
Prediction of Thermostability of Enzymes Based on the Amino Acid Index (AAindex) Database and Machine Learning

Abstract
:1. Introduction
2. Results and Discussion
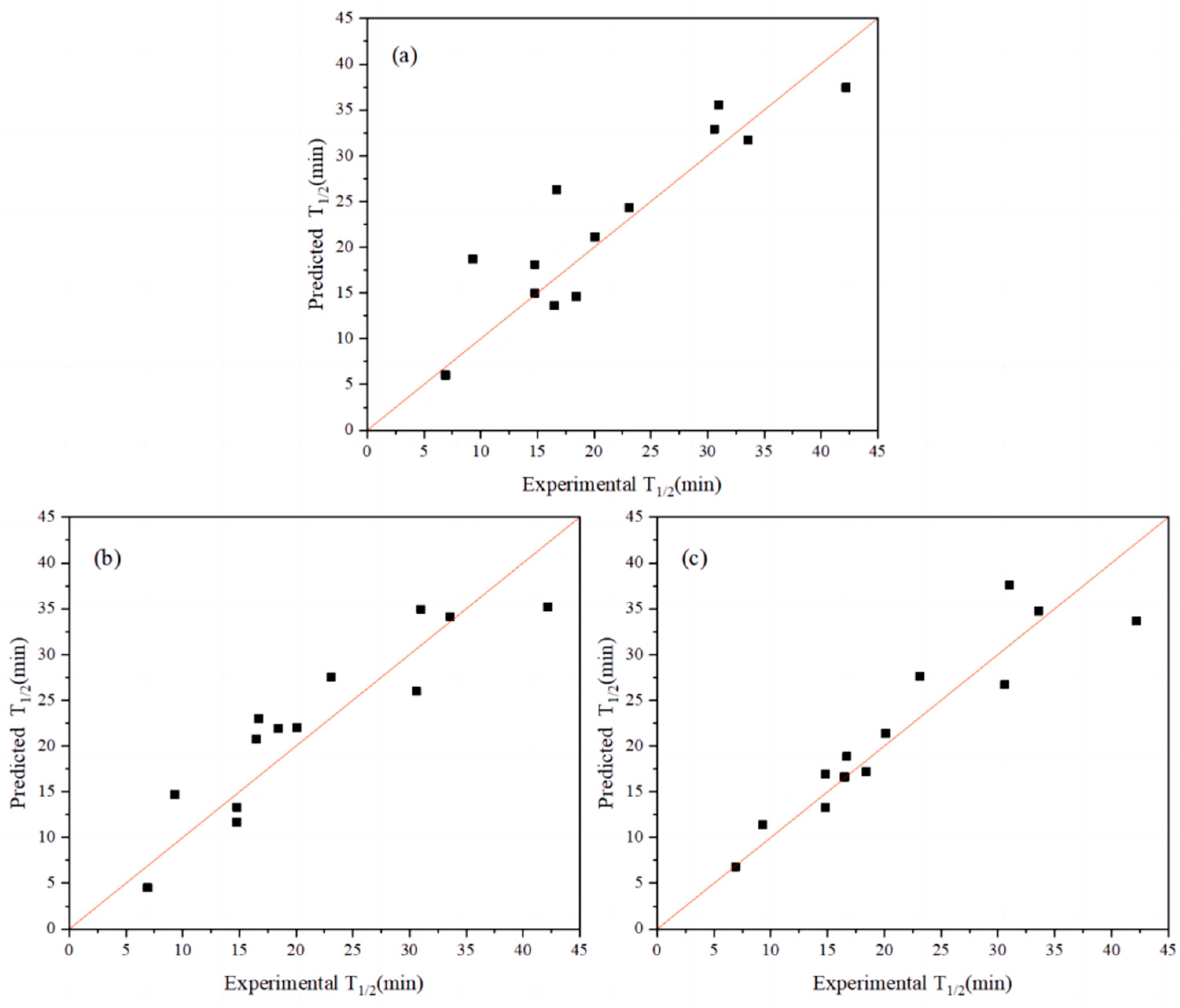
2.1. Combination Strategy Using Multiple Index Codes
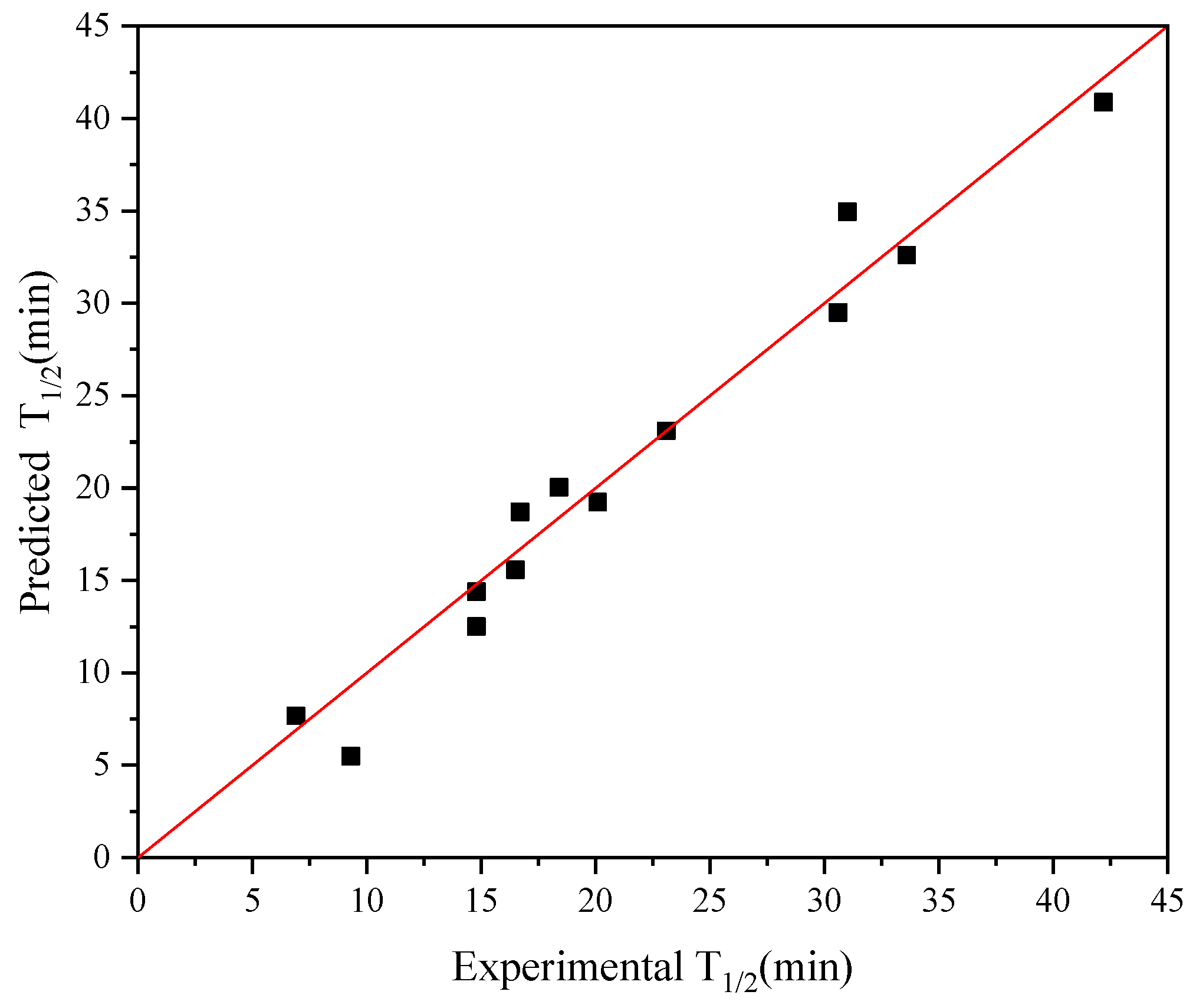
2.2. Iterative Connection Strategy
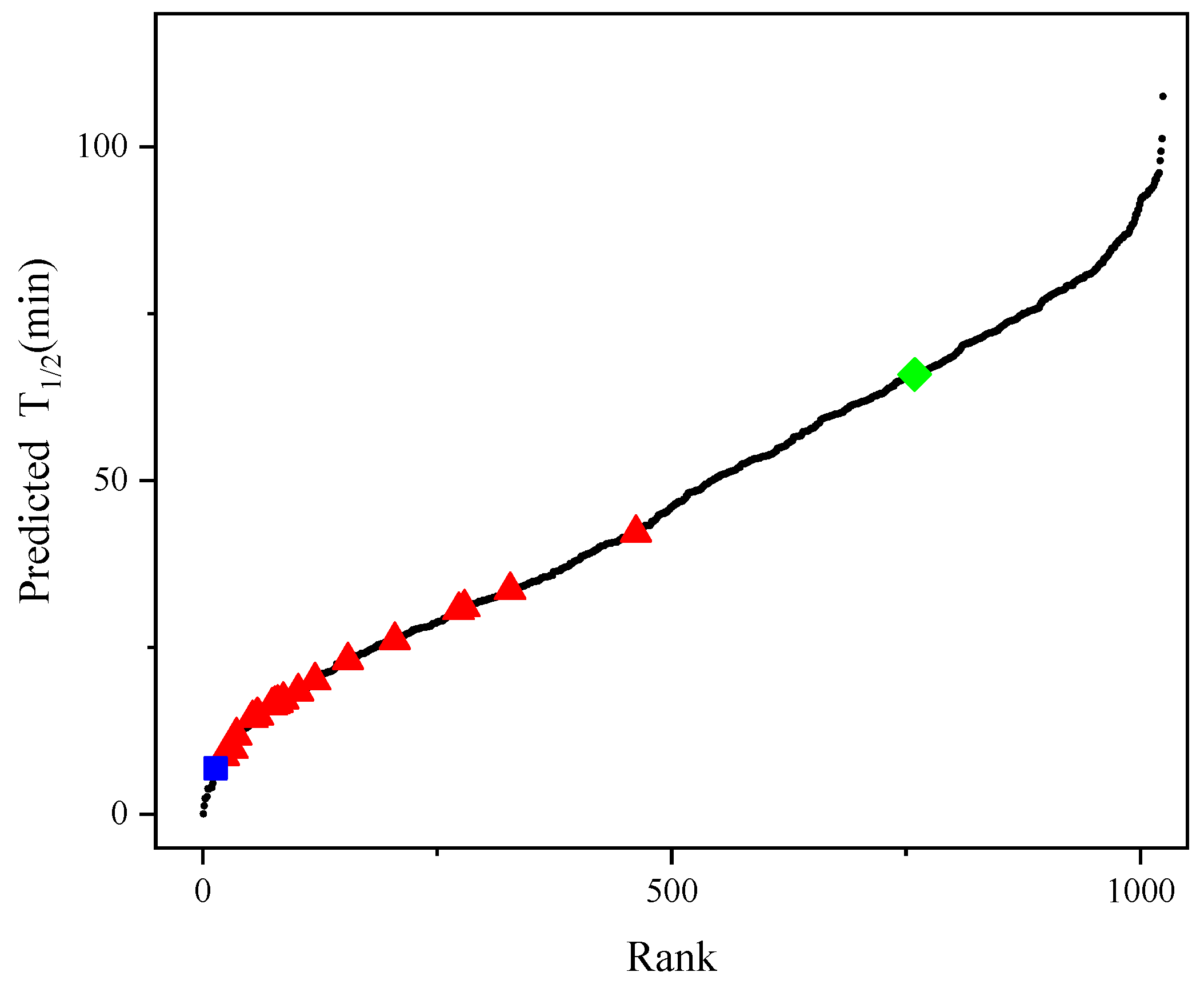
2.3. Prediction of Newly Improved AT-ATA Mutants
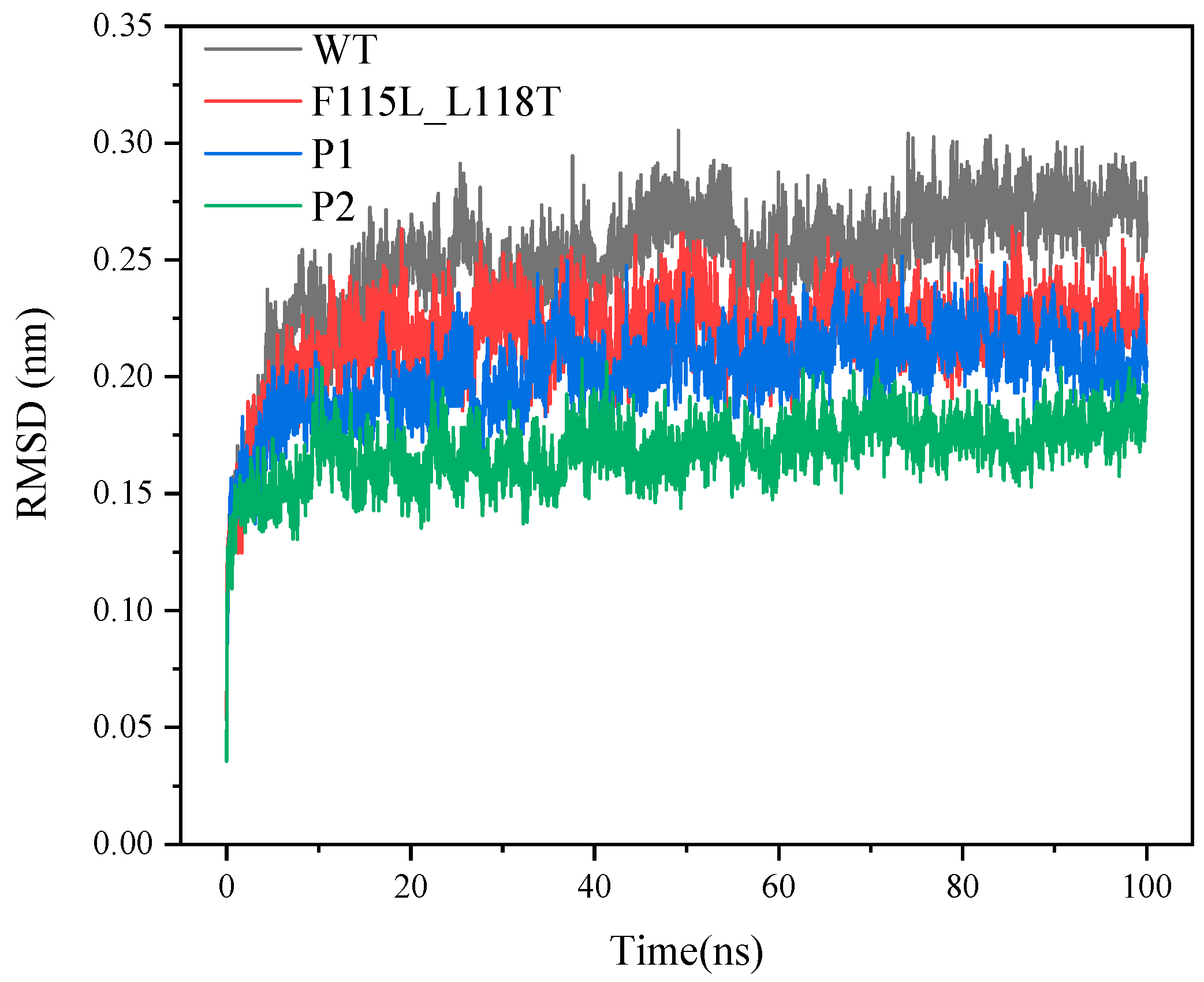
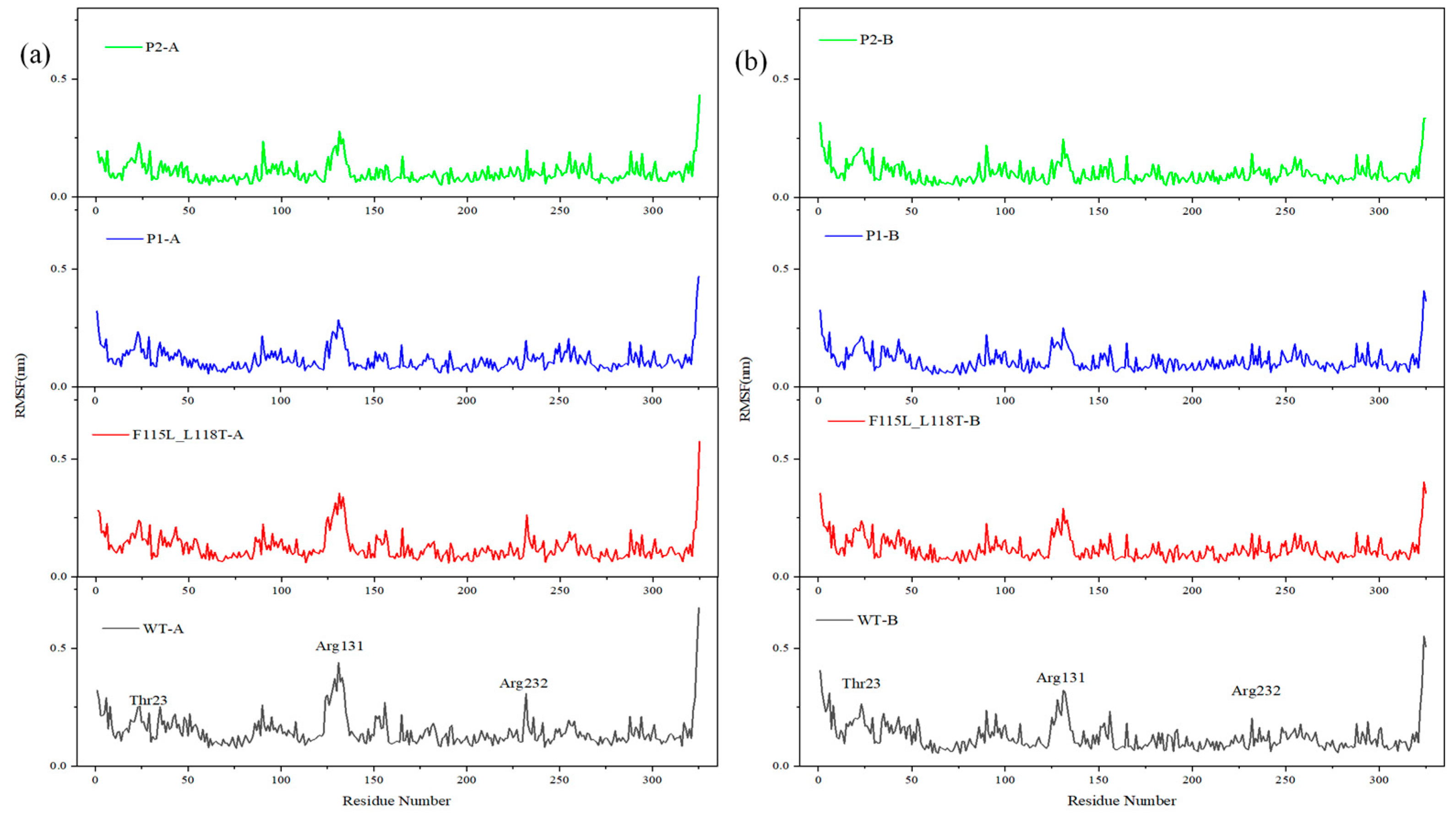
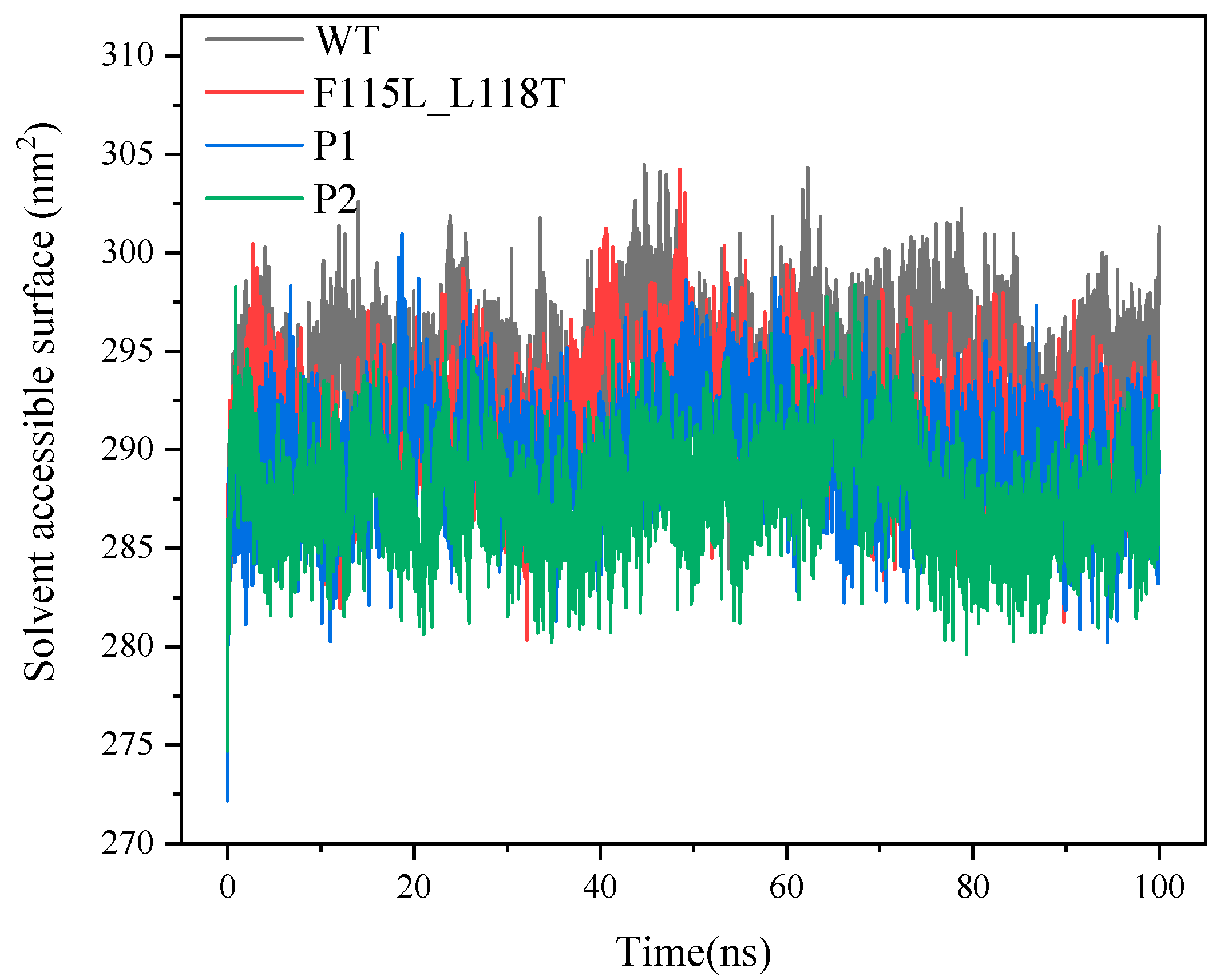
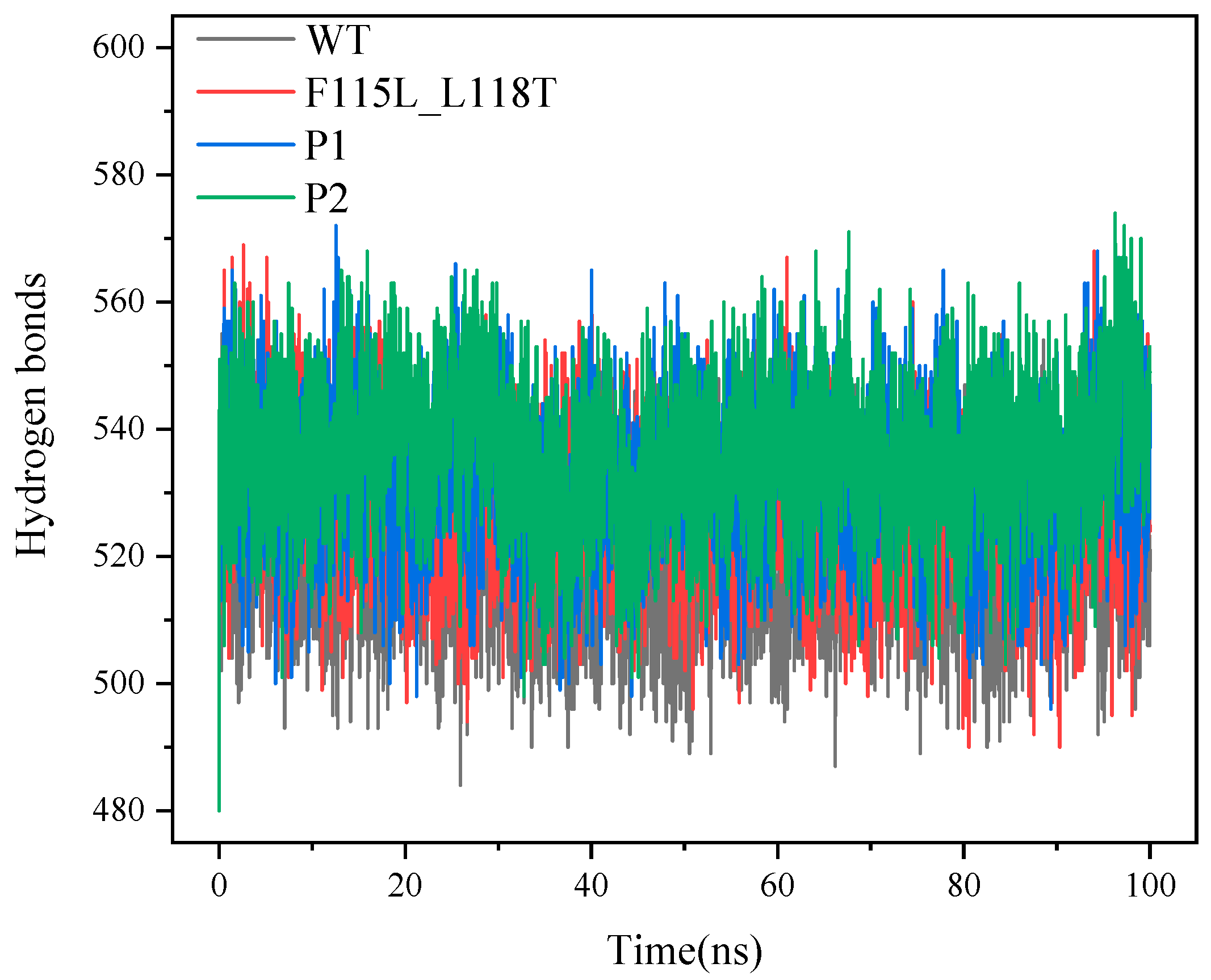
2.4. Analysis of MD Simulation for Predicting AT- ATA Mutants, Experimental Mutants, and WT
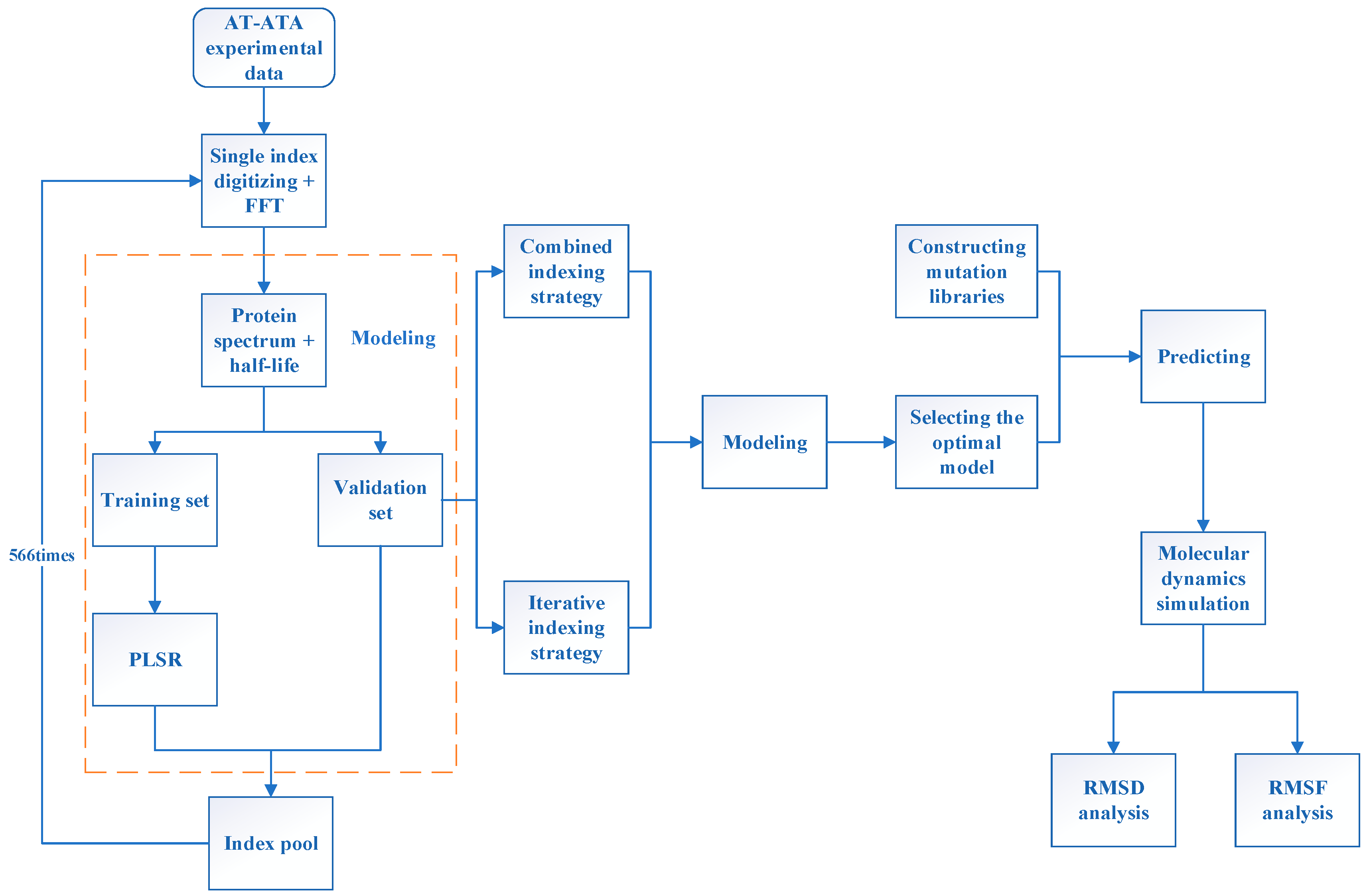
3. Materials and Methods
3.1. Aspergillus terreus Dataset
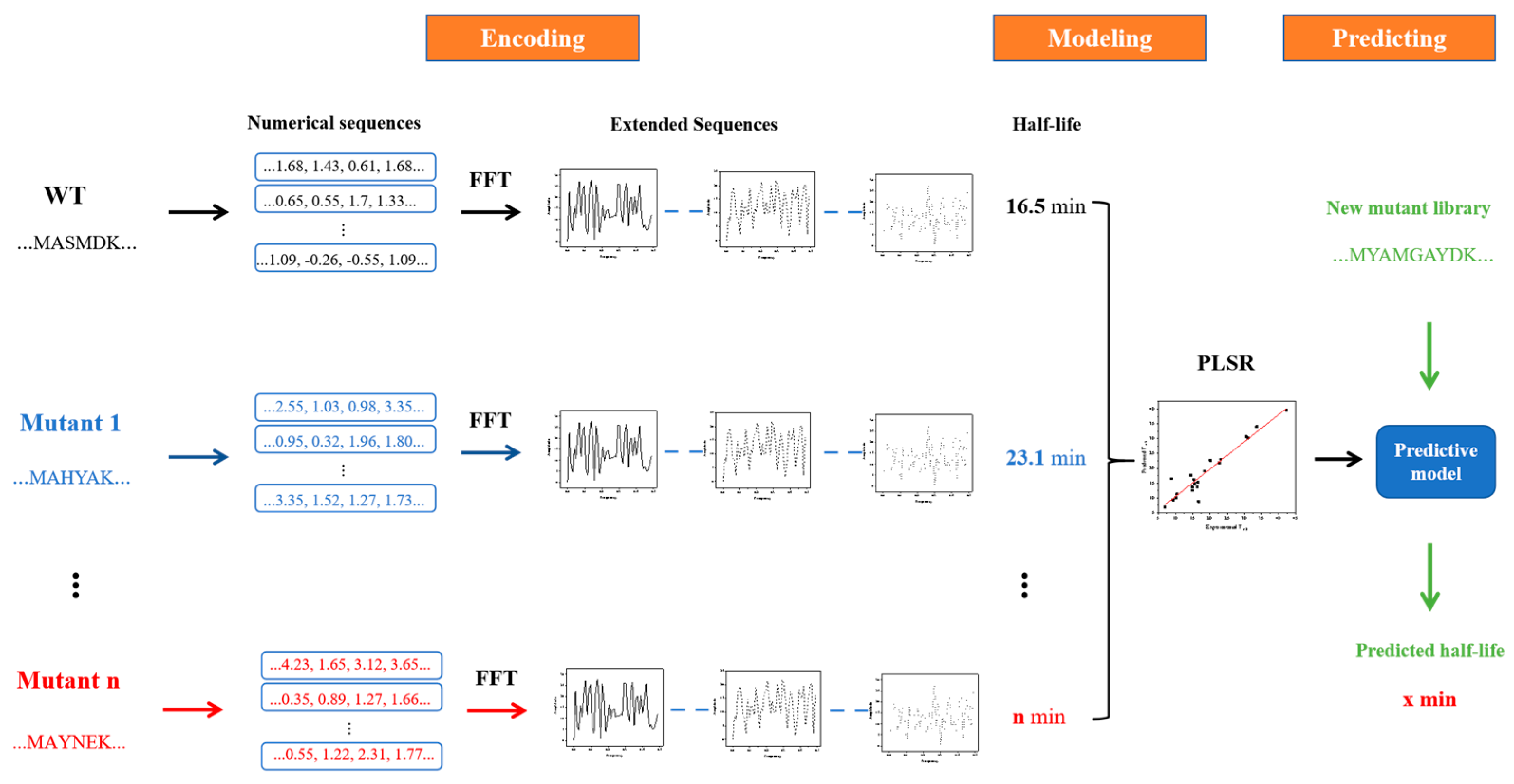
3.2. innov’SAR
3.3. Evaluation of Modeling Performance
3.4. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Romero, P.A.; Arnold, F.H. Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the Directed Evolution of Proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T. Recent Advances in Directed Evolution of Stereoselective Enzymes. In Directed Enzyme Evolution: Advances and Applications; Springer: Cham, Switzerland, 2017; pp. 69–99. [Google Scholar] [CrossRef]
- Reetz, M.T. Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.; Singh, W.; Arkin, M.; Moody, T.S.; Huang, M.; Zhou, J.; Wu, Q.; Reetz, M.T. Artificial Cysteine-Lipases with High Activity and Altered Catalytic Mechanism Created by Laboratory Evolution. Nat. Commun. 2019, 10, 3198–4208. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Wang, B.; Ilie, A.; Dubey, K.D.; Bange, G.; Korendovych, I.V.; Shaik, S.; Reetz, M.T. A Redox-Mediated Kemp Eliminase. Nat. Commun. 2017, 8, 14876. [Google Scholar] [CrossRef]
- Schwander, T.; von Borzyskowski, L.S.; Burgener, S.; Cortina, N.S.; Erb, T.J. A Synthetic Pathway for the Fixation of Carbon Dioxide in Vitro. Science 2016, 354, 900–904. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef]
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine Learning in Enzyme Engineering. ACS Catal. 2019, 10, 1210–1223. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef]
- Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Machine Learning Applications in Systems Metabolic Engineering. Curr. Opin. Biotechnol. 2020, 64, 1–9. [Google Scholar] [CrossRef]
- Woodley, J.M. Accelerating the Implementation of Biocatalysis in Industry. Appl. Microbiol. Biotechnol. 2019, 103, 4733–4739. [Google Scholar] [CrossRef]
- Wu, Z.; Kan, S.J.; Lewis, R.D.; Wittmann, B.J.; Arnold, F.H. Machine Learning-Assisted Directed Protein Evolution with Combinatorial Libraries. Proc. Natl. Acad. Sci. USA 2019, 116, 8852–8858. [Google Scholar] [CrossRef] [PubMed]
- Muggleton, S.; King, R.D.; Stenberg, M.J. Protein Secondary Structure Prediction Using Logic-Based Machine Learning. Protein Eng. Des. Sel. 1992, 5, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Folkman, L.; Stantic, B.; Sattar, A.; Zhou, Y. EASE-MM: Sequence-Based Prediction of Mutation-Induced Stability Changes with Feature-Based Multiple Models. J. Mol. Biol. 2016, 428, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Srivastava, A.K.; Wang, L. Sequence Feature-Based Prediction of Protein Stability Changes upon Amino Acid Substitutions. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef]
- Huang, L.T.; Gromiha, M.M.; Ho, S.Y. IPTREE-STAB: Interpretable Decision Tree Based Method for Predicting Protein Stability Changes upon Mutations. Bioinformatics 2007, 23, 1292–1293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Niroula, A.; Shen, B.; Vihinen, M. PON-Sol: Prediction of Effects of Amino Acid Substitutions on Protein Solubility. Bioinformatics 2016, 32, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, P.; Törönen, P.; Nokso-Koivisto, J.; Holm, L. PANNZER: High-Throughput Functional Annotation of Uncharacterized Proteins in an Error-Prone Environment. Bioinformatics 2015, 31, 1544–1552. [Google Scholar] [CrossRef]
- Cadet, F.; Fontaine, N.; Vetrivel, I.; Chong, M.N.F.; Savriama, O.; Cadet, X.; Charton, P. Application of Fourier Transform and Proteochemometrics Principles to Protein Engineering. BMC Bioinform. 2018, 19, 382. [Google Scholar] [CrossRef]
- Cadet, F.; Fontaine, N.; Li, G.; Sanchis, J.; Chong, M.N.F.; Pandjaitan, R.; Vetrivel, I.; Offmann, B.; Reetz, M.T. A Machine Learning Approach for Reliable Prediction of Amino Acid Interactions and Its Application in the Directed Evolution of Enantioselective Enzymes. Sci. Rep. 2018, 8, 16757–16772. [Google Scholar] [CrossRef]
- Ferrandi, E.E.; Monti, D. Amine Transaminases in Chiral Amines Synthesis: Recent Advances and Challenges. World J. Microbiol. Biotechnol. 2018, 34, 13. [Google Scholar] [CrossRef]
- Gao, S.; Su, Y.; Zhao, L.; Li, G.; Zheng, G. Characterization of a (R)-Selective Amine Transaminase from Fusarium Oxysporum. Process Biochem. 2017, 63, 130–136. [Google Scholar] [CrossRef]
- Kelly, S.A.; Mix, S.; Moody, T.S.; Gilmore, B.F. Transaminases for Industrial Biocatalysis: Novel Enzyme Discovery. Appl. Microbiol. Biotechnol. 2020, 104, 4781–4794. [Google Scholar] [CrossRef]
- Lyskowski, A.; Gruber, C.; Steinkellner, G.; Schürmann, M.; Schwab, H.; Gruber, K.; Steiner, K. Crystal Structure of an (R)-Selective ω-Transaminase from Aspergillus terreus. PLoS ONE 2014, 9, e87350. [Google Scholar] [CrossRef]
- Liu, C.Y.; Cecylia Severin, L.; Lyu, C.J.; Zhu, W.L.; Wang, H.P.; Jiang, C.J.; Mei, L.H.; Liu, H.G.; Huang, J. Improving Thermostability of (R)-Selective Amine Transaminase from Aspergillus terreus by Evolutionary Coupling Saturation Mutagenesis. Biochem. Eng. J. 2021, 167, 107926. [Google Scholar] [CrossRef]
- Cao, J.R.; Fan, F.F.; Lv, C.J.; Wang, H.P.; Li, Y.; Hu, S.; Zhao, W.R.; Chen, H.B.; Huang, J.; Mei, L.H. Improving the Thermostability and Activity of Transaminase from Aspergillus terreus by Charge-Charge Interaction. Front. Chem. 2021, 9, 664156. [Google Scholar] [CrossRef]
- Jia, L.; Sun, T.T.; Wang, Y.; Shen, Y. A Machine Learning Study on the Thermostability Prediction of (R)-ω-Selective Amine Transaminase from Aspergillus terreus. Biomed Res. Int. 2021, 2021, 2593748. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino Acid Index Database, Progress Report 2008. Nucleic Acids Res. 2008, 36, 202–205. [Google Scholar] [CrossRef]
- Fontaine, N.; Cadet, X.; Vetrivel, I. Novel Descriptors and Digital Signal Processing- Based Method for Protein Sequence Activity Relationship Study. Int. J. Mol. Sci. 2019, 20, 5640. [Google Scholar] [CrossRef]
- Veljković, V.; Cosić, I.; Dimitrijević, B.; Lalović, D. Is It Possible to Analyze DNA and Protein Sequences by the Methods of Digital Signal Processing? IEEE Trans. Biomed. Eng. 1985, 32, 337–341. [Google Scholar] [CrossRef]
- Benson, D.C. Digital Signal Processing Methods for Biosequence Comparison. Nucleic Acids Res. 1990, 18, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, Small Molecules and a New Graphical Interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef] [PubMed]
- Buss, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comp. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View-Molecular Graphics for All Devices-from Smartphones to Workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Index Number | cvRMSE | R2 |
