

Generation of Aurachin Derivatives by Whole-Cell Biotransformation and Evaluation of Their Antiprotozoal Properties

 , , and
, , and
Abstract
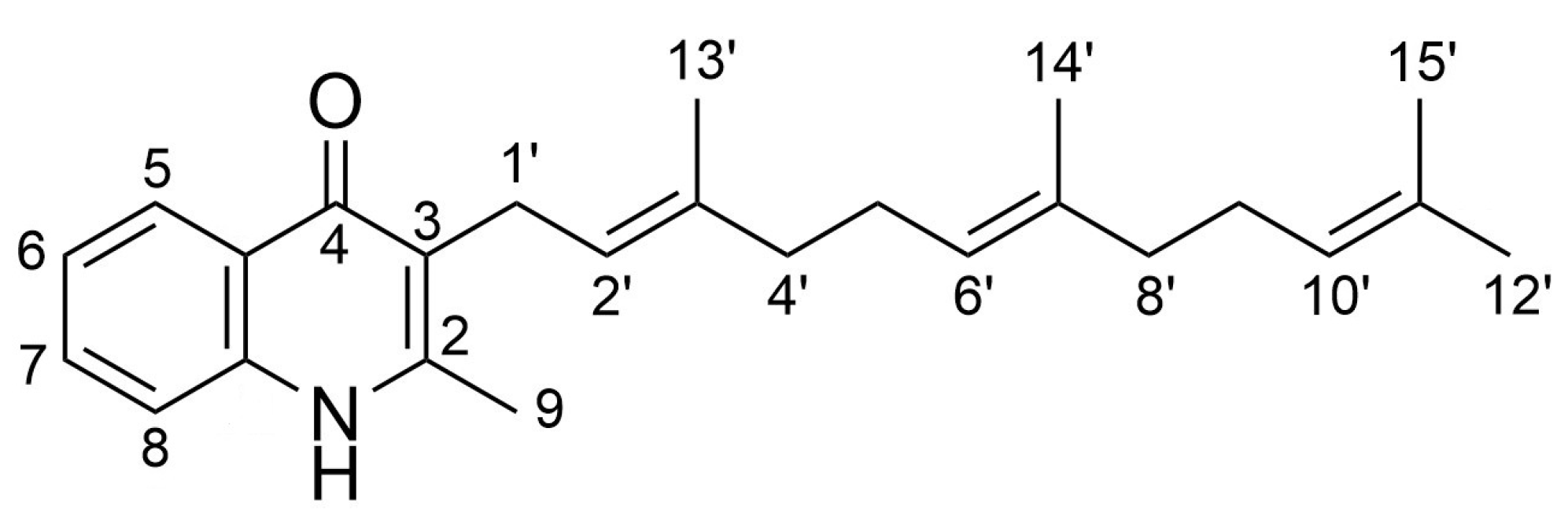
1. Introduction
2. Results
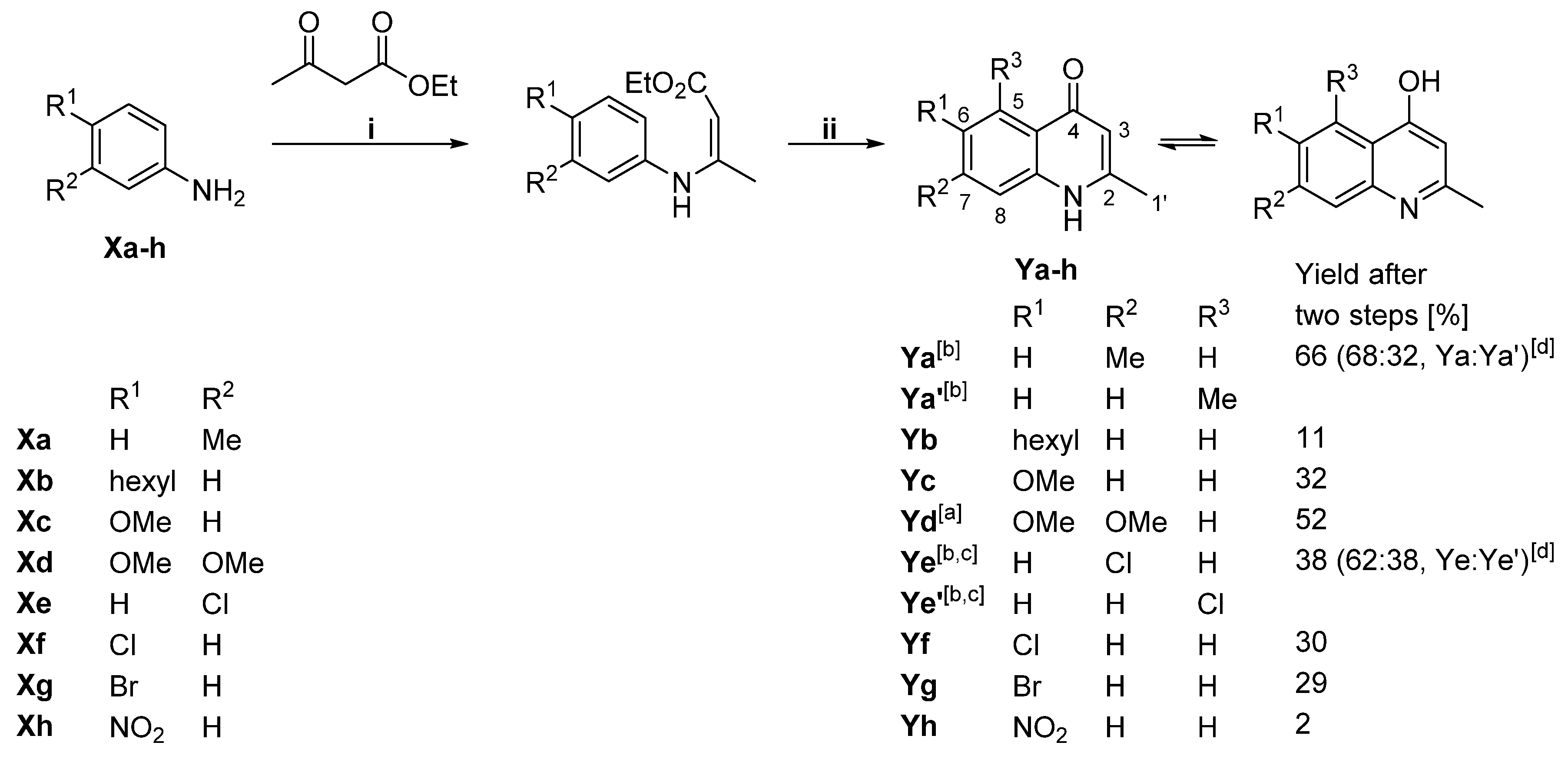
2.1. Generation of Aryl-Substituted Aurachin D Analogs by Whole-Cell Biotransformation
2.2. Spectroscopic Analysis of the Generated Aurachin D Analogs
2.3. Evaluation of Antiprotozoal Properties
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Synthesis of 1H-Quinolin-4-One Substrates
4.3. Analytical Data of 1H-Quinolin-4-One Substrates
4.4. Whole-Cell Biotransformation and Product Recovery
4.5. Spectroscopic Data of Aurachin Derivatives
4.6. Biological Tests
4.6.1. Antiplasmodial Assay
4.6.2. Antitrypanosomal Assays
4.6.3. Antileishmanial Assay
4.6.4. Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Talapko, J.; Škrlec, I.; Alebić, T.; Jukić, M.; Včev, A. Malaria: The past and the present. Microorganisms 2019, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, D.H.; Savioli, L.; Engels, D. Neglected tropical diseases: Progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.R.; Simarro, P.P.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clin. Epidemiol. 2014, 6, 257–275. [Google Scholar] [PubMed]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas disease: From discovery to a worldwide health problem. Front. Public Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Kumari, D.; Mahajan, S.; Kour, P.; Singh, K. Virulence factors of Leishmania parasite: Their paramount importance in unraveling novel vaccine candidates and therapeutic targets. Life Sci. 2022, 306, 120829. [Google Scholar] [CrossRef]
- Varikuti, S.; Jha, B.K.; Volpedo, G.; Ryan, N.M.; Halsey, G.; Hamza, O.M.; McGwire, B.S.; Satoskar, A.R. Host-directed drug therapies for neglected tropical diseases caused by protozoan parasites. Front. Microbiol. 2018, 9, 2655. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef]
- Dorababu, A. Quinoline: A promising scaffold in recent antiprotozoal drug discovery. ChemistrySelect 2021, 6, 2164–2177. [Google Scholar] [CrossRef]
