

Transporter-Mediated Drug Delivery


Abstract
:1. Introduction
2. Strategies for Utilizing Transporters for Drug Delivery
3. Targeting Transporters
3.1. Facilitative Glucose Transporters (GLUTs)
3.2. Amino Acid Transporters
3.3. Bile Acid Transporters
3.4. Choline Transporters
3.5. Vitamin Transporters
3.6. Oligopeptide Transporters (PEPT1/PEPT2)
3.7. Organic Cation Transporters (OCTNs)
3.8. Organic Anion Transporters (OATPs)
3.9. Monocarboxylate Transporters (MCTs)
4. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gyimesi, G.; Pujol-Giménez, J.; Kanai, Y.; Hediger, M.A. Sodium-Coupled Glucose Transport, the SLC5 Family, and Therapeutically Relevant Inhibitors: From Molecular Discovery to Clinical Application. Pflugers Arch. 2020, 472, 1177–1206. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Moitra, K.; Allikmets, R. The Human ATP-Binding Cassette (ABC) Transporter Superfamily. Hum. Mutat. 2022, 43, 1162–1182. [Google Scholar] [CrossRef] [PubMed]
- Nwabufo, C.K. Relevance of ABC Transporters in Drug Development. Curr. Drug Metab. 2022, 23, 434–446. [Google Scholar] [CrossRef]
- Hediger, M.A.; Clémençon, B.; Burrier, R.E.; Bruford, E.A. The ABCs of Membrane Transporters in Health and Disease (SLC Series): Introduction. Mol. Asp. Med. 2013, 34, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Gyimesi, G.; Hediger, M.A. Systematic in Silico Discovery of Novel Solute Carrier-like Proteins from Proteomes. PLoS One 2022, 17, e0271062. [Google Scholar] [CrossRef]
- Lee, S.-C.; Arya, V.; Yang, X.; Volpe, D.A.; Zhang, L. Evaluation of Transporters in Drug Development: Current Status and Contemporary Issues. Adv. Drug Deliv. Rev. 2017, 116, 100–118. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Yee, S.W.; Koleske, M.L.; Zou, L.; Matsson, P.; Chen, E.; Kroetz, D.L.; Miller, M.A.; Gozalpour, E.; Chu, X. New and Emerging Research on Solute Carrier (SLC) and ABC Transporters in Drug Discovery and Development: Outlook from the International Transporter Consortium. Clin. Pharmacol. Ther. 2022, 112, 540–561. [Google Scholar] [CrossRef]
- Kou, L.; Bhutia, Y.D.; Yao, Q.; He, Z.; Sun, J.; Ganapathy, V. Transporter-Guided Delivery of Nanoparticles to Improve Drug Permeation across Cellular Barriers and Drug Exposure to Selective Cell Types. Front. Pharmacol. 2018, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.W.; Gallo, L.; Jadhav, A.; Hawkins, R.; Parker, C.G. The Druggability of Solute Carriers. J. Med. Chem. 2020, 63, 3834–3867. [Google Scholar] [CrossRef]
- Deng, F.; Bae, Y.H. Bile Acid Transporter-Mediated Oral Drug Delivery. J. Control. Release 2020, 327, 100–116. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, J.; Huang, Z.; Wang, M. Transporter-Mediated Tissue Targeting of Therapeutic Molecules in Drug Discovery. BioOrg. Med. Chem. Lett. 2015, 25, 993–997. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T. A Minireview: Usefulness of Transporter-Targeted Prodrugs in Enhancing Membrane Permeability. J. Pharm. Sci. 2016, 105, 2515–2526. [Google Scholar] [CrossRef] [Green Version]
- Szakács, G.; Váradi, A.; Ozvegy-Laczka, C.; Sarkadi, B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef]