|---|---|---|---|
| AURR980108 | 396 | 4.56 | 0.81 |
| OOBM770102 | 201 | 5.33 | 0.76 |
| MUNV940102 | 416 | 6.01 | 0.67 |
| CORJ870102 | 507 | 6.10 | 0.67 |
| GEOR030108 | 484 | 6.58 | 0.59 |
| Index Number | cvRMSE | R2 |
|---|---|---|
| 396,201,507,484 | 3.64 | 0.86 |
| 396,507,484 | 3.71 | 0.86 |
| 396,201,507 | 3.91 | 0.85 |
| 201,507 | 4.36 | 0.84 |
| 396,416,507 | 4.22 | 0.84 |
| 396,201,416,507 | 4.17 | 0.83 |
| 201,507,484 | 4.20 | 0.83 |
| 396,201 | 4.25 | 0.82 |
| 396,507 | 4.33 | 0.82 |
| 396,201,416 | 4.37 | 0.81 |
| Variant | Mutations | Predicted T1/2 (min) |
|---|---|---|
| P1 | Q97E_F115L_L118T_E133A_H210N_N245D_E253A_G292D | 107.59 |
| P2 | I77L_F115L_L118T_E133A_H210N_N245D_E253A | 101.25 |
| Mutations | T1/2 (min) | Note |
|---|---|---|
| WT | 6.9 | Dataset 1/Dataset 2 |
| I77L | 20.1 | Dataset 1/Dataset 2 |
| Q97E | 16.5 | Dataset 1/Dataset 2 |
| F115L | 17.2 | Dataset 2 |
| L118T | 26.1 | Dataset 2 |
| E133A | 9.8 | Dataset 2 |
| H210N | 23.1 | Dataset 1/Dataset 2 |
| N245D | 14.8 | Dataset 1/Dataset 2 |
| E253A | 11.8 | Dataset 2 |
| G292D | 14.8 | Dataset 1/Dataset 2 |
| I295V | 9.3 | Dataset 1/Dataset 2 |
| F115L_L118T | 65.9 | Dataset 2 |
| I77L_H210N | 42.2 | Dataset 1/Dataset 2 |
| Q97E_H210N | 30.6 | Dataset 1/Dataset 2 |
| H210N_N245D | 18.4 | Dataset 1/Dataset 2 |
| H210N_G292D | 33.6 | Dataset 1/Dataset 2 |
| I77L_Q97E_H210N | 31 | Dataset 1/Dataset 2 |
| I77L_H210N_G292D | 16.7 | Dataset 1/Dataset 2 |
| I77L_Q97E_H210N_N245D | 14.4 | Dataset 2 |
| I77L_H210N_N245D_G292D | 16.3 | Dataset 2 |
| I77L_Q97E_H210N_N245D_G292D | 8.7 | Dataset 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Jia, L.; Wang, K.; Sun, T.; Huang, J. Prediction of Thermostability of Enzymes Based on the Amino Acid Index (AAindex) Database and Machine Learning. Molecules 2023, 28, 8097. https://doi.org/10.3390/molecules28248097
Li G, Jia L, Wang K, Sun T, Huang J. Prediction of Thermostability of Enzymes Based on the Amino Acid Index (AAindex) Database and Machine Learning. Molecules. 2023; 28(24):8097. https://doi.org/10.3390/molecules28248097
Chicago/Turabian StyleLi, Gaolin, Lili Jia, Kang Wang, Tingting Sun, and Jun Huang. 2023. "Prediction of Thermostability of Enzymes Based on the Amino Acid Index (AAindex) Database and Machine Learning" Molecules 28, no. 24: 8097. https://doi.org/10.3390/molecules28248097