- Höfle, G.; Böhlendorf, B.; Fecker, T.; Sasse, F.; Kunze, B. Semisynthesis and antiplasmodial activity of the quinoline alkaloid aurachin E. J. Nat. Prod. 2008, 71, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-W.; Herrmann, J.; Zang, Y.; Grellier, P.; Prado, S.; Müller, R.; Nay, B. Synthesis and biological activities of the respiratory chain inhibitor aurachin D and new ring versus chain analogues. Beilstein J. Org. Chem. 2013, 9, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Kruth, S.; Schibajew, L.; Nett, M. Biocatalytic production of the antibiotic aurachin D in Escherichia coli. AMB Express 2022, 12, 138. [Google Scholar] [CrossRef]
- Sester, A.; Stüer-Patowsky, K.; Hiller, W.; Kloss, F.; Lütz, S.; Nett, M. Biosynthetic plasticity enables production of fluorinated aurachins. ChemBioChem 2020, 21, 2268–2273. [Google Scholar] [CrossRef]
- Winand, L.; Sester, A.; Nett, M. Bioengineering of anti-inflammatory natural products. ChemMedChem 2021, 16, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Stec, E.; Pistorius, D.; Müller, R.; Li, S.-M. AuaA, a membrane-bound farnesyltransferase from Stigmatella aurantiaca catalyzes the prenylation of 2-methyl-4-hydroxyquinoline in the biosynthesis of aurachins. ChemBioChem 2011, 12, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Kunze, B.; Höfle, G.; Reichenbach, H. The aurachins, new quinoline antibiotics from myxobacteria: Production, physico-chemical and biological properties. J. Antibiot. 1987, 40, 258–265. [Google Scholar] [CrossRef]
- Meunier, B.; Madgwick, S.A.; Reil, E.; Oettmeier, W.; Rich, P.R. New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry 1995, 34, 1076–1083. [Google Scholar] [CrossRef]
- Radloff, M.; Elamri, I.; Grund, T.N.; Witte, L.F.; Hohmann, K.F.; Nakagaki, S.; Goojani, H.G.; Nasiri, H.; Miyoshi, H.; Bald, D.; et al. Short-chain aurachin D derivatives are selective inhibitors of E. coli cytochrome bd-I and bd-II oxidases. Sci. Rep. 2021, 11, 23852. [Google Scholar] [CrossRef]
- Lawer, A.; Tyler, C.; Hards, K.; Keighley, L.M.; Cheung, C.-Y.; Kierek, F.; Su, S.; Matikonda, S.S.; McInnes, T.; Tyndall, J.D.A.; et al. Synthesis and biological evaluation of aurachin D analogues as inhibitors of Mycobacterium tuberculosis cytochrome bd oxidase. ACS Med. Chem. Lett. 2022, 13, 1663–1669. [Google Scholar] [CrossRef]
- Vollmann, D.J.; Winand, L.; Nett, M. Emerging concepts in the semisynthetic and mutasynthetic production of natural products. Curr. Opin. Biotechnol. 2022, 77, 102761. [Google Scholar] [CrossRef] [PubMed]
- Dias-Lopes, G.; Zabala-Peñafiel, A.; de Albuquerque-Melo, B.C.; Souza-Silva, F.; do Canto, L.M.; Cysne-Finkelstein, L.; Alves, C.R. Axenic amastigotes of Leishmania species as a suitable model for in vitro studies. Acta Trop. 2021, 220, 105956. [Google Scholar] [CrossRef] [PubMed]
- Tidman, R.; Abela-Ridder, B.; De Castañeda, R.R. The impact of climate change on neglected tropical diseases: A systematic review. Trans. R. Soc. Trop. Med. Hyg. 2021, 115, 147–168. [Google Scholar] [CrossRef] [PubMed]
- Khoshbakht, M.; Srey, J.; Adpressa, D.A.; Jagels, A.; Loesgen, S. Precursor-directed biosynthesis of aminofulvenes: New chalanilines from endophytic fungus Chalara sp. Molecules 2021, 26, 4418. [Google Scholar] [CrossRef]
- Winand, L.; Schneider, P.; Kruth, S.; Greven, N.-J.; Hiller, W.; Kaiser, M.; Pietruszka, J.; Nett, M. Mutasynthesis of physostigmines in Myxococcus xanthus. Org. Lett. 2021, 23, 6563–6567. [Google Scholar] [CrossRef]
- Aryal, N.; Chen, J.; Bhattarai, K.; Hennrich, O.; Handayani, I.; Kramer, M.; Straetener, J.; Wommer, T.; Berschied, A.; Peter, S.; et al. High plasticity of the amicetin biosynthetic pathway in Streptomyces sp. SHP 22-7 led to the discovery of streptcytosine P and cytosaminomycins F and G and facilitated the production of 12F-plicacetin. J. Nat. Prod. 2022, 85, 530–539. [Google Scholar] [CrossRef]
- Pistorius, D.; Li, Y.; Sandmann, A.; Müller, R. Completing the puzzle of aurachin biosynthesis in Stigmatella aurantiaca Sg a15. Mol. Biosyst. 2011, 7, 3308–3315. [Google Scholar] [CrossRef]
- Walia, M.; Teijaro, C.N.; Gardner, A.; Tran, T.; Kang, J.; Zhao, S.; O’Connor, S.E.; Courdavault, V.; Andrade, R.B. Synthesis of (-)-melodinine K: A case study of efficiency in natural product synthesis. J. Nat. Prod. 2020, 83, 2425–2433. [Google Scholar] [CrossRef]
- Dejon, L.; Speicher, A. Synthesis of aurachin D and isoprenoid analogues from the myxobacterium Stigmatella aurantiaca. Tetrahedron Lett. 2013, 54, 6700–6702. [Google Scholar] [CrossRef]
- Cao, X.; You, Q.-D.; Li, Z.-Y.; Yang, Y.; Wang, X.-J. Microwave-assisted simple synthesis of substituted 4-quinolone derivatives. Synth. Commun. 2009, 39, 4375–4383. [Google Scholar] [CrossRef]
- Cross, R.M.; Manetsch, R. Divergent route to access structurally diverse 4-quinolones via mono or sequential cross-couplings. J. Org. Chem. 2010, 75, 8654–8657. [Google Scholar] [CrossRef] [PubMed]
- Pitta, E.; Rogacki, M.K.; Balabon, O.; Huss, S.; Cunningham, F.; Lopez-Roman, E.M.; Joossens, J.; Augustyns, K.; Ballell, L.; Bates, R.H.; et al. Searching for new leads for tuberculosis: Design, synthesis and biological evaluation of novel 2-quinolin-4-yloxyacetamides. J. Med. Chem. 2016, 59, 6709–6728. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Roy, K.K.; Khan, S.R.; Kashyap, V.K.; Sharma, A.; Jaiswal, S.; Sharma, S.K.; Krishnan, M.Y.; Chaturvedi, V.; Lal, J.; et al. Novel, potent, orally bioavailable and selective mycobacterial ATP synthase inhibitors that demonstrated activity against both replicating and non-replicating M. tuberculosis. Bioorg. Med. Chem. 2015, 23, 742–752. [Google Scholar] [CrossRef]
- Upadhayaya, R.S.; Vandavasi, J.K.; Kardile, R.A.; Lahore, S.V.; Dixit, S.S.; Deokar, H.D.; Shinde, P.D.; Sarmah, M.P.; Chattopadhyaya, J. Novel quinoline and naphthalene derivatives as potent antimycobacterial agents. Eur. J. Med. Chem. 2010, 45, 1854–1867. [Google Scholar] [CrossRef]
- Bender, S.L.; Bhumralkar, D.; Collins, M.R.; Cripps, S.J.; Deal, J.G.; Jia, L.; Nambu, M.D.; Palmer, C.L.; Peng, Z.; Varney, M.D. Amide Compounds and Pharmaceutical Compositions for Inhibiting Protein Kinases, and Methods for Their Use. U.S. Patent 6635641B2, 21 October 2003. [Google Scholar]
- Peralta-Yahya, P.P.; Ouellet, M.; Chan, R.; Mukhopadhyay, A.; Keasling, J.D.; Lee, T.S. Identification and microbial production of a terpene-based advanced biofuel. Nat. Commun. 2011, 2, 483. [Google Scholar] [CrossRef] [PubMed]
- Tartoff, K.D.; Hobbs, C.A. Improved media for growing plasmids and cosmid clones. Bethesda Res. Lab. Focus 1987, 9, 12. [Google Scholar]
- Dolensky, J.; Hinteregger, C.; Leitner, A.; Seebacher, W.; Saf, R.; Belai, F.; Mäser, P.; Kaiser, M.; Weis, R. Antiprotozoal activity of azabicyclo-nonanes linked to tetazole or sulfonamide cores. Molecules 2022, 27, 6217. [Google Scholar] [CrossRef]
- Mahmoud, A.B.; Danton, O.; Kaiser, M.; Khalid, S.; Hamburger, M.; Mäser, P. HPLC-based activity profiling for antiprotozoal compounds in Croton gratissimus and Cuscuta hyaline. Front. Pharmacol. 2020, 11, 1246. [Google Scholar] [CrossRef]
- Räz, B.; Iten, M.; Grether-Bühler, Y.; Kaminsky, R.; Brun, R. The Alamar Blue® assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997, 68, 139–147. [Google Scholar] [CrossRef]
- Buckner, F.S.; Verlinde, C.L.M.J.; La Flamme, A.C.; Van Voorhis, W.C. Efficient techniques for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef]
- Cunningham, I. New culture medium for maintenance of tsetse tissues and growth of trypanosomatids. J. Protozool. 1977, 24, 325–329. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Functional Groups on 1H-Quinolin-4-One Substrate | Product | Molar Yield Coefficient YP/S [%] | Relative Activity [%] |
|---|---|---|---|
| 2-methyl | aurachin D (1) | 11 | 100.0 |
| - | 2-desmethyl aurachin D (2) | 27 | 245.5 |
| 2,6-dimethyl | 6-methyl aurachin D (3) | 5 | 45.5 |
| 2,7-dimethyl (Ya) | 7-methyl aurachin D (4) | 14 | 127.3 |
| 2-methyl, 6-hexyl (Yb) | - | 0 | 0 |
| 2-methyl, 6-methoxy (Yc) | 6-methoxy aurachin D (5) | 2 | 18.2 |
| 2-methyl, 7-methoxy | 7-methoxy aurachin D (6) | 1 | 9.1 |
| 2-methyl, 6,7-dimethoxy (Yd) | - | 0 | 0 |
| 2-methyl, 6-fluoro | 6-fluoro aurachin D (7) | 13 | 118.2 |
| 2-methyl, 6-chloro (Yf) | 6-chloro aurachin D (8) | 8 | 72.7 |
| 2-methyl, 7-chloro (Ye) | 7-chloro aurachin D (9) | 19 | 172.7 |
| 2-methyl, 6-bromo (Yg) | 6-bromo aurachin D (10) | 9 | 81.8 |
| 2-methyl, 6-nitro (Yh) | - | 0 | 0 |
| Test or Reference Compound | IC50 P. falciparum NF54 | IC50 T. brucei rhodesiense STIB 900 | IC50 T. cruzi Tulahuen C4 | IC50 L. donovani MHOM-ET-67/L82 | IC50 Rat Myoblast L6 Cells | Antileishmanial S.I. |
|---|---|---|---|---|---|---|
| 1 | 0.012 | 4.5 | 1.3 | 0.044 | 130.7 | 2969.5 |
| 2 | 0.006 | 9.5 | 2.4 | 1.5 | 20.4 | 13.2 |
| 3 | 0.514 | 43.2 | 7.6 | 4.3 | 18.6 | 4.3 |
| 4 | 0.007 | 163.8 | 45.7 | 9.1 | 65.3 | 7.2 |
| 5 | 0.866 | 40.3 | 19.3 | 7.3 | 118.3 | 16.2 |
| 6 | 0.089 | 44.8 | 21.9 | 17.1 | 80.8 | 4.7 |
| 7 | 0.061 | 37.2 | 1.8 | 0.617 | 125.8 | 203.9 |
| 8 | 0.021 | 37.4 | 11.2 | 6.2 | 117.5 | 18.9 |
| 9 | 0.070 | 104.9 | 41.8 | 9.2 | 54.7 | 5.9 |
| 10 | 0.119 | 40.2 | 22.6 | 13.9 | 107.7 | 7.7 |
| Chloroquine | 0.008 | |||||
| Melarsoprol | 0.013 | |||||
| Benznidazole | 2.5 | |||||
| Miltefosine | 0.732 | |||||
| Podophyllotoxin | 0.022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruth, S.; Zimmermann, C.J.-M.; Kuhr, K.; Hiller, W.; Lütz, S.; Pietruszka, J.; Kaiser, M.; Nett, M. Generation of Aurachin Derivatives by Whole-Cell Biotransformation and Evaluation of Their Antiprotozoal Properties. Molecules 2023, 28, 1066. https://doi.org/10.3390/molecules28031066
Kruth S, Zimmermann CJ-M, Kuhr K, Hiller W, Lütz S, Pietruszka J, Kaiser M, Nett M. Generation of Aurachin Derivatives by Whole-Cell Biotransformation and Evaluation of Their Antiprotozoal Properties. Molecules. 2023; 28(3):1066. https://doi.org/10.3390/molecules28031066
Chicago/Turabian StyleKruth, Sebastian, Cindy J.-M. Zimmermann, Katharina Kuhr, Wolf Hiller, Stephan Lütz, Jörg Pietruszka, Marcel Kaiser, and Markus Nett. 2023. "Generation of Aurachin Derivatives by Whole-Cell Biotransformation and Evaluation of Their Antiprotozoal Properties" Molecules 28, no. 3: 1066. https://doi.org/10.3390/molecules28031066
APA StyleKruth, S., Zimmermann, C. J.-M., Kuhr, K., Hiller, W., Lütz, S., Pietruszka, J., Kaiser, M., & Nett, M. (2023). Generation of Aurachin Derivatives by Whole-Cell Biotransformation and Evaluation of Their Antiprotozoal Properties. Molecules, 28(3), 1066. https://doi.org/10.3390/molecules28031066