- Hillgren, K.M.; Keppler, D.; Zur, A.A.; Giacomini, K.M.; Stieger, B.; Cass, C.E.; Zhang, L. International Transporter Consortium Emerging Transporters of Clinical Importance: An Update from the International Transporter Consortium. Clin. Pharmacol. Ther. 2013, 94, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Shitara, Y.; Horie, T.; Sugiyama, Y. Transporters as a Determinant of Drug Clearance and Tissue Distribution. Eur. J. Pharm. Sci. 2006, 27, 425–446. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-Brain Barrier Delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug Transport across the Blood-Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Enerson, B.E.; Drewes, L.R. The Rat Blood-Brain Barrier Transcriptome. J. Cereb. Blood Flow Metab. 2006, 26, 959–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, V.J. Prodrugs: Some Thoughts and Current Issues. J. Pharm. Sci. 2010, 99, 4755–4765. [Google Scholar] [CrossRef]
- Hu, M.; Subramanian, P.; Mosberg, H.I.; Amidon, G.L. Use of the Peptide Carrier System to Improve the Intestinal Absorption of L-Alpha-Methyldopa: Carrier Kinetics, Intestinal Permeabilities, and in Vitro Hydrolysis of Dipeptidyl Derivatives of L-Alpha-Methyldopa. Pharm. Res. 1989, 6, 66–70. [Google Scholar] [CrossRef]
- Sai, Y.; Tsuji, A. Transporter-Mediated Drug Delivery: Recent Progress and Experimental Approaches. Drug Discov. Today 2004, 9, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, M.; Leake, B.; Fromm, M.F.; Wilkinson, G.R.; Kim, R.B. OATP and P-Glycoprotein Transporters Mediate the Cellular Uptake and Excretion of Fexofenadine. Drug Metab. Dispos. 1999, 27, 866–871. [Google Scholar]
- Chen, C.; Hanson, E.; Watson, J.W.; Lee, J.S. P-Glycoprotein Limits the Brain Penetration of Nonsedating but Not Sedating H1-Antagonists. Drug Metab. Dispos. 2003, 31, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conen, S.; Theunissen, E.L.; Vermeeren, A.; van Ruitenbeek, P.; Stiers, P.; Mehta, M.A.; Toennes, S.W.; Ramaekers, J.G. The Role of P-Glycoprotein in CNS Antihistamine Effects. Psychopharmacology 2013, 229, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Kido, Y.; Yamashita, J.; Sai, Y.; Tsuji, A. Blood-Brain Barrier Transport of H1-Antagonist Ebastine and Its Metabolite Carebastine. J. Drug Target. 2000, 8, 383–393. [Google Scholar] [CrossRef]
- Zhang, L.; Sui, C.; Yang, W.; Luo, Q. Amino Acid Transporters: Emerging Roles in Drug Delivery for Tumor-Targeting Therapy. Asian J. Pharm. Sci. 2020, 15, 192–206. [Google Scholar] [CrossRef]
- Yuan, H.; Li, J.; Bao, G.; Zhang, S. Variable Nanoparticle-Cell Adhesion Strength Regulates Cellular Uptake. Phys. Rev. Lett. 2010, 105, 138101. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.A.; Feng, S.-S. Effects of Particle Size and Surface Modification on Cellular Uptake and Biodistribution of Polymeric Nanoparticles for Drug Delivery. Pharm. Res. 2013, 30, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Wang, Y.; Liu, S.; Wang, Y.; Liu, Q.; Liu, G.; Chen, Q. Emerging Transporter-Targeted Nanoparticulate Drug Delivery Systems. Acta Pharm. Sin. B 2019, 9, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Gong, P.; Sun, M.; Kou, L.; Ganapathy, V.; Jing, Y.; He, Z.; Sun, J. Transporter Occluded-State Conformation-Induced Endocytosis: Amino Acid Transporter ATB0,+-Mediated Tumor Targeting of Liposomes for Docetaxel Delivery for Hepatocarcinoma Therapy. J. Control. Release 2016, 243, 370–380. [Google Scholar] [CrossRef]
- Ouyang, Q.; Meng, Y.; Zhou, W.; Tong, J.; Cheng, Z.; Zhu, Q. New Advances in Brain-Targeting Nano-Drug Delivery Systems for Alzheimer’s Disease. J. Drug Target. 2021, 30, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Asghar, S.; Wu, Y.; Chen, Z.; Jin, X.; Yin, L.; Huang, L.; Ping, Q.; Xiao, Y. Improving Intestinal Absorption and Oral Bioavailability of Curcumin via Taurocholic Acid-Modified Nanostructured Lipid Carriers. Int. J. Nanomed. 2017, 12, 7897–7911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul Razzak, R.; Florence, G.J.; Gunn-Moore, F.J. Approaches to CNS Drug Delivery with a Focus on Transporter-Mediated Transcytosis. Int. J. Mol. Sci. 2019, 20, E3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.R.; Yang, X.; Fu, M.; Zhai, G. Recent Progress of Drug Nanoformulations Targeting to Brain. J. Control. Release 2018, 291, 37–64. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Gonzalez-Carter, D.; Tockary, T.A.; Nakamura, N.; Xue, Y.; Nakakido, M.; Akiba, H.; Dirisala, A.; Liu, X.; Toh, K.; et al. Dual-Sensitive Nanomicelles Enhancing Systemic Delivery of Therapeutically Active Antibodies Specifically into the Brain. ACS Nano 2020, 14, 6729–6742. [Google Scholar] [CrossRef]
- Jiang, X.; Xin, H.; Ren, Q.; Gu, J.; Zhu, L.; Du, F.; Feng, C.; Xie, Y.; Sha, X.; Fang, X. Nanoparticles of 2-Deoxy-D-Glucose Functionalized Poly(Ethylene Glycol)-Co-Poly(Trimethylene Carbonate) for Dual-Targeted Drug Delivery in Glioma Treatment. Biomaterials 2014, 35, 518–529. [Google Scholar] [CrossRef]
- Park, J.-H.; Cho, H.-J.; Kim, D.-D. Poly((D,L)Lactic-Glycolic)Acid-Star Glucose Nanoparticles for Glucose Transporter and Hypoglycemia-Mediated Tumor Targeting. Int. J. Nanomed. 2017, 12, 7453–7467. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, Y.; Li, J.; Zhang, Y.; Lu, Y.; Jiang, X.; He, X.; Ma, H.; An, S.; Jiang, C. Cell Microenvironment-Controlled Antitumor Drug Releasing-Nanomicelles for GLUT1-Targeting Hepatocellular Carcinoma Therapy. ACS Appl. Mater. Interfaces 2015, 7, 5444–5453. [Google Scholar] [CrossRef]
- Sze, L.P.; Li, H.Y.; Lai, K.L.A.; Chow, S.F.; Li, Q.; KennethTo, K.W.; Lam, T.N.T.; Lee, W.Y.T. Oral Delivery of Paclitaxel by Polymeric Micelles: A Comparison of Different Block Length on Uptake, Permeability and Oral Bioavailability. Colloids Surf. B Biointerfaces 2019, 184, 110554. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, J.; Cai, Q.; Fan, S.; Xu, Q.; Zang, J.; Yang, H.; Yu, W.; Li, Z.; Zhang, Z. Acetic Acid Transporter-Mediated, Oral, Multifunctional Polymer Liposomes for Oral Delivery of Docetaxel. Colloids Surf. B Biointerfaces 2021, 198, 111499. [Google Scholar] [CrossRef]
- Mintz, K.J.; Mercado, G.; Zhou, Y.; Ji, Y.; Hettiarachchi, S.D.; Liyanage, P.Y.; Pandey, R.R.; Chusuei, C.C.; Dallman, J.; Leblanc, R.M. Tryptophan Carbon Dots and Their Ability to Cross the Blood-Brain Barrier. Colloids Surf. B Biointerfaces 2019, 176, 488–493. [Google Scholar] [CrossRef]
- Porta, F.; Lamers, G.E.M.; Morrhayim, J.; Chatzopoulou, A.; Schaaf, M.; den Dulk, H.; Backendorf, C.; Zink, J.I.; Kros, A. Folic Acid-Modified Mesoporous Silica Nanoparticles for Cellular and Nuclear Targeted Drug Delivery. Adv. Healthc. Mater. 2013, 2, 281–286. [Google Scholar] [CrossRef]
- Bharti, C.; Nagaich, U.; Pal, A.K.; Gulati, N. Mesoporous Silica Nanoparticles in Target Drug Delivery System: A Review. Int. J. Pharm. Investig. 2015, 5, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, S.K.; Han, H.-S.; Subedi, L.; Pangeni, R.; Chung, J.Y.; Kweon, S.; Choi, J.U.; Byun, Y.; Kim, Y.-H.; Park, J.W. Enhanced Oral Bioavailability of an Etoposide Multiple Nanoemulsion Incorporating a Deoxycholic Acid Derivative-Lipid Complex. Drug Deliv. 2020, 27, 1501–1513. [Google Scholar] [CrossRef] [PubMed]
- Kou, L.; Hou, Y.; Yao, Q.; Guo, W.; Wang, G.; Wang, M.; Fu, Q.; He, Z.; Ganapathy, V.; Sun, J. L-Carnitine-Conjugated Nanoparticles to Promote Permeation across Blood-Brain Barrier and to Target Glioma Cells for Drug Delivery via the Novel Organic Cation/Carnitine Transporter OCTN2. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, G.; Chen, S.; Luo, S.; Zhang, J. Biomimetic and Bioinspired Strategies for Oral Drug Delivery. Biomater. Sci. 2020, 8, 1020–1044. [Google Scholar] [CrossRef]
- Cui, Y.; Shan, W.; Zhou, R.; Liu, M.; Wu, L.; Guo, Q.; Zheng, Y.; Wu, J.; Huang, Y. The Combination of Endolysosomal Escape and Basolateral Stimulation to Overcome the Difficulties of “Easy Uptake Hard Transcytosis” of Ligand-Modified Nanoparticles in Oral Drug Delivery. Nanoscale 2018, 10, 1494–1507. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The Endosomal-Lysosomal System: From Acidification and Cargo Sorting to Neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) Family of Membrane Transporters. Mol. Aspects Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [Green Version]
- Klepper, J.; Akman, C.; Armeno, M.; Auvin, S.; Cervenka, M.; Cross, H.J.; De Giorgis, V.; Della Marina, A.; Engelstad, K.; Heussinger, N.; et al. Glut1 Deficiency Syndrome (Glut1DS): State of the Art in 2020 and Recommendations of the International Glut1DS Study Group. Epilepsia Open 2020, 5, 354–365. [Google Scholar] [CrossRef]
- Nishioka, T.; Oda, Y.; Seino, Y.; Yamamoto, T.; Inagaki, N.; Yano, H.; Imura, H.; Shigemoto, R.; Kikuchi, H. Distribution of the Glucose Transporters in Human Brain Tumors. Cancer Res. 1992, 52, 3972–3979. [Google Scholar]
- Battaglia, G.; La Russa, M.; Bruno, V.; Arenare, L.; Ippolito, R.; Copani, A.; Bonina, F.; Nicoletti, F. Systemically Administered D-Glucose Conjugates of 7-Chlorokynurenic Acid Are Centrally Available and Exert Anticonvulsant Activity in Rodents. Brain Res. 2000, 860, 149–156. [Google Scholar] [CrossRef]
- Bonina, F.P.; Arenare, L.; Ippolito, R.; Boatto, G.; Battaglia, G.; Bruno, V.; de Caprariis, P. Synthesis, Pharmacokinetics and Anticonvulsant Activity of 7-Chlorokynurenic Acid Prodrugs. Int. J. Pharm. 2000, 202, 79–88. [Google Scholar] [CrossRef]
- Bonina, F.; Puglia, C.; Rimoli, M.G.; Melisi, D.; Boatto, G.; Nieddu, M.; Calignano, A.; La Rana, G.; De Caprariis, P. Glycosyl Derivatives of Dopamine and L-Dopa as Anti-Parkinson Prodrugs: Synthesis, Pharmacological Activity and in Vitro Stability Studies. J. Drug Target. 2003, 11, 25–36. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Filosa, R.; de Caprariis, P.; Conte, G.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Prasad, P.D.; Pavan, B. Molecular Mechanism Involved in the Transport of a Prodrug Dopamine Glycosyl Conjugate. Int. J. Pharm. 2007, 336, 133–139. [Google Scholar] [CrossRef]
- Halmos, T.; Santarromana, M.; Antonakis, K.; Scherman, D. Synthesis of Glucose-Chlorambucil Derivatives and Their Recognition by the Human GLUT1 Glucose Transporter. Eur. J. Pharmacol. 1996, 318, 477–484. [Google Scholar] [CrossRef]
- Eary, J.F.; Conrad, E.U. Positron Emission Tomography in Grading Soft Tissue Sarcomas. Semin. Musculoskelet. Radiol. 1999, 3, 135–138. [Google Scholar] [CrossRef]
- Larson, S.M. Positron Emission Tomography-Based Molecular Imaging in Human Cancer: Exploring the Link between Hypoxia and Accelerated Glucose Metabolism. Clin. Cancer Res. 2004, 10, 2203–2204. [Google Scholar] [CrossRef] [Green Version]
- Shan, X.H.; Hu, H.; Xiong, F.; Gu, N.; Geng, X.D.; Zhu, W.; Lin, J.; Wang, Y.F. Targeting Glut1-Overexpressing MDA-MB-231 Cells with 2-Deoxy-D-G1ucose Modified SPIOs. Eur. J. Radiol. 2012, 81, 95–99. [Google Scholar] [CrossRef]
- Xie, F.; Yao, N.; Qin, Y.; Zhang, Q.; Chen, H.; Yuan, M.; Tang, J.; Li, X.; Fan, W.; Zhang, Q.; et al. Investigation of Glucose-Modified Liposomes Using Polyethylene Glycols with Different Chain Lengths as the Linkers for Brain Targeting. Int. J. Nanomed. 2012, 7, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Xin, H.; Gu, J.; Du, F.; Feng, C.; Xie, Y.; Fang, X. Enhanced Antitumor Efficacy by D-Glucosamine-Functionalized and Paclitaxel-Loaded Poly(Ethylene Glycol)-Co-Poly(Trimethylene Carbonate) Polymer Nanoparticles. J. Pharm. Sci. 2014, 103, 1487–1496. [Google Scholar] [CrossRef]
- Shao, K.; Zhang, Y.; Ding, N.; Huang, S.; Wu, J.; Li, J.; Yang, C.; Leng, Q.; Ye, L.; Lou, J.; et al. Functionalized Nanoscale Micelles with Brain Targeting Ability and Intercellular Microenvironment Biosensitivity for Anti-Intracranial Infection Applications. Adv. Healthc. Mater. 2015, 4, 291–300. [Google Scholar] [CrossRef]
- Shao, K.; Ding, N.; Huang, S.; Ren, S.; Zhang, Y.; Kuang, Y.; Guo, Y.; Ma, H.; An, S.; Li, Y.; et al. Smart Nanodevice Combined Tumor-Specific Vector with Cellular Microenvironment-Triggered Property for Highly Effective Antiglioma Therapy. ACS Nano 2014, 8, 1191–1203. [Google Scholar] [CrossRef]
- Agus, D.B.; Gambhir, S.S.; Pardridge, W.M.; Spielholz, C.; Baselga, J.; Vera, J.C.; Golde, D.W. Vitamin C Crosses the Blood-Brain Barrier in the Oxidized Form through the Glucose Transporters. J. Clin. Invest. 1997, 100, 2842–2848. [Google Scholar] [CrossRef] [Green Version]
- Qu, B.; Li, X.; Guan, M.; Li, X.; Hai, L.; Wu, Y. Design, Synthesis and Biological Evaluation of Multivalent Glucosides with High Affinity as Ligands for Brain Targeting Liposomes. Eur. J. Med. Chem. 2014, 72, 110–118. [Google Scholar] [CrossRef]
- Venturelli, L.; Nappini, S.; Bulfoni, M.; Gianfranceschi, G.; Dal Zilio, S.; Coceano, G.; Del Ben, F.; Turetta, M.; Scoles, G.; Vaccari, L.; et al. Glucose Is a Key Driver for GLUT1-Mediated Nanoparticles Internalization in Breast Cancer Cells. Sci. Rep. 2016, 6, 21629. [Google Scholar] [CrossRef] [Green Version]
- Anraku, Y.; Kuwahara, H.; Fukusato, Y.; Mizoguchi, A.; Ishii, T.; Nitta, K.; Matsumoto, Y.; Toh, K.; Miyata, K.; Uchida, S.; et al. Glycaemic Control Boosts Glucosylated Nanocarrier Crossing the BBB into the Brain. Nat. Commun. 2017, 8, 1001. [Google Scholar] [CrossRef] [Green Version]
- Ung, P.M.-U.; Song, W.; Cheng, L.; Zhao, X.; Hu, H.; Chen, L.; Schlessinger, A. Inhibitor Discovery for the Human GLUT1 from Homology Modeling and Virtual Screening. ACS Chem. Biol. 2016, 11, 1908–1916. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhu, F.; Liu, Y.; Zheng, M.; Wang, Y.; Zhang, D.; Anraku, Y.; Zou, Y.; Li, J.; Wu, H.; et al. Blood-Brain Barrier-Penetrating SiRNA Nanomedicine for Alzheimer’s Disease Therapy. Sci. Adv. 2020, 6, eabc7031. [Google Scholar] [CrossRef]
- Gromnicova, R.; Davies, H.A.; Sreekanthreddy, P.; Romero, I.A.; Lund, T.; Roitt, I.M.; Phillips, J.B.; Male, D.K. Glucose-Coated Gold Nanoparticles Transfer across Human Brain Endothelium and Enter Astrocytes in Vitro. PLoS One 2013, 8, e81043. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Jeong, D.C.; Pak, K.; Han, M.-E.; Kim, J.-Y.; Liangwen, L.; Kim, H.J.; Kim, T.W.; Kim, T.H.; Hyun, D.W.; et al. SLC2A2 (GLUT2) as a Novel Prognostic Factor for Hepatocellular Carcinoma. Oