-Leu-Ile-Pro-Pro-Phe-Phe-Val-Ile | 9 | 20.48% |
| 3 | [1–8-NαC],[1-(Rs,Ss)-MetO]-linusorb B1 | [(R,S)-MetO]-Leu-Val-Phe-Pro-Leu-Phe-Ile | 8 | 55.28% |
| 4 | [1–9-NαC],[1-(Rs,Ss)-MetO]-linusorb B2 | [(R,S)-MetO]-Leu-Ile-ProPro-Phe-Phe-Val-Ile | 9 | 29.43% |
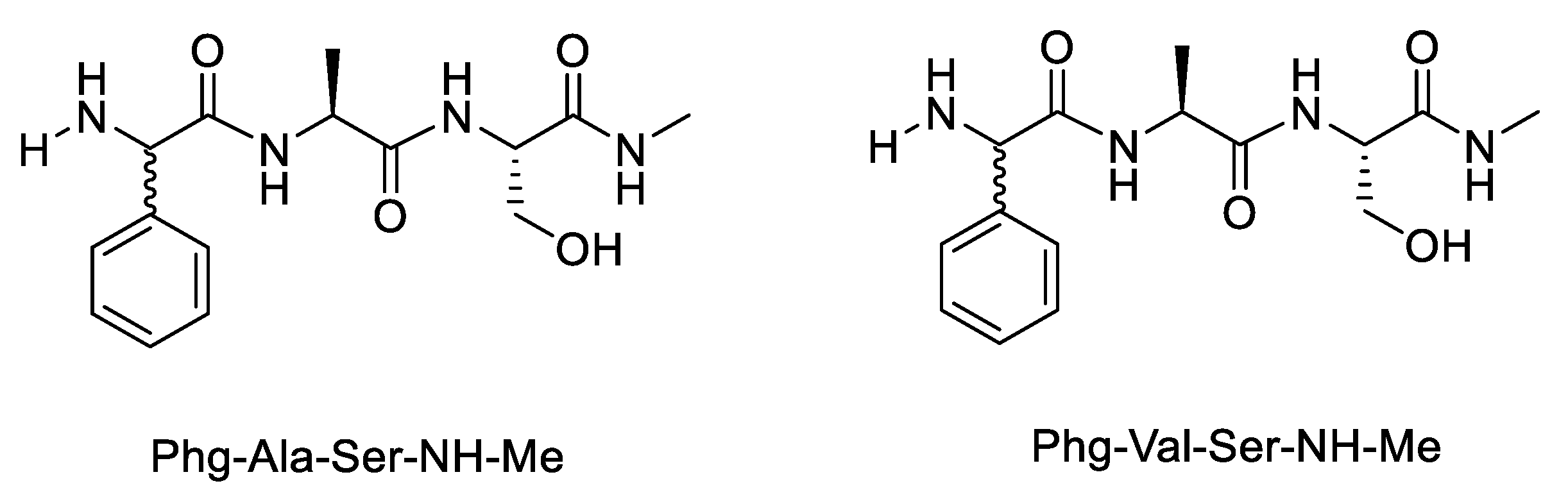
| Variable | Phg-Ala-OH | Phg-Val-OH | Phg-Ala-Ser-NH-Me | Phg-Val-Ser-NH-Me | Phg-Ala-NH |
|---|---|---|---|---|---|
| deL (∞) (%) | −13 | −42 | −1 | −16 | −15 |
| krDL (10−5 s−1) | 0.54 | 0.19 | 0.50 | - | 3.9 |
| krLL (10−5 s−1) | 0.70 | 0.46 | 0.51 | - | 5.3 |
| LO | Area under | LO | Area under | % D to L Conversion |
|---|---|---|---|---|
| Curve | Curve | |||
| 2 | 574.834 | 8 | 31.1244 | 5.14 |
| 3 | 2314.17 | 9 | 964.877 | 29.43 |
| 4 | 1251.1 | 10 | 322.223 | 20.48 |
| 5 | 767.894 | 11 | 227.048 | 22.82 |
| 6 | 411.138 | 12 | 508.19 | 55.28 |
| 7 | 319.425 | 13 | 502.883 | 61.16 |
| LOCode | Amino acid sequence (NaC-) | Molecular formula | MW (Da) | |
| 1–9-NαC]-linusorb B3 (1) | Ile-Leu-Val-Pro-Pro-Phe-Phe-Leu-Ile | C57H85N9O9 | 1040.34 | |
| [1–9-NαC]-linusorb B2 (2) | Met-Leu-Ile-Pro-Pro-Phe-Phe-Val-Ile | C56H83N9O9S | 1058.38 | |
| [1–9-NαC],[1-(Rs,Ss)-MetO]-linusorb B2 (3) | [(Rs,Ss)-MetO]-Leu-Ile-Pro-Pro-Phe-Phe-Val-Ile | C56H83N9O10S | 1074.38 | |
| [1–9-NαC],[1-MetO2]-linusorb B2 (4) | MetO2-Leu-Ile-Pro-Pro-Phe-Phe-Val-Ile | C56H83N9O11S | 1090.38 | |
| [1–8-NαC]-linusorb B1 (5) | Met-Leu-Val-Phe-Pro-Leu-Phe-Ile | C51H76N8O8S | 961.26 | |
| [1–8-NαC],[1-(Rs,Ss)-MetO]-linusorb B1 (6) | [(Rs,Ss)-MetO]-Leu-Val-Phe-Pro-Leu-Phe-Ile | C51H76N8O9S | 977.26 | |
| [1–8-NαC],[1-MetO2]-linusorb B1 (7) | MetO2-Leu-Val-Phe-Pro-Leu-Phe-Ile | C51H76N8O10S | 993.26 | |
| [1–9-NαC], DMet-linusorb B2 (8) | DMet-Leu-Ile-Pro-Pro-Phe-Phe-Val-Ile | C56H83N9O9S | 1058.38 | |
| [1–9-NαC],[1-(Rs,Ss)-DMetO]-linusorb B2 (9) | [(Rs,Ss)-DMetO]-Leu-Ile-Pro-Pro-Phe-Phe-Val-Ile | C56H83N9O10S | 1074.38 | |
| [1–9-NαC],[1-DMetO2]-linusorb B2 (10) | DMetO2-Leu-Ile-Pro-Pro-Phe-Phe-Val-Ile | C56H83N9O11S | 1090.38 | |
| [1–8-NαC], DMet-linusorb B1 (11) | DMet-Leu-Val-Phe-Pro-Leu-Phe-Ile | C51H76N8O8S | 961.26 | |
| [1–8-NαC],[1-(Rs,Ss)-DMetO]-linusorb B1 (12) | [(Rs,Ss)-DMetO]-Leu-Val-Phe-Pro-Leu-Phe-Ile | C51H76N8O9S | 977.26 | |
| [1–8-NαC],[1-DMetO2]-linusorb B1 (13) | DMetO2-Leu-Val-Phe-Pro-Leu-Phe-Ile | C51H76N8O10S | 993.26 | |
| Entry | Coupling Model | Coupling Reagent | DL/LL (%) |
|---|---|---|---|
| 1 | H-Gly-Ser-Phe-NH2 | DIC/HOBt | 3.3 |
| 2 | DIC/HOAt | 0.4 | |
| 3 | DIC/OxymaPure | 0.4 | |
| 4 | DIC/Oxyma-B | 0.4 | |
| 5 | H-Gly-Cys-Phe-NH2 | DIC/HOBt | 0.5 |
| 6 | DIC/HOAt | 0.4 | |
| 7 | DIC/OxymaPure | 0.3 | |
| 8 | DIC/Oxyma-B | 0.3 | |
| 9 | H-Gly-Cys(Acm)-Phe-NH2 | DIC/HOBt | 0.4 |
| 10 | DIC/HOAt | 0.3 | |
| 11 | DIC/OxymaPure | 0.3 | |
| 12 | DIC/Oxyma-B | 0.3 | |
| 13 | H-Gly-His-Phe-NH2 | DIC/HOBt | 1.1 |
| 14 | DIC/HOAt | 1.9 | |
| 15 | DIC/OxymaPure | 3.0 | |
| 16 | DIC/Oxyma-B | 1.0 |
 | |||||
|---|---|---|---|---|---|
| Entry | Coupling Reagent | Additive | Time | Yield | Dr |
| 1 | HBTU | DIEA | 10 min | 90% | 82:18 |
| 2 | HATU | DIEA | 10 min | 70% | 87:13 |
| 3 | PyBop | DIEA | 10 min | 91% | 88:12 |
| 4 | DCC | -- | 10 min | 98% | 91:9 |
| 5 | DEPBT | DIEA | 20 min | 61% | 99:1 |
| 6 | MYMsA | -- | 22 h | 99% | 100:0 |
| 7 | MYTsA | -- | 22 h | 94% | 100:0 |
 | |||||
|---|---|---|---|---|---|
| Entry | PG | R1 | R2 | Yield | De (%) |
| 1 | Boc | Gly | Bn | 88 | 99 |
| 2 | Fmoc | Gly | Et | 81 | 99 |
| 3 | Cbz | Ala | tBu | 79 | 99 |
| 4 | Boc | Leu | Me | 99 | 99 |
| 5 | Cbz | Leu | Me | 98 | 99 |
| 6 | Boc | Ile | Me | 96 | 98 |
| 7 | Fmoc | Ile | Me | 74 | 98 |
| 8 | Boc | Val | Me | 99 | 99 |
| 9 | Cbz | Val | Me | 88 | 99 |
| 10 | Boc | tBu | Me | 97 | 98 |
| 11 | Boc | Me2 | Bn | 21 | 93 |
| 12 | Boc | (N-Me) Gly | Bn | 47 | 96 |
| 13 | Cbz | Tyr (O-tBu) | Me | 79 | 99 |
| 14 | Boc | Tyr (O-tBu) | Me | 85 | 99 |
| 15 | Fmoc | Tyr (O-tBu) | Me | 79 | 99 |
| 16 | Fmoc | Tyr | Me | 68 | 98 |
| 17 | Boc | Asp (O-tBu | Me | 97 | 99 |
| 18 | Fmoc | Trp | Me | 79 | 96 |
| 19 | Boc | Lys (Boc) | Me | 99 | 96 |
| 20 | Boc | Arg (Pbf) | Me | 72 | 99 |
| 21 | Boc | His (Trt) | Me | 64 | 98 |
| 22 | Boc | Met (Me) | Me | 81 | 99 |
| 23 | Boc | Cys (Bn) | Me | 75 | 99 |
 | |||||
|---|---|---|---|---|---|
| Entry | Peptide Acid | R = SMe | R = SO2Me | ||
| Yield (%) | de (%) | Yield (%) | de (%) | ||
| 1 | Boc-Trp-Phe-OH | 57 | n.d. * | - | - |
| 2 | Boc-Phe-Phe-OH | 77 | 99.9 | 97 | 99.9 |
| 3 | Boc-Phe-D-Phe-OH | 75 | 99.9 | quant | 99.9 |
| 4 | Boc-Phe-Phg-OH | 70 | 99.9 | quant | 99.9 |
| 5 | Boc-Phe-D-Phg-OH | 73 | 99.9 | 95 | 99.9 |
| 6 | Boc-Phe-Val-OH | 68 | n.d. | quant | n.d. |
| 7 | Boc-Trp(Boc)-Val-OH | 70 | n.d. | quant | n.d. |
| 8 | Boc-Phe-Ala-OH | 83 | n.d. | quant | n.d. |
| 9 | Boc-Asp-(tBu)-Phe-Phe-OH | 55 | n.d. | quant | n.d. |
| Inorganic Additive (Equiv.) | Z-I-Ala-D-Phe-Gly-OBzl (%) | Z-I-Ala-D-Phe-Gly-OH (%) | ||||
|---|---|---|---|---|---|---|
| 0.5 | 1 | 2 | 0.5 | 1 | 2 | |
| None | 3.0 | 2.7 | ||||
| LiCl | 2.7 | 1.2 | 5.4 | 2.0 | 1.0 | 4.8 |
| NaI | 1.7 | <1.0 | 2.4 | 2.2 | 1.5 | 4.8 |
| RbClO4 | 1.5 | <1.0 | 1.8 | 2.7 | <1.0 | 1.6 |
| CsF | 3.3 | 2.5 | 5.2 | 3.0 | 2.7 | 5 |
| Mg(ClO4)2 | 4.0 | 3.5 | 5.4 | 4.7 | 4 | 6.6 |
| CaCl2 | 3.6 | 2.8 | 4.2 | 4.3 | 3.0 | 5.7 |
| BaI2 | 3.7 | 3.0 | 5.2 | 4.3 | 4.0 | 6 |
| BF3gt;Et2O | 1.6 | <1.0 | 1.4 | 2.0 | <1.0 | 1.7 |
| ZnCl2 | 2.4 | <1.0 | 3.1 | 2.0 | <1.0 | 3.0 |
| SnCl4 | 1.8 | <1.0 | 2.1 | 1.5 | <1.0 | 2.2 |
| AlCl3 | 1.8 | <1.0 | 1.5 | 1.4 | <1.0 | 1.2 |
| CdI2 | 2.3 | 2.0 | 2.7 | 3.0 | 1.6 | 3.4 |
| CuCl2 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
 | |||
|---|---|---|---|
| Entry | [Si]-Reagent | Yield of 1 (%) | dr |
| 1 | ClSi(OEt)3 | 43 | 97:3 |
| 2 | Si(OCH2CF3)4 | 53 | >99:1 |
| 3 | Si(OCH(CF3)2)4 | 66 | >99:1 |
| 4 | HSi(OCH2CF3)3 | 63 | >99:1 |
| 5 | HSi(OCH2CCl3)3 | 53 | >99:1 |
| 6 | HSi(OCH(CF3)2)3 | 78 | >99:1 |
| 7 | HSi(OCH2CF2CHF2)3 | 53 | >99:1 |
| 8 | HSi(OCH(CF3)2)3 | 95 | >99:1 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Reagent | Temperature (°C) | Time, t (min) | Yield (%) | Purity (%) | LDL (%) |
| 1 | EDC.HCl/Oxyma | 33 (33) | 10 (30) | 93(88) | >99 (32) | <1 (9) |
| 2 | EDC.HCl/HOBt.H2O | 34 (34) | 10 (30) | 90 (90) | 70 (48) | 25 (35) |
| 3 | EDC.HCl/HOAt | 34 (34) | 10 (30) | 88 (90) | 95 (59) | <1 (26) |
| 4 | DIC/HOAt | 30 (31) | 10 (40) | n.d (n.d) | 67 (39) | 17 (33) |
| 5 | DIC/Oxyma | 31 (31) | 10 (40) | n.d (n.d) | 46 (<10) | <1 (n.d) |
| 6 | HATU/Et3N | 34 (34) | 10 (60) | 85 (88) | 88 (58) | 1 (<1) |
| 7 | HBTU/Et3N | 33 (33) | 10 (20) | 86 (82) | 71 (55) | 2 (9) |
| 8 | EDC.HCl/Oxyma | n.d | 30 | 96 | >99 | <1 |
| Amino Acid | Synthesis Condition | |
|---|---|---|
| Conventional HBTU/DIPEA in DMF | Microwave HBTU/HOBT in DMF (80 °C) | |
| D-Asp | 1.19 | No detection |
| D-Ala | 0.21 | No detection |
| D-Arg | 0.18 | No detection |
| D-Cys | 1.09 | <1.14 |
| D-Glu | 1.46 | No detection |
| D-His | 0.65 | 0.67 |
| D-Ile | <0.1 | No detection |
| L-allo Ile | <0.1 | No detection |
| D-allo Ile | <0.1 | No detection |
| D-Leu | 0.17 | No detection |
| D-Lys | 0.1 | No detection |
| D-Met | 0.48 | No detection |
| D-Phe | 0.28 | No detection |
| D-Pro | <0.1 | No detection |
| D-Ser | 0.46 | No detection |
| D-Thr | <0.1 | No detection |
| L-allo Thr | <0.1 | No detection |
| D-allo Thr | <0.1 | No detection |
| D-Trp | 0.19 | No detection |
| D-Tyr | 0.42 | No detection |
| D-Val | <0.1 | No detection |
| Crude product purity | 68.4% | 84.0% |
| Synthesis time (h) | 23.3 | 10.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duengo, S.; Muhajir, M.I.; Hidayat, A.T.; Musa, W.J.A.; Maharani, R. Epimerisation in Peptide Synthesis. Molecules 2023, 28, 8017. https://doi.org/10.3390/molecules28248017
Duengo S, Muhajir MI, Hidayat AT, Musa WJA, Maharani R. Epimerisation in Peptide Synthesis. Molecules. 2023; 28(24):8017. https://doi.org/10.3390/molecules28248017
Chicago/Turabian StyleDuengo, Suleman, Muhamad Imam Muhajir, Ace Tatang Hidayat, Weny J. A. Musa, and Rani Maharani. 2023. "Epimerisation in Peptide Synthesis" Molecules 28, no. 24: 8017. https://doi.org/10.3390/molecules28248017
APA StyleDuengo, S., Muhajir, M. I., Hidayat, A. T., Musa, W. J. A., & Maharani, R. (2023). Epimerisation in Peptide Synthesis. Molecules, 28(24), 8017. https://doi.org/10.3390/molecules28248017