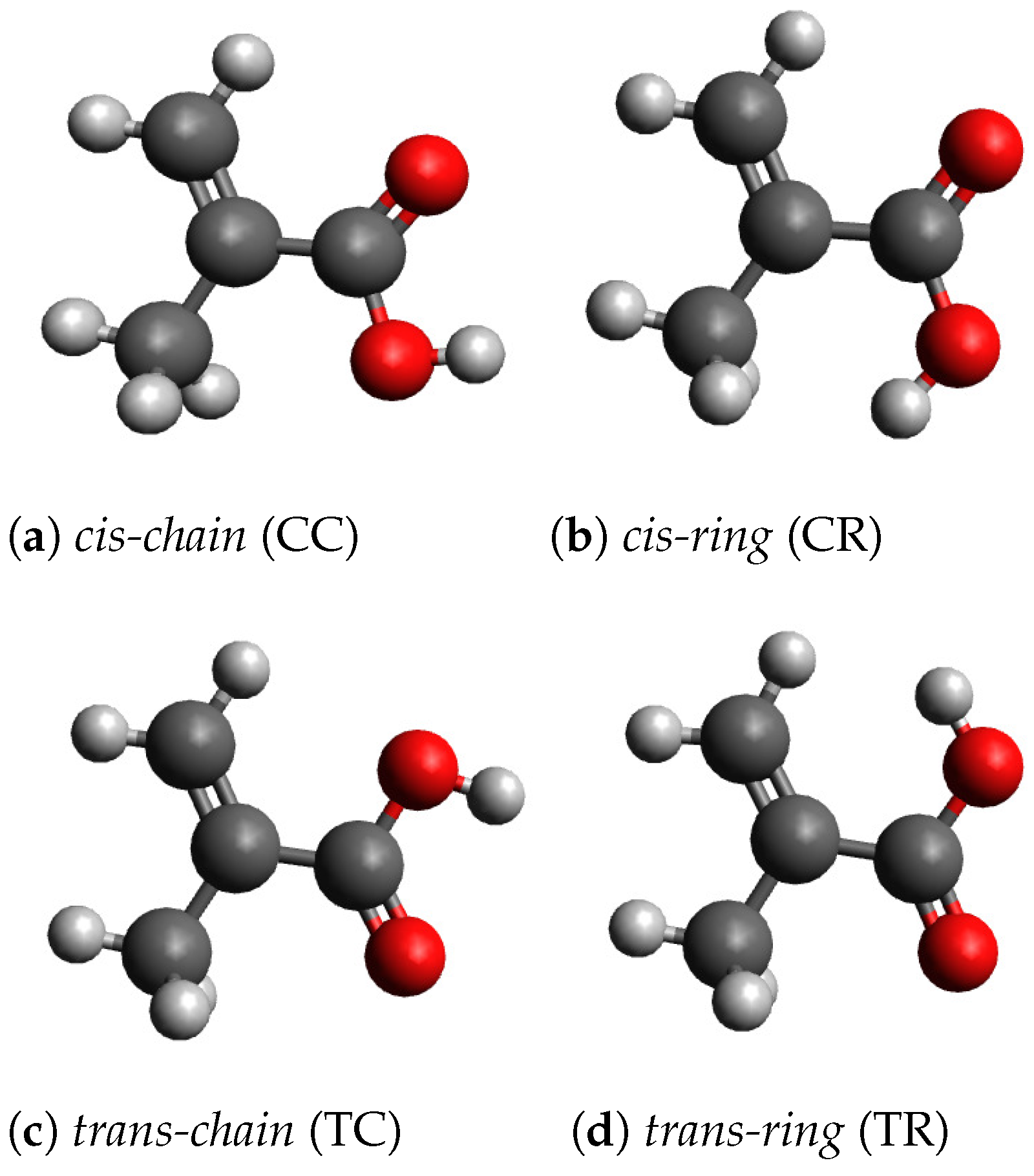
With the geometry optimization, we found four stable conformers, which are shown in
Figure 1. For the geometry optimizations, the Berny algorithm [
29] is used. For each conformer the vibrational frequencies have been calculated and no imaginary frequencies have been found. In order to distinguish the conformers, we call them
cis, if the OH-group and the CH
-group are on the same side of two carbons atoms, which connect the two groups. Otherwise the conformers are called
trans. Furthermore, we distinguish the conformers by the relative orientation of the OH-group. If the Hydrogen atom of the OH-group is rotated to the outside of the molecule, we call the conformer
chain-like, otherwise
ring-like. With this naming convention, we can derive four names:
cis-chain (CC),
cis-ring (CR),
trans-chain (TC) and
trans-ring (TR). The cartesian coordinates of the four conformers are given in
Appendix A. The two chain-like conformers CC and TC have been also found by Badawi et al. [
9] and by Frighetto et al. [
10]. The two ring-like conformers have not been discussed previously. The conformers CC, CR and TC belong to the symmetry point group
. These conformers have one mirror plane, which contains all carbon and oxygen atoms. The geometry of the conformer TR has no mirror plane and the molecule transforms as the point group
. In this case, the COOH-group is rotated out of the plane, which reduces the static interaction between the hydrogen atoms, which carry a small positive charge, in the OH-group and the CH
-group. In the conformer CR, the static interaction between the OH-group and the CH
-group is smaller, because the hydrogen atoms of the CH
-group have larger distances from the hydrogen atom of the OH-group.
Table 1 shows the symmetry groups, the total electronic energies
, the enthalpies
and the Gibbs free enthalpies
of the four conformers. TC is the most stable conformer. The conformer CC is about 2.5 kJ/mol less stable. The two
ring-like conformers, CR and TR, are more than 20 kJ/mol higher in energy. The large energy difference between chain-like and ring-like conformers imply that under equilibrium conditions (at room temperature) less than 1 percent of the molecules will be found in the ring-like conformation. In the condensed phase, the energy difference could be smaller, because the larger dipole moment of the ring-like conformers causes a larger solvation energy. Furthermore, the molecules can be frozen in their conformation.
The rotational constants for the conformer TC are 5.40, 3.49 and 2.15 GHz, which is in good agreement with the results of the calculations, which have been performed by Badawi et al. [
9] (5.374, 3.484 and 2.141 GHz with B3LYP/6-311+G**). For the conformer CC, the rotational constants are 5.33, 3.52 and 2.15 GHz. This is again in good agreement with calculations by Badawi et al. [
9] (5.308, 3.517 and 2.143 GHz with B3LYP/6-311+G**).
4.1. Cross Sections
In
Figure 2, we show the elastic cross sections for the four conformers of MAA. For energies above a few eV, the elastic cross sections are proportional to the inverse of the collision energy and show as straight lines in the double-logarithmic plot in
Figure 2. The elastic cross sections for the conformer CR is slightly larger than that for the conformer TR. The elastic cross sections for the two ring-like conformers CR and TR are roughly five times larger than those of the chain-like conformers CC and TC. The relative size of the elastic cross sections is due to the square of the absolute values of the molecular dipole moments in Equation (
11). The absolute values of the dipole moments of the two ring-like conformers CR and TR are 4.71 D and 4.27 D, respectively. These values are about 2.5 times larger than those of the two chain-like conformers CC and TC (1.91 D and 1.79 D, respectively).
In
Figure 3, we show the electronically inelastic cross sections
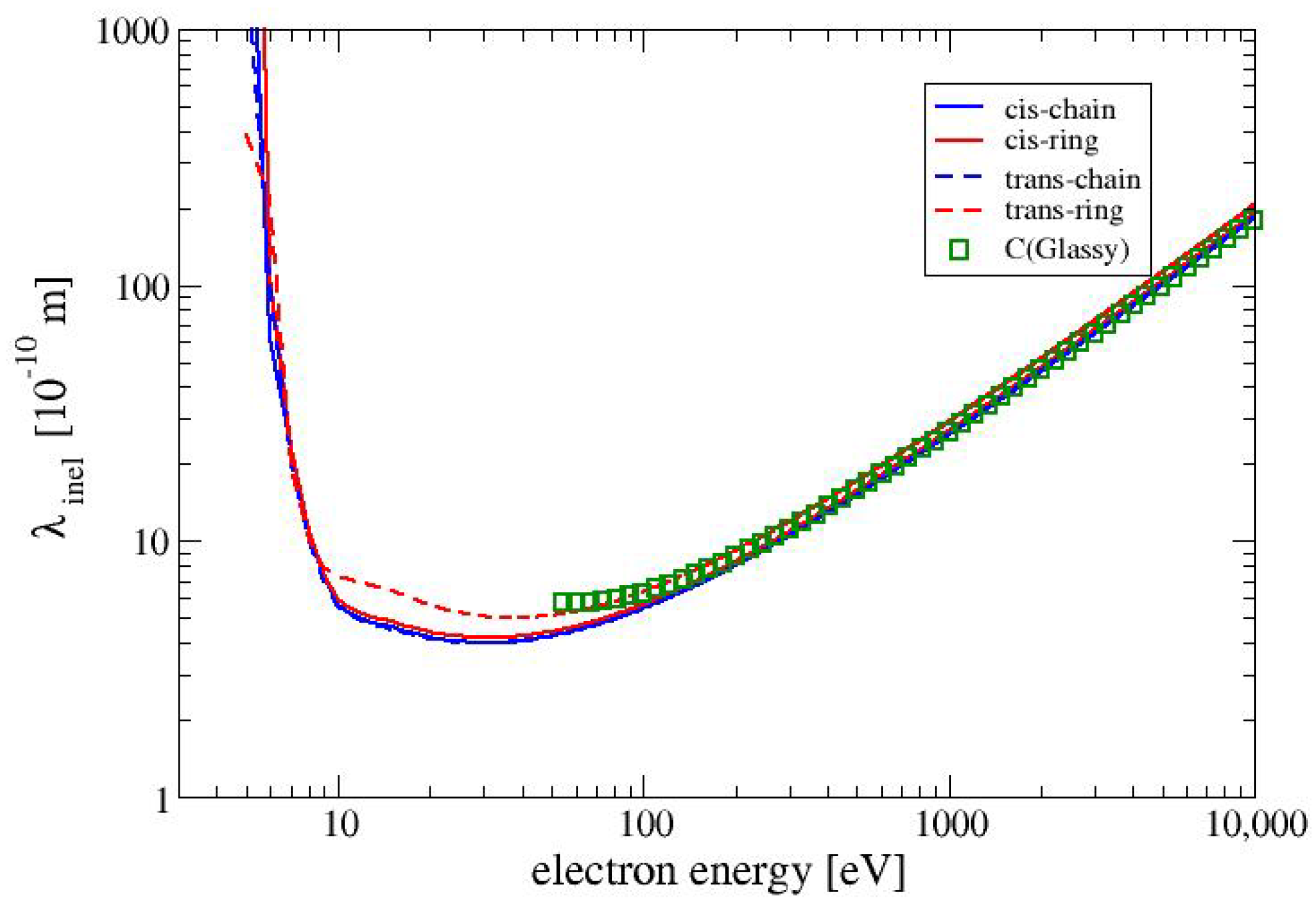
for the four conformers. The cross sections for the three conformers CC, CR and TC are nearly identical to each other, whereas the cross section for the conformer TR is slightly lower in the region between 10 and 100 eV. The values of the cross sections for electron–phonon interactions for all MAA conformers are the same and for the examined range of electron energies (1 to 10,000 eV) are in the range between
and
. When the electron energy increases the cross section for electron-phonon interaction decreases as
. For all conformers of MAA, the values of the cross sections for polaronic effect are the same. and they exhibit the smallest values of among all cross sections which were taken into account in this study. It is worth noticing that the values of the cross section obtained for electron–phonon and electron–polaron interactions are much more smaller than those received for the electronically inelastic cross sections. Therefore,
Figure 2 and
Figure 3 present only elastic and electronically inelastic cross sections.
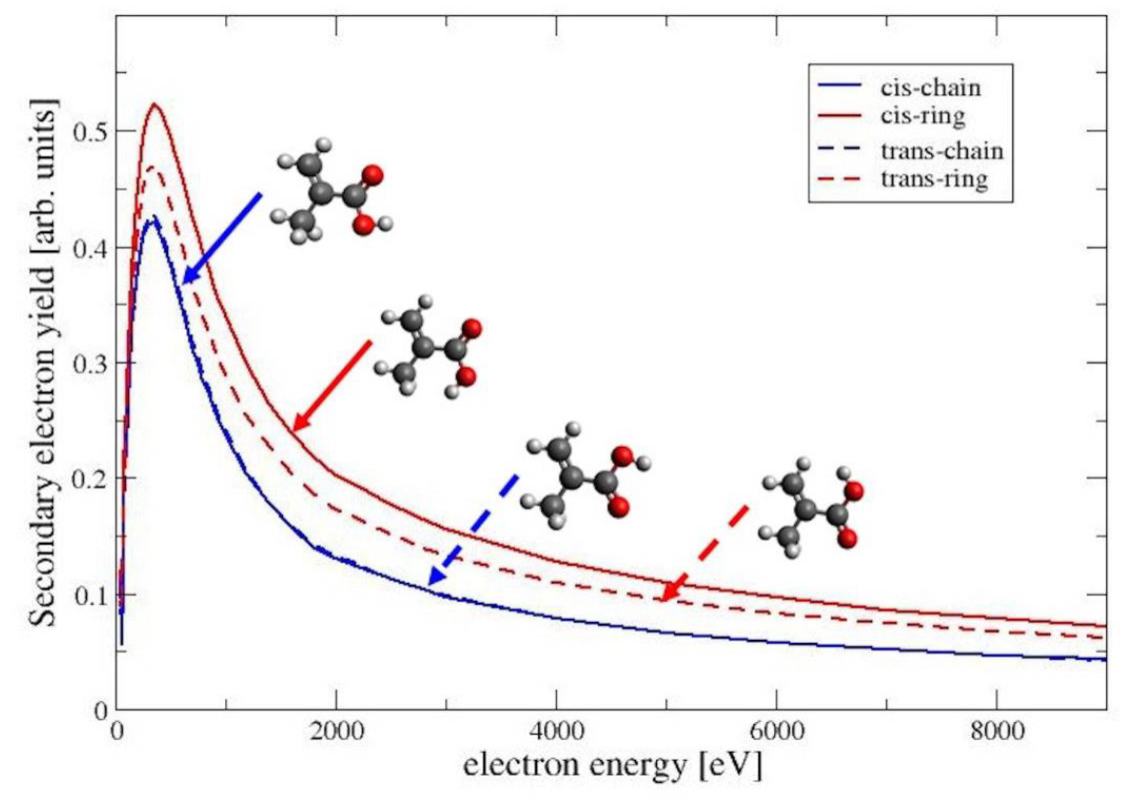
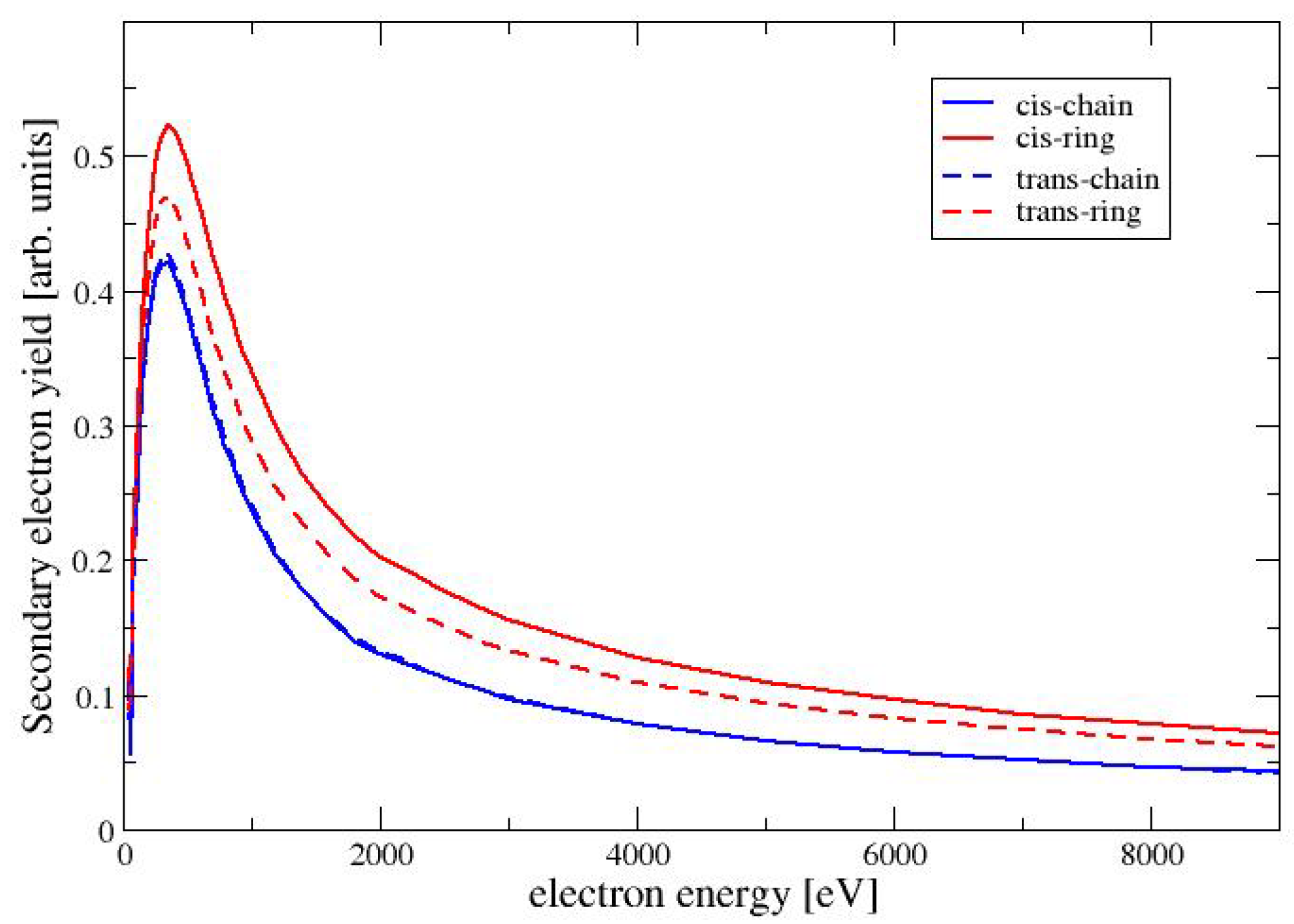
4.4. Secondary Electron Yield
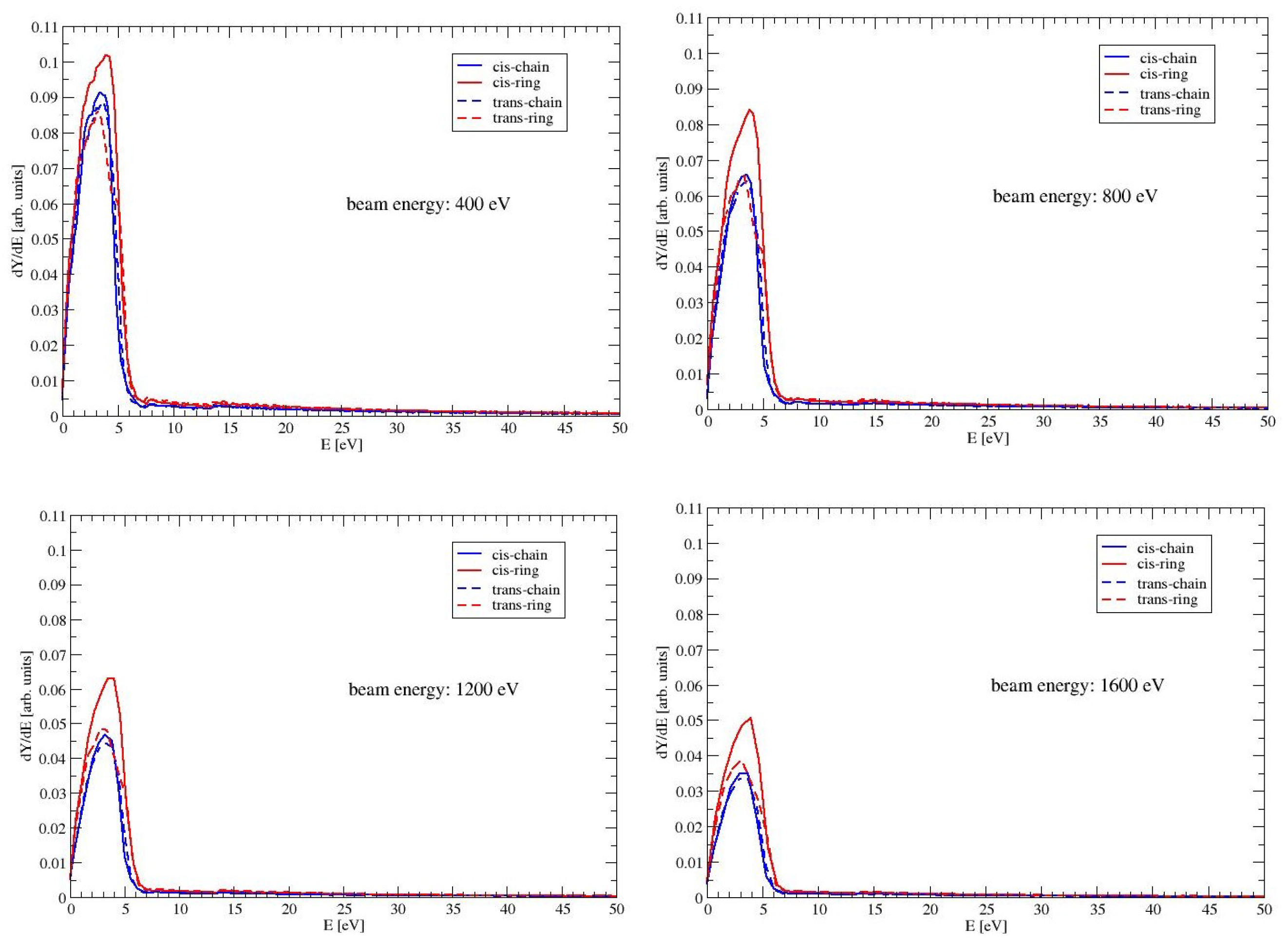
The secondary electron yield (SEY) for a given beam energy
is defined as integral of the distribution of emitted electrons with energies below 50 eV (see, e.g., Chapter 9 in Dapor [
12]). The SEY can be expressed as follows
where we have used
as the number of electrons, which are ejected with an energy between
E and
. In the Monte Carlo code, a discrete value of
is chosen and the number of ejected electrons in each interval is recorded in a histogram. Routines from the GNU Scientific Library [
32] are used for this purpose. It should be noted that we use the expression for the secondary electron yield as a measure for the low-energy electrons, that are emitted from the surface. This should not be confused with the total number of secondary electrons, which are generated by ionization events inside the material and cannot reach the surface. In
Figure 6, we show the SEY as a function of the beam energy for the four conformers of MAA. For all conformers, the SEY has its maximum at a beam energy of 350 eV. For higher energies the SEY decreases, because the secondary electrons are generated at a larger depth inside of the material and are trapped within the target. Among the four conformers has the largest SEY, followed by TR and the two chain-like conformers CC and TC. This is the same trend as seen for the elastic cross sections in
Figure 2. A larger elastic cross section increases the probability for the deflection of the primary electron beam, also increasing the backscattering of the electrons. The backscattered primary electrons generate secondary electrons before leaving the material. These secondary electons are generated in a layer close enough to the surface to be able to escape. This situation is discussed in more detail in Chapter 3 of the book by Goldstein et al. [
11].

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






