The Discovery of Indole-2-carboxylic Acid Derivatives as Novel HIV-1 Integrase Strand Transfer Inhibitors

 and
and
Abstract
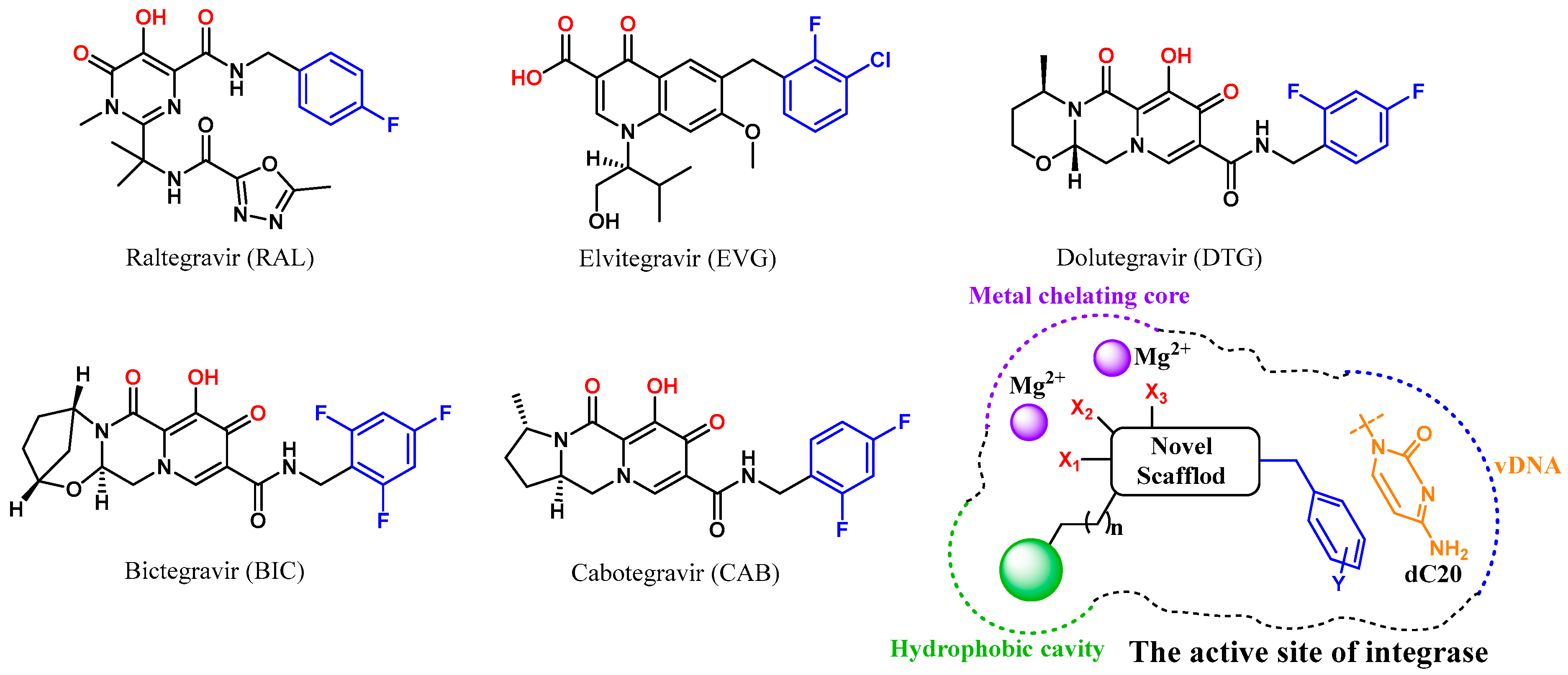
:1. Introduction
2. Results and Discussion
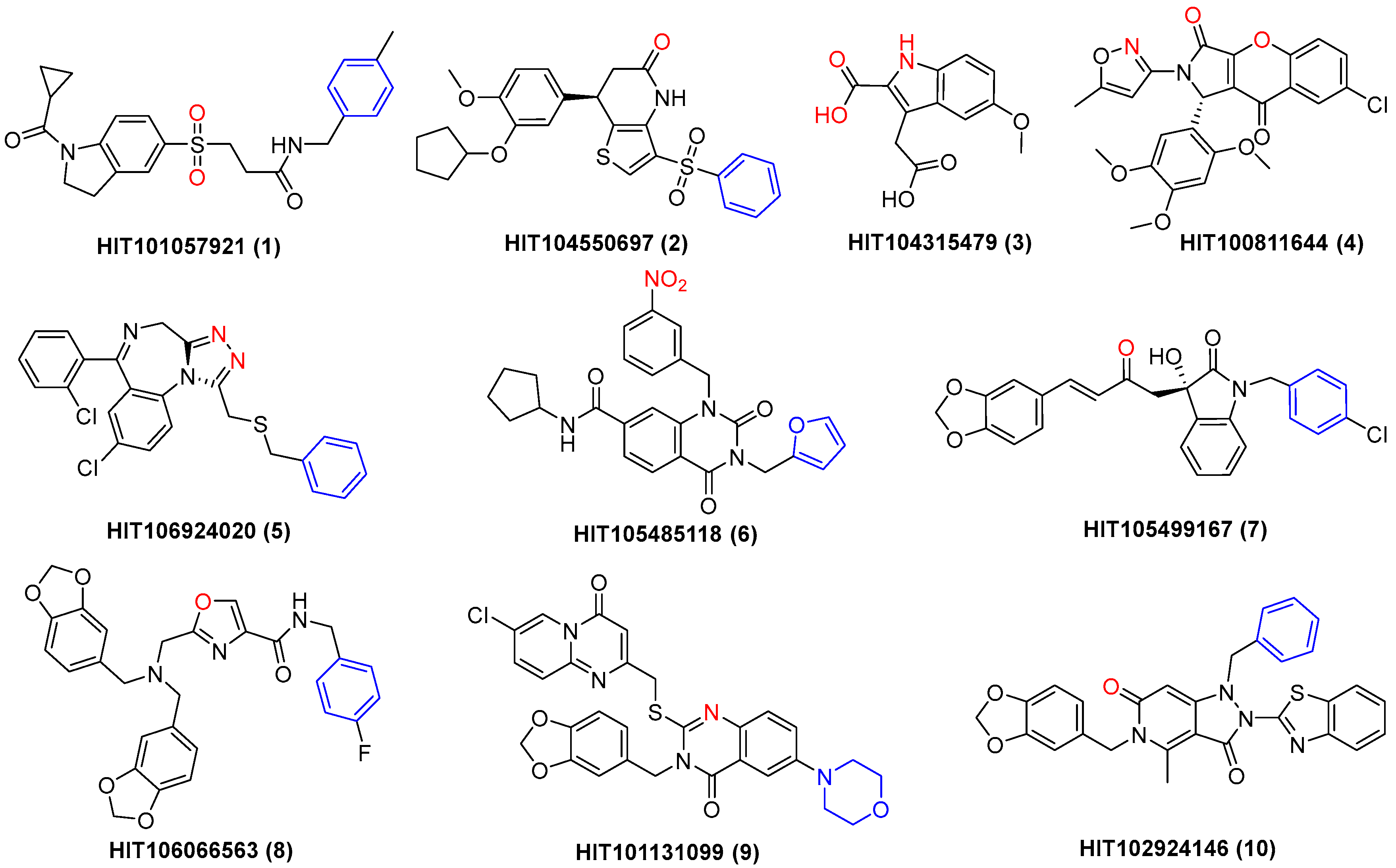
2.1. Molecular Docking-Based Virtual Screening
2.2. Evaluation of Integrase Strand Transfer Inhibitory Effect
2.3. Optimization Strategies
2.4. Synthesis route of Compound 3 Derivatives
2.5. Evaluation of Biological Activity
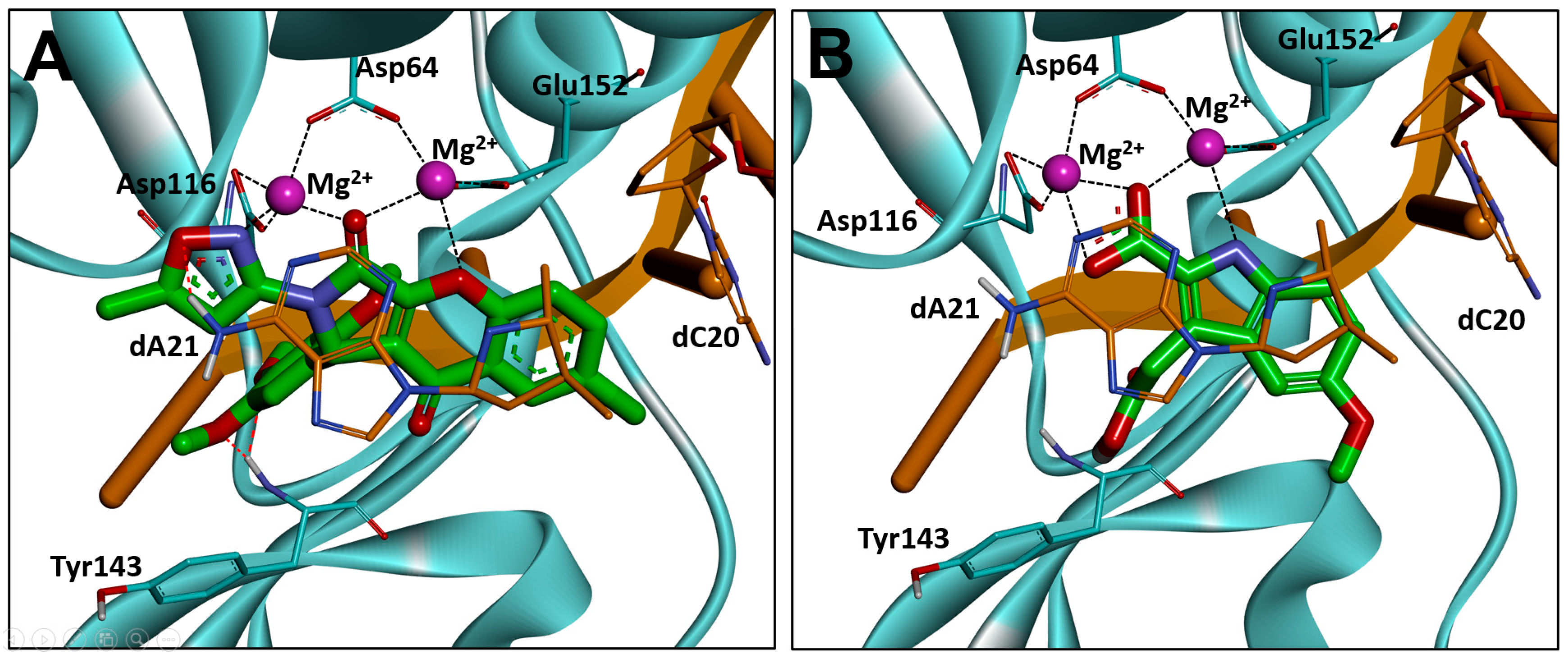
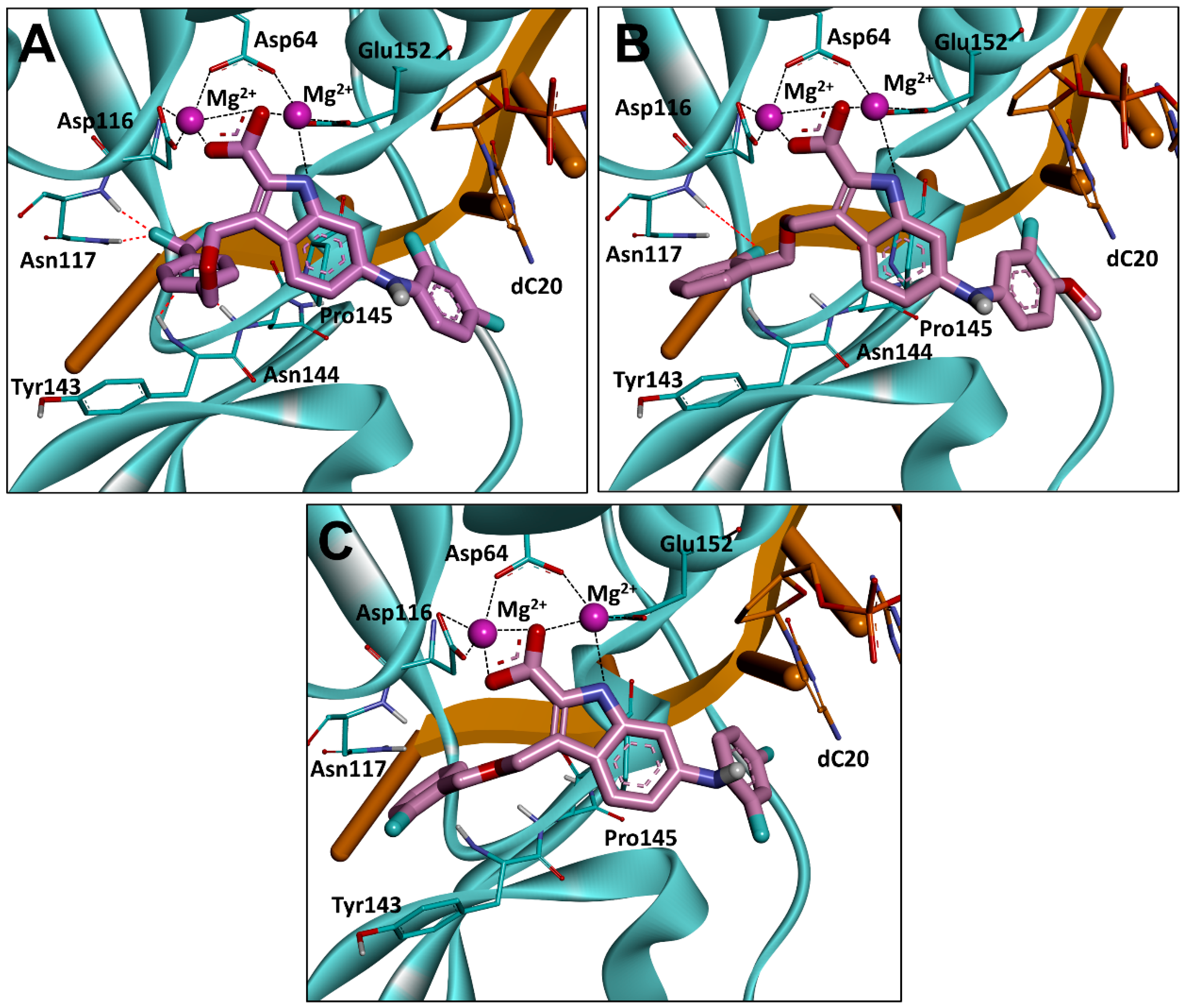
2.6. Binding Mode Analysis
3. Conclusions
4. Experimental Section
4.1. General Methods and Materials
4.2. LibDock Based Virtual Screening
4.3. Molecular Docking
4.4. PAINS Remover
4.5. General Procedure for the Synthesis of Compounds 12–21
4.5.1. Synthesis of Ethyl 6-bromo-1H-indole-2-carboxylate (12)
4.5.2. Synthesis of Ethyl 6-bromo-3-formyl-1H-indole-2-carboxylate (13)
4.5.3. Synthesis of Isopropyl 6-bromo-3-(hydroxymethyl)-1H-indole-2-carboxylate (14)
4.5.4. Synthesis of Isopropyl 6-bromo-3-(((4-(trifluoromethyl)benzyl)oxy)methyl)-1H-indole-2-carboxylate (15)
4.5.5. Synthesis of Isopropyl 6-((3-fluoro-4-methoxyphenyl)amino)-3-(((4-(trifluoromethyl)benzyl) oxy)methyl)-1H-indole-2-carboxylate (16a)
4.5.6. Isopropyl 6-((2,4-difluorophenyl)amino)-3-(((4-(trifluoromethyl)benzyl)oxy) methyl)-1H-indole-2-carboxylate (16b)
4.5.7. Synthesis of 6-((3-fluoro-4-methoxyphenyl)amino)-3-(((4-(trifluoromethyl) benzyl)oxy)methyl) -1H-indole-2-carboxylic acid (17a)
4.5.8. 6-((2,4-difluorophenyl)amino)-3-(((4-(trifluoromethyl)benzyl)oxy)methyl)-1H-indole-2-carboxylic acid (17b)
4.5.9. Isopropyl 6-bromo-3-(((2-fluorobenzyl)oxy)methyl)-1H-indole-2-carboxylate (18)
4.5.10. Isopropyl 6-((3-fluoro-4-methoxyphenyl)amino)-3-(((2-fluorobenzyl)oxy) methyl)-1H-indole-2-carboxylate (19a)
4.5.11. Isopropyl 6-((2,4-difluorophenyl)amino)-3-(((2-fluorobenzyl)oxy)methyl)-1H-indole-2-carboxylate (19b)
4.5.12. 6-((3-fluoro-4-methoxyphenyl)amino)-3-(((2-fluorobenzyl)oxy)methyl)-1H-indole-2-carboxylic acid (20a)
4.5.13. 6-((2,4-difluorophenyl)amino)-3-(((2-fluorobenzyl)oxy)methyl)-1H-indole-2-carboxylic acid (20b)
4.5.14. 6-bromo-3-(((2-fluorobenzyl)oxy)methyl)-1H-indole-2-carboxylic acid (21)
4.6. Biochemistry
4.6.1. Cell Lines, and Culture Conditions
4.6.2. Strand Transfer Inhibition Assay
4.6.3. Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoshimura, K. Current status of HIV/AIDS in the ART era. J. Infect. Chemother. 2017, 23, 12–16. [Google Scholar] [CrossRef]
- Pomerantz, R.J.; Horn, D.L. Twenty years of therapy for HIV-1 infection. Nat. Med. 2003, 9, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Amariles, P.; Rivera-Cadavid, M.; Ceballos, M. Clinical relevance of drug interactions in people living with human immunodeficiency virus on antiretroviral therapy-update 2022: Systematic review. Pharmaceutics 2023, 15, 2488. [Google Scholar] [CrossRef] [PubMed]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef]
- Eid, A.; Qi, Y. Prime editor integrase systems boost targeted DNA insertion and beyond. Trends Biotechnol. 2022, 40, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hazuda, D.J. HIV integrase as a target for antiretroviral therapy. Curr. Opin. HIV AIDS 2012, 7, 383–389. [Google Scholar] [CrossRef]
- Chiu, K.T.; Davies, R.D. Structure and function of HIV-1 integrase. Curr. Top. Med. Chem. 2004, 4, 965–977. [Google Scholar] [CrossRef]
- Gill, M.S.A.; Hassan, S.S.; Ahemad, N. Evolution of HIV-1 reverse transcriptase and integrase dual inhibitors: Recent advances and developments. Eur. J. Med. Chem. 2019, 179, 423–448. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Smith, S.J.; Maskell, D.P.; Métifiot, M.; Pye, V.E.; Fesen, K.; Marchand, C.; Pommier, Y.; Cherepanov, P.; Hughes, S.H.; et al. Structure-guided optimization of HIV integrase strand transfer inhibitors. J. Med. Chem. 2017, 60, 7315–7332. [Google Scholar] [CrossRef]
- Aknin, C.; Smith, E.A.; Marchand, C.; Andreola, M.-L.; Pommier, Y.; Metifiot, M. Discovery of novel integrase inhibitors acting outside the active site through high-throughput screening. Molecules 2019, 24, 3675. [Google Scholar] [CrossRef]
- Ramkumar, K.; Yarovenko, V.N.; Nikitina, A.S.; Zavarzin, I.V.; Krayushkin, M.M.; Kovalenko, L.V.; Esqueda, A.; Odde, S.; Neamati, N. Design, Synthesis and structure-activity studies of rhodanine derivatives as HIV-1 integrase inhibitors. Molecules 2010, 15, 3958–3992. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, S.-X.; He, Q.; Fan, R. Advances in the development of HIV integrase strand transfer inhibitors. Eur. J. Med. Chem. 2021, 225, 113787. [Google Scholar] [CrossRef]
- Mahajan, P.S.; Smith, S.J.; Hughes, S.H.; Zhao, X.; Burke, T.R. A practical approach to bicyclic carbamoyl pyridones with application to the synthesis of HIV-1 integrase strand transfer inhibitors. Molecules 2023, 28, 1428. [Google Scholar] [CrossRef]
