
New ψ-Santonin Derivatives from Crossostephium chinense and Their Anti-Proliferative Activities against Leishmania major and Human Cancer Cells A549

 , , , ,
, , , ,
Abstract
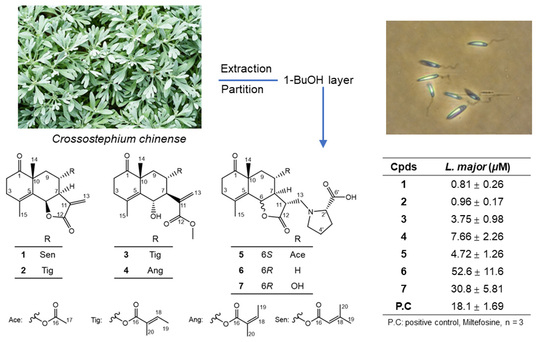
:
1. Introduction
2. Results and Discussion
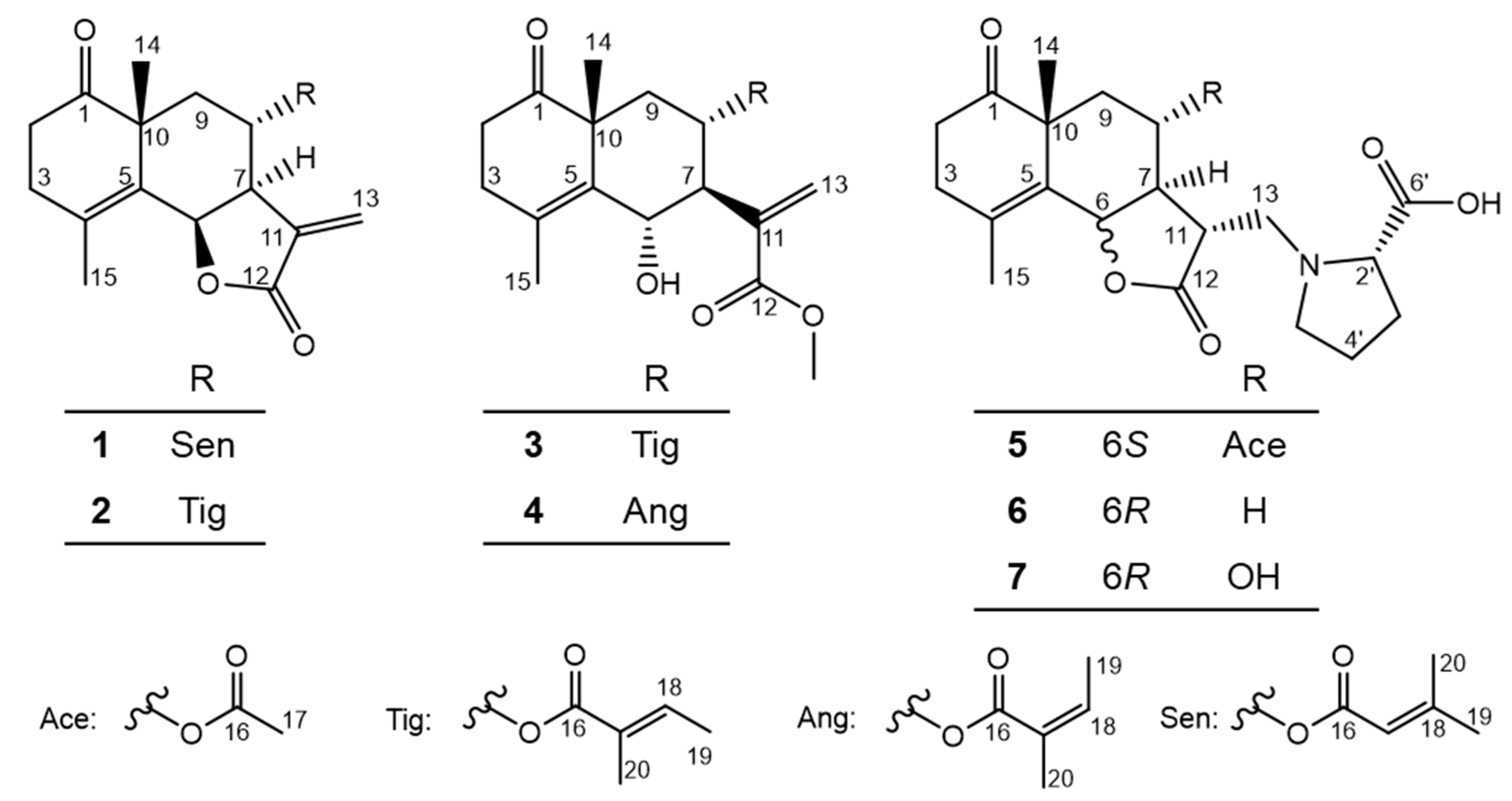
2.1. Structure Determination of New Compounds 1–7
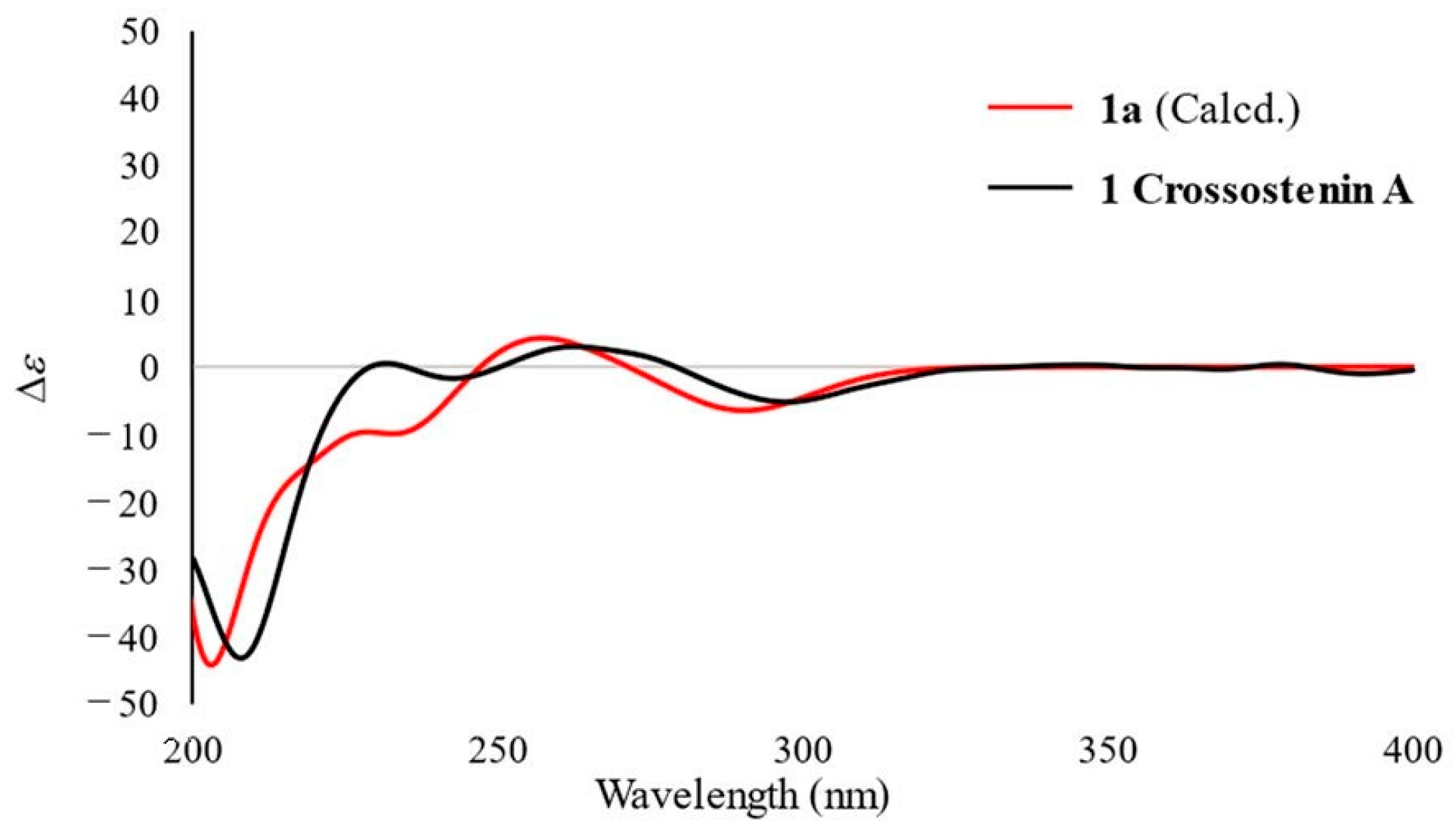
2.1.1. Structure of Crossostenin A (1)
2.1.2. Structure of Crossostenin B (2)
2.1.3. Structure of Crossostenin C (3)
2.1.4. Structure of Crossostenin D (4)
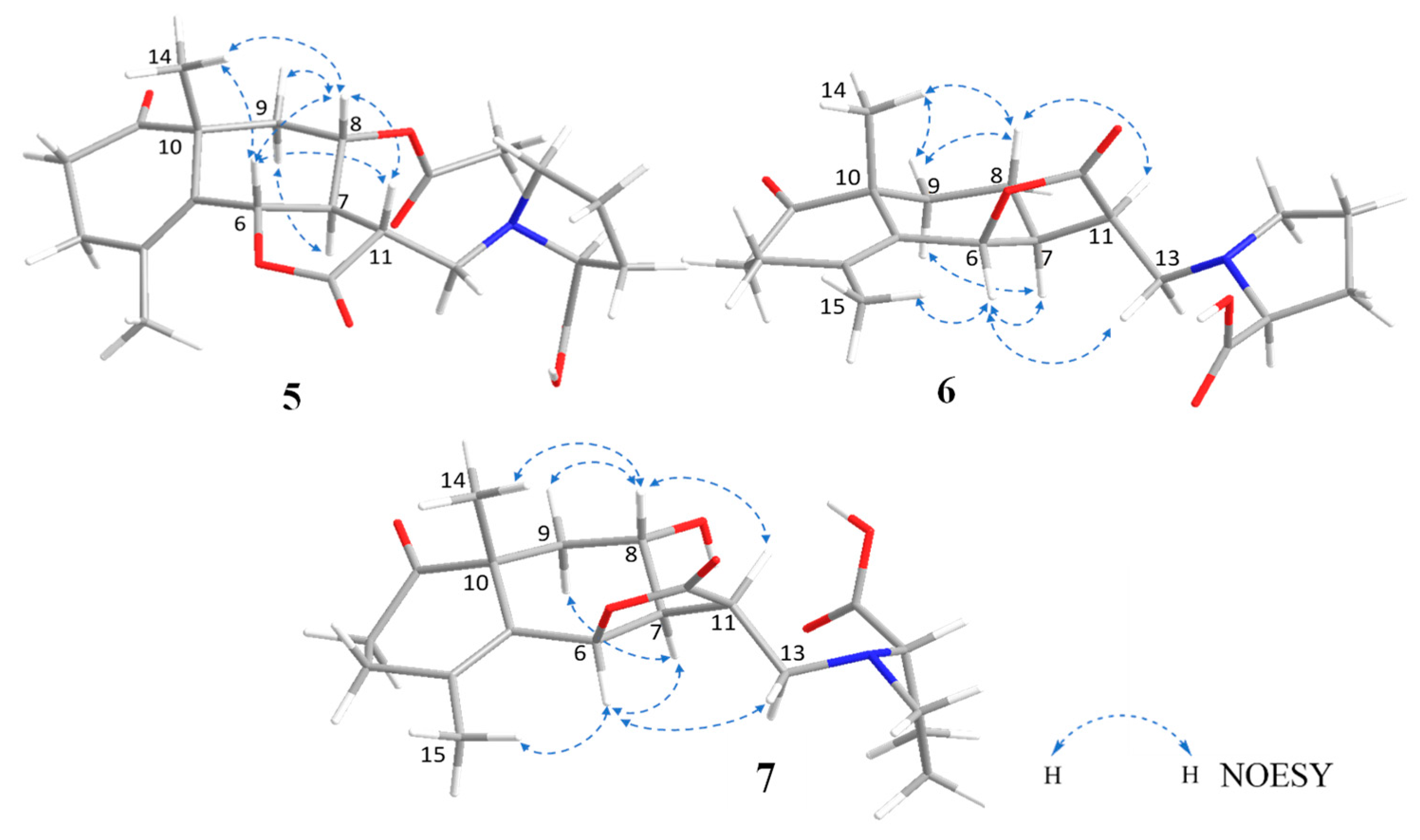
2.1.5. Structure of Crossoseamine C (5)
2.1.6. Structure of Crossoseamine D (6)
2.1.7. Structure of Crossoseamine E (7)
2.2. Cytotoxic Activities of the Isolated Compounds
3. Experiments
3.1. General Experimental Procedure
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Acid Hydrolysis of Compounds 5–7
3.5. Computational Calculations
3.6. Anti-Proliferative Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abirami, M.; Kumar, B.K.; Faheem; Dey, S.; Johri, S.; Reguera, R.M.; Balaña-Fouce, R.; Sekhar, K.V.G.C.; Sankaranarayanan, M. Molecular-level strategic goals and repressors in Leishmaniasis—Integrated data to accelerate target-based heterocyclic scaffolds. Eur. J. Med. Chem. 2023, 257, 115471. [Google Scholar] [CrossRef]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Sasidharan, S.; Saudagar, P. Leishmaniasis: Where are we and where are we heading? Parasitol. Res. 2021, 120, 1541–1554. [Google Scholar] [CrossRef]