- Rashamuse, T.J.; Fish, M.Q.; Coyanis, E.M.; Bode, M.L. Studies towards the design and synthesis of novel 1,5-diaryl-1H-imidazole-4-carboxylic acids and 1,5-diaryl-1H-imidazole-4-carbohydrazides as host LEDGF/p75 and HIV-1 integrase interaction inhibitors. Molecules 2021, 26, 6203. [Google Scholar] [CrossRef]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of Raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef]
- Shimura, K.; Kodama, E.N. Elvitegravir: A new HIV integrase inhibitor. Antivir. Chem. Chemother. 2009, 20, 79–85. [Google Scholar] [CrossRef]
- Dow, D.E.; Bartlett, J.A. Dolutegravir, the second-generation of integrase strand transfer inhibitors (INSTIs) for the treatment of HIV. Infect. Dis. Ther. 2014, 3, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Rowan-Nash, A.D.; Korry, B.J.; Mylonakis, E.; Belenky, P. Cross-domain and viral interactions in the microbiome. Microbiol. Mol. Biol. Rev. 2019, 83, 10–1128. [Google Scholar] [CrossRef]
- Swindells, S.; Andrade-Villanueva, J.-F.; Richmond, G.J.; Rizzardini, G.; Baumgarten, A.; Masiá, M.; Latiff, G.; Pokrovsky, V.; Bredeek, F.; Smith, G.; et al. Long-acting Cabotegravir and Rilpivirine for maintenance of HIV-1 suppression. N. Engl. J. Med. 2020, 382, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Métifiot, M.; Johnson, B.C.; Kiselev, E.; Marler, L.; Zhao, X.Z.; Burke, T.R., Jr.; Marchand, C.; Hughes, S.H.; Pommier, Y. Selectivity for strand-transfer over 3′-processing and susceptibility to clinical resistance of HIV-1 integrase inhibitors are driven by key enzyme–DNA interactions in the active site. Nucleic Acids Res. 2016, 44, 6896–6906. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Maertens, G.N.; Cherepanov, P. 3′-Processing and strand transfer catalysed by retroviral integrase in crystallo. EMBO J. 2012, 31, 3020–3028. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.A.; Zaharatos, G.J.; Brenner, B.G. Development of antiretroviral drug resistance. N. Engl. J. Med. 2011, 365, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Rhee, S.-Y.; Chu, C.; Avalos, A.; Ahluwalia, A.K.; Gupta, R.K.; Jordan, M.R.; Shafer, R.W. Treatment emergent Dolutegravir resistance mutations in individuals naïve to HIV-1 integrase inhibitors: A rapid scoping review. Viruses 2023, 15, 1932. [Google Scholar] [CrossRef]
- Hurt, C.B.; Sebastian, J.; Hicks, C.B.; Eron, J.J. Resistance to HIV integrase strand transfer inhibitors among clinical specimens in the United States, 2009–2012. Clin. Infect. Dis. 2014, 58, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Zhao, X.Z.; Burke, T.R.; Hughes, S.H. Efficacies of Cabotegravir and Bictegravir against drug-resistant HIV-1 integrase mutants. Retrovirology 2018, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.J.; Li, W.; Berta, D.; Badaoui, M.; Ballandras-Colas, A.; Nans, A.; Kotecha, A.; Rosta, E.; Engelman, A.N.; Cherepanov, P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 2020, 367, 806–810. [Google Scholar] [CrossRef]
- Passos, D.O.; Li, M.; Jóźwik, I.K.; Zhao, X.Z.; Santos-Martins, D.; Yang, R.; Smith, S.J.; Jeon, Y.; Forli, S.; Hughes, S.H.; et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367, 810–814. [Google Scholar] [CrossRef]
- Zhang, C.; Xie, Q.; Wan, C.C.; Jin, Z.; Hu, C. Recent advances in small-molecule HIV-1 integrase inhibitors. Curr. Med. Chem. 2021, 28, 4910–4934. [Google Scholar] [CrossRef]
- Diller, D.J.; Merz, K.M., Jr. High throughput docking for library design and library prioritization. Proteins 2001, 43, 113–124. [Google Scholar] [CrossRef]
- Rao, S.N.; Head, M.S.; Kulkarni, A.; LaLonde, J.M. Validation studies of the site-directed docking program LibDock. J. Chem. Inf. Model. 2007, 47, 2159–2171. [Google Scholar] [CrossRef]
- Wu, J.; Ma, Y.; Zhou, H.; Zhou, L.; Du, S.; Sun, Y.; Li, W.; Dong, W.; Wang, R. Identification of protein tyrosine phosphatase 1B (PTP1B) inhibitors through De Novo Evoluton, synthesis, biological evaluation and molecular dynamics simulation. Biochem. Biophys. Res. Commun. 2020, 526, 273–280. [Google Scholar] [CrossRef]
- Chang, T.T.; Sun, M.F.; Chen, H.Y.; Tsai, F.J.; Fisher, M.; Lin, J.G.; Chen, C.Y. Screening from the world’s largest TCM database against H1N1 virus. J. Biomol. Struct. Dyn. 2011, 28, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jiang, J.H.; Li, R.Y.; Deng, P. Docking-based virtual screening of TβR1 inhibitors: Evaluation of pose prediction and scoring functions. BMC Chem. 2020, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Zhang, Y.; Liu, Z.; Song, M.; Xing, Y.; Xiang, Q.; Wang, Z.; Tu, Z.; Zhou, Y.; Ding, K.; et al. Discovery of benzo[cd]indol-2(1H)-ones as potent and specific BET bromodomain inhibitors: Structure-based virtual screening, optimization, and biological evaluation. J. Med. Chem. 2016, 59, 1565–1579. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; You, Q.; Zhang, X. Medicinal chemistry of metal chelating fragments in metalloenzyme active sites: A perspective. Eur. J. Med. Chem. 2019, 165, 172–197. [Google Scholar] [CrossRef] [PubMed]
- Mbhele, N.; Chimukangara, B.; Gordon, M. HIV-1 integrase strand transfer inhibitors: A review of current drugs, recent advances and drug resistance. Int. J. Antimicrob. Agents. 2021, 57, 106343. [Google Scholar] [CrossRef] [PubMed]
- Kapewangolo, P.; Tawha, T.; Nawinda, T.; Knott, M.; Hans, R. Sceletium tortuosum demonstrates in vitro anti-HIV and free radical scavenging activity. S. Afr. J. Bot. 2016, 106, 140–143. [Google Scholar] [CrossRef]
- Jiang, F.; Chen, W.; Yi, K.; Wu, Z.; Si, Y.; Han, W.; Zhao, Y. The evaluation of catechins that contain a galloyl moiety as potential HIV-1 integrase inhibitors. Clin. Immunol. 2010, 137, 347–356. [Google Scholar] [CrossRef]
- Ivashchenko, A.A.; Ivanenkov, Y.A.; Koryakova, A.G.; Karapetian, R.N.; Mitkin, O.D.; Aladinskiy, V.A.; Kravchenko, D.V.; Savchuk, N.P.; Ivashchenko, A.V. Synthesis, biological evaluation and in silico modeling of novel integrase strand transfer inhibitors (INSTIs). Eur. J. Med. Chem. 2020, 189, 112064. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | HIT ID | m.p. (°C) a | Lowest Binding Energy (kcal/mol) | Highest Binding Energy (kcal/mol) | Most Binding Energy (kcal/mol) | IC50 (μM) b |
|---|---|---|---|---|---|---|
| 1 | HIT101057921 | 173.4–175.3 | −15.8 | −11.4 | −15.8 | 39.06 ± 1.21 |
| 2 | HIT104550697 | 113.0–116.7 | −15.3 | −11.3 | −15.3 | 33.01 ± 1.29 |
| 3 | HIT104315479 | 159.7–163.2 | −15.1 | −14.5 | −15.1 | 12.41 ± 0.07 |
| 4 | HIT100811644 | 271.4–279.0 | −13.4 | −11.0 | −13.4 | 18.52 ± 1.06 |
| 5 | HIT106924020 | 164.7–167.8 | −13.2 | −8.8 | −13.2 | 47.44 ± 1.45 |
| 6 | HIT105485118 | 235.3–241.2 | −13.2 | −11.7 | −13.1 | 22.63 ± 0.58 |
| 7 | HIT105499167 | 147.0–148.2 | −13.2 | −9.4 | −11.9 | 21.45 ± 0.26 |
| 8 | HIT106066563 | 171.5–176.8 | −13.1 | −8.5 | −12.7 | 28.16 ± 1.07 |
| 9 | HIT101131099 | 239.0–240.1 | −13.1 | −9.7 | −11.7 | 32.75 ± 0.83 |
| 10 | HIT102924146 | 139.9–143.6 | −12.9 | −11.4 | −12.9 | 22.25 ± 0.43 |
| RAL | - c | 154.3–157.9 | −13.6 | −8.9 | −12.0 | 0.08 ± 0.04 |
| Compd. | IC50 (μM) | CC50 (μM) a | Compd. | IC50 (μM) | CC50 (μM) |
|---|---|---|---|---|---|
| 15 | 2.34 ± 0.31 | >80 | 19a | 1.05 ± 0.32 | 37.58 ± 3.64 |
| 16a | 3.47 ± 0.82 | >80 | 19b | 1.70 ± 0.15 | 29.37 ± 1.76 |
| 16b | 1.06 ± 0.25 | >80 | 20a | 0.13 ± 0.02 | >80 |
| 17a | 0.93 ± 0.07 | >80 | 20b | 0.64 ± 0.06 | >80 |
| 17b | 0.39 ± 0.09 | >80 | 21 | 6.85 ± 0.66 | >80 |
| 18 | 1.92 ± 0.07 | 45.29 ± 2.73 | RAL | 0.06 ± 0.04 | >80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-C.; Zhang, W.-L.; Zhang, R.-H.; Liu, C.-H.; Zhao, Y.-L.; Yan, G.-Y.; Liao, S.-G.; Li, Y.-J.; Zhou, M. The Discovery of Indole-2-carboxylic Acid Derivatives as Novel HIV-1 Integrase Strand Transfer Inhibitors. Molecules 2023, 28, 8020. https://doi.org/10.3390/molecules28248020
Wang Y-C, Zhang W-L, Zhang R-H, Liu C-H, Zhao Y-L, Yan G-Y, Liao S-G, Li Y-J, Zhou M. The Discovery of Indole-2-carboxylic Acid Derivatives as Novel HIV-1 Integrase Strand Transfer Inhibitors. Molecules. 2023; 28(24):8020. https://doi.org/10.3390/molecules28248020
Chicago/Turabian StyleWang, Yu-Chan, Wen-Li Zhang, Rong-Hong Zhang, Chun-Hua Liu, Yong-Long Zhao, Guo-Yi Yan, Shang-Gao Liao, Yong-Jun Li, and Meng Zhou. 2023. "The Discovery of Indole-2-carboxylic Acid Derivatives as Novel HIV-1 Integrase Strand Transfer Inhibitors" Molecules 28, no. 24: 8020. https://doi.org/10.3390/molecules28248020
APA StyleWang, Y.-C., Zhang, W.-L., Zhang, R.-H., Liu, C.-H., Zhao, Y.-L., Yan, G.-Y., Liao, S.-G., Li, Y.-J., & Zhou, M. (2023). The Discovery of Indole-2-carboxylic Acid Derivatives as Novel HIV-1 Integrase Strand Transfer Inhibitors. Molecules, 28(24), 8020. https://doi.org/10.3390/molecules28248020