- Abadías-Granado, I.; Diago, A.; Cerro, P.A.; Palma-Ruiz, A.M.; Gilaberte, Y. Leishmaniasis cutánea y mucocutánea. Actas Dermosifiliogr. 2021, 112, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Falcão, G.; Lins-Kusterer, L.; Leite-Ribeiro, P.; Sarmento, V. Orofacial manifestations of mucocutaneous leishmaniasis: A case series from Brazil. F1000Research 2020, 8, 756. [Google Scholar] [CrossRef] [PubMed]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Research 2017, 6, 750. [Google Scholar] [CrossRef]
- Wang, Z.; Chitama, B.-Y.A.; Suganuma, K.; Yamano, Y.; Sugimoto, S.; Kawakami, S.; Kaneko, O.; Otsuka, H.; Matsunami, K. Two New Cytotoxic Sesquiterpene-Amino Acid Conjugates and a Coumarin-Glucoside from Crossostephium chinense. Molecules 2023, 28, 4696. [Google Scholar] [CrossRef]
- Zhao, X.; Alper, H.; Yu, Z. Stereoselective Hydroxycarbonylation of Vinyl Bromides to α,β-Unsaturated Carboxylic Acids in the Ionic Liquid [BMIM]PF6. J. Org. Chem. 2006, 71, 3988–3990. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Muranaka, A.; Mack, J. Chapter 2: Empirical Rules in CD Spectra and Absolute Configuration of Molecules. In Circular Dichroism and Magnetic Circular Dichroism Spectroscopy for Organic Chemists; The Royal Society of Chemistry: London, UK, 2011; pp. 42–52. [Google Scholar] [CrossRef]
- Joseph-Nathan, P.; Wesener, J.R.; Günther, H. A Two-Dimensional NMR study of Angelic and Tiglic Acid. Org. Magn. Reson. 1984, 22, 190–191. [Google Scholar] [CrossRef]
- Matsuda, H.; Kageura, T.; Inoue, Y.; Morikawa, T.; Yoshikawa, M. Absolute Stereostructures and Syntheses of Saussureamines A, B, C, D and E, Amino Acid–Sesquiterpene Conjugates with Gastroprotective Effect, from the Roots of Saussurea lappa. Tetrahedron 2000, 56, 7763–7777. [Google Scholar] [CrossRef]
- Sugimoto, S.; Yamano, Y.; Desoukey, S.Y.; Katakawa, K.; Wanas, A.S.; Otsuka, H.; Matsunami, K. Isolation of Sesquiterpene–Amino Acid Conjugates, Onopornoids A–D, and a Flavonoid Glucoside from Onopordum alexandrinum. J. Nat. Prod. 2019, 82, 1471–1477. [Google Scholar] [CrossRef]
- Pauli, N.; Séquin, U.; Walter, A. Boscialin and Boscialin 4′-O-Glucoside, Two New Compounds Isolated from the Leaves of Boscia salicifolia OLIV. Helv. Chim. Acta 1990, 73, 578–582. [Google Scholar] [CrossRef]
- Yu, Q.; Otsuka, H.; Hirata, E.; Shinzato, T.; Takeda, Y. Turpinionosides A-E: Megastigmane Glucosides from Leaves of Turpinia ternata NAKAI. Chem. Pharm. Bull. 2002, 50, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, M.; Magre, M.; Zubar, V.; Rueping, M. Reduction of Cyclic and Linear Organic Carbonates Using a Readily Available Magnesium Catalyst. ACS Catal. 2019, 9, 11634–11639. [Google Scholar] [CrossRef]
- Miyase, T.; Ueno, A.; Takizawa, N.; Kobayashi, H.; Karasawa, H. Studies on the Glycosides of Epimedium grandiflorum MORR. Var. thunbergianum (MIQ.) NAKAI. I. Chem. Pharm. Bull. 1987, 35, 1109–1117. [Google Scholar] [CrossRef]
- Jeon, S.H.; Chun, W.; Choi, Y.J.; Kwon, Y.S. Cytotoxic constituents from the bark of Salix hulteni. Arch. Pharmacal Res. 2008, 31, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Domondon, D.L.; He, W.; De Kimpe, N.; Höfte, M.; Poppe, J. β-Adenosine, a bioactive compound in grass chaff stimulating mushroom production. Phytochemistry 2004, 65, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-Q.; Yue, J.-M. Biologically active phenols from Saussurea medusa. Biorg. Med. Chem. 2003, 11, 703–708. [Google Scholar] [CrossRef]
- Kazuma, K.; Noda, N.; Suzuki, M. Malonylated flavonol glycosides from the petals of Clitoria ternatea. Phytochemistry 2003, 62, 229–237. [Google Scholar] [CrossRef]
- Lee, E.H.; Kim, H.J.; Song, Y.S.; Jin, C.; Lee, K.-T.; Cho, J.; Lee, Y.S. Constituents of the stems and fruits of Opuntia ficus-indica var. saboten. Arch. Pharmacal Res. 2003, 26, 1018–1023. [Google Scholar] [CrossRef]
- Beck, M.-A.; Häberlein, H. Flavonol glycosides from Eschscholtzia californica. Phytochemistry 1999, 50, 329–332. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Asaumi, S.; Kawakami, S.; Sugimoto, S.; Matsunami, K.; Otsuka, H.; Shinzato, T. Alkylated Benzoquinones: Ardisiaquinones A–H from the Leaves of Ardisia quinquegona and Their Anti-Leishmania Activity. Chem. Pharm. Bull. 2018, 66, 757–763. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crossostenin A (1) | Crossostenin B (2) | Crossostenin C (3) | Crossostenin D (4) | |||||
|---|---|---|---|---|---|---|---|---|
| Position | 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C |
| 1 | 216.4 | 214.9 | 216.0 | 216.0 | ||||
| 2 | 2.34 (m) α 2.60 (dd, 14.5, 1.6) β | 35.9 | 2.31 (dd, 16.6, 2.2) α 2.64 (dd, 16.6, 1.6) β | 35.7 | 2.43 (m) α 2.64 (ddd, 12.8, 6.3, 5.5) β | 36.7 | 2.43 (ddd, 13.4, 7.1, 6.1) α 2.65 (ddd, 13.4, 8.0, 6.1) β | 36.7 |
| 3 | 2.37 (m) α 2.75 (m) β | 30.3 | 2.39 (ddd, 16.1, 7.5, 2.2) α 2.79 (m) β | 30.5 | 2.35 (m) α 2.51 (m) β | 34.4 | 2.34 (br dd, 13.1, 8.0) α 2.45 (br d, 13.1) β | 34.4 |
| 4 | 128.1 | 129.7 | 131.2 | 131.3 | ||||
| 5 | 141.1 | 141.1 | 135.0 | 135.0 | ||||
| 6 | 5.68 (d, 7.1) | 77.5 | 5.70 (d, 7.2) | 77.5 | 4.90 (br d, 10.1) | 71.5 | 4.90 (m) | 71.6 |
| 7 | 3.29 (m) | 45.6 | 3.34 (m) | 45.5 | 2.72 (t, 10.1) | 57.9 | 2.69 (t, 10.2) | 58.1 |
| 8 | 4.81 (m) | 72.7 | 4.83 (m) | 73.9 | 5.44 (ddd, 11.0, 10.1, 4.6) | 71.8 | 5.49 (ddd, 11.1, 10.2, 4.7) | 71.4 |
| 9 | 1.49 (dd, 14.5, 2.1) β 2.52 (ddd, 14.5, 6.6, 1.8) α | 33.8 | 1.49 (dd, 14.5, 2.2) β 2.61 (ddd, 14.5, 6.4, 1.6) α | 33.2 | 1.51 (dd, 12.7, 11.0) α 2.15 (dd, 12.7, 4.6) β | 39.9 | 1.52 (dd, 12.7, 11.1), α 2.13 (dd, 12.7, 4.7) β | 40.0 |
| 10 | 46.8 | 46.6 | 49.8 | 49.8 | ||||
| 11 | 139.1 | 139.2 | 140.1 | 140.1 | ||||
| 12 | 171.9 | 171. | 168.5 | 168.4 | ||||
| 12-OCH3 | 3.75 (s) | 52.4 | 3.74 (s) | 52.4 | ||||
| 13 | 5.90 (d, 2.0) 6.34 (d, 2.0) | 125.8 | 5.91 (d, 2.0) 6.35 (d, 2.0) | 125.8 | 5.70 (d, 1.1) 6.26 (d, 1.1) | 129.3 | 5.69 (d, 1.1) 6.27 (d, 1.1) | 129.5 |
| 14 | 1.21 (s) | 26.2 | 1.23 (s) | 26.3 | 1.40 (s) | 23.9 | 1.36 (s) | 23.9 |
| 15 | 1.99 (d, 1.0) | 20.0 | 2.00 (d, 0.9) | 19.9 | 2.0 (br s) | 20.9 | 2.03 (br s) | 20.9 |
| 16 | 166.9 | 168.6 | 168.8 | 168.8 | ||||
| 17 | 5.49 (sept, 1.4) | 116.2 | 127.8 | 129.8 | 129.3 | |||
| 18 | 160.1 | 6.67 (qq, 7.0, 1.4) | 139.5 | 6.77 (qq, 7.1, 1.3) | 138.9 | 6.04 (qq, 7.2, 1.6) | 138.8 | |
| 19 | 1.88 (d, 1.4) | 27.6 | 1.77 (dq, 7.0, 1.4) | 14.5 | 1.77 (dq, 7.1, 1.1) | 14.5 | 1.88 (dq, 7.2, 1.6) | 16.1 |
| 20 | 2.12 (d, 1.4) | 20.5 | 1.74 (quint-like, 1.4) | 12.2 | 1.75 (dq, 1.3, 1.1) | 12.2 | 1.78 (dq, 1.6, 1.6) | 20.8 |
| Position | Crossoseamine C (5) | Crossoseamine D (6) | Crossoseamine E (7) | |||
|---|---|---|---|---|---|---|
| 1H | 13C | 1H | 13C | 1H | 13C | |
| 1 | 211.7 | 213.6 | 213.1 | |||
| 2 | 2.35–2.44 (m) α 2.67 (m) β | 34.9 | 2.27–2.35 (m) α 2.57–2.66 (m) β | 34.9 | 2.27–2.36 (m) α 2.57–2.62 (m) β | 34.6 |
| 3 | 2.35–2.44 (m) | 32.5 | 2.27–2.35 (m) α 2.57–2.66 (m) β | 30.0 | 2.27–2.36 (m) α 2.57–2.62 (m) β | 29.7 |
| 4 | 126.6 | 127.2 | 126.8 | |||
| 5 | 127.9 | 139.1 | 138.7 | |||
| 6 | 4.97 (dq, 11.2, 1.3) | 77.5 | 5.56 (d, 5.6) | 76.9 | 5.67 (d, 5.8) | 78.2 |
| 7 | 2.42 (t, 11.2) | 49.9 | 2.47 (dd-like, 8.8, 5.6) | 49.2 | 2.15 (dd, 8.9, 5.8) | 47.6 |
| 8 | 5.12 (td, 11.2, 4.4) | 70.4 | 1.35 (m) α 1.56 (m) β | 22.8 | 3.54 (ddd, 10.8, 8.9, 3.3) | 65.2 |
| 9 | 1.38 (dd, 12.9, 11.2) α 1.99 (dd, 12.9, 4.4) β | 40.0 | 1.47–1.55 (m) | 30.4 | 1.46 (dd, 13.2, 10.8) α 1.75 (m) β | 39.4 |
| 10 | 46.9 | 45.3 | 45.8 | |||
| 11 | 2.95 (ddd, 11.2, 5.9, 3.3) | 44.0 | 2.43 (br t, 6.6) | 38.5 | 2.79 (br t, 6.4) | 46.5 |
| 12 | 176.7 | 177.9 | 177.9 | |||
| 13 | 2.64 (m) 3.05 (m) | 53.8 | 2.98 (dd, 12.9, 6.6) 3.08 (dd, 12.9, 6.6) | 53.3 | 3.08 (dd, 12.7, 6.4) 3.02 (dd, 12.7, 6.4) | 53.3 |
| 14 | 1.31 (s) | 24.4 | 1.12 (s) | 23.9 | 1.12 (s) | 24.6 |
| 15 | 1.89 (s) | 19.2 | 1.87 (s) | 19.2 | 1.86 (s) | 19.4 |
| 16 | 169.9 | |||||
| 17 | 2.02 (s) | 20.9 | ||||
| 2′ | 3.38 (dd, 9.1, 3.8) | 66.9 | 3.30 (dd, 8.9, 4.5) | 66.1 | 3.35 (dd, 8.7, 4.4) | 66.1 |
| 3′ | 1.83 (m) 2.05 (m) | 29.2 | 1.82 (m) 2.06 (m) | 29.0 | 1.76 (m) 2.03 (m) | 28.6 |
| 4′ | 1.70 (m) 1.77 (m) | 23.7 | 1.74 (m) | 22.5 | 1.74 (m) | 23.4 |
| 5′ | 3.02 (m) | 51.7 | 2.57–2.66 (m) 3.13 (ddd, 11.0, 7.4, 3.4) | 53.7 | 2.57–2.62 (m) 3.06 (m) | 52.5 |
| 6′ | 174.1 | 174.3 | 174.3 | |||
| Compounds | L. major (μM) | A549 (μM) | Vero Cell (μM) | SI1 | SI2 |
|---|---|---|---|---|---|
| 1 | 0.81 ± 0.26 | 2.97 ± 0.58 | 5.99 ± 0.76 | 7.4 | 2.0 |
| 2 | 0.96 ± 0.17 | 2.30 ± 0.26 | 7.84 ± 0.61 | 8.2 | 3.4 |
| 3 | 3.75 ± 0.98 | 5.59 ± 0.72 | 6.20 ± 1.36 | 3.3 | 1.7 |
| 4 | 7.66 ± 2.26 | 17.1 ± 1.86 | 17.2 ± 1.36 | 1.7 | 1.1 |
| 5 | 4.72 ± 1.26 | 17.4 ± 1.86 | 11.4 ± 0.98 | 2.2 | 1.0 |
| 6 | 52.6 ± 11.6 | 83.9 ± 12.1 | 48.5 ± 4.07 | 2.4 | 0.7 |
| 7 | 30.8 ± 5.81 | 58.9 ± 13.6 | 23.8 ± 2.47 | 0.9 | 0.6 |
| P.C. | 18.1 ± 1.69 | 0.45 ± 0.13 | 3.82 ± 0.79 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Yamano, Y.; Kawakami, S.; Al-Hamoud, G.A.; Sugimoto, S.; Otsuka, H.; Matsunami, K. New ψ-Santonin Derivatives from Crossostephium chinense and Their Anti-Proliferative Activities against Leishmania major and Human Cancer Cells A549. Molecules 2023, 28, 8108. https://doi.org/10.3390/molecules28248108
Wang Z, Yamano Y, Kawakami S, Al-Hamoud GA, Sugimoto S, Otsuka H, Matsunami K. New ψ-Santonin Derivatives from Crossostephium chinense and Their Anti-Proliferative Activities against Leishmania major and Human Cancer Cells A549. Molecules. 2023; 28(24):8108. https://doi.org/10.3390/molecules28248108
Chicago/Turabian StyleWang, Zhichao, Yoshi Yamano, Susumu Kawakami, Gadah Abdulaziz Al-Hamoud, Sachiko Sugimoto, Hideaki Otsuka, and Katsuyoshi Matsunami. 2023. "New ψ-Santonin Derivatives from Crossostephium chinense and Their Anti-Proliferative Activities against Leishmania major and Human Cancer Cells A549" Molecules 28, no. 24: 8108. https://doi.org/10.3390/molecules28248108
APA StyleWang, Z., Yamano, Y., Kawakami, S., Al-Hamoud, G. A., Sugimoto, S., Otsuka, H., & Matsunami, K. (2023). New ψ-Santonin Derivatives from Crossostephium chinense and Their Anti-Proliferative Activities against Leishmania major and Human Cancer Cells A549. Molecules, 28(24), 8108. https://doi.org/10.3390/molecules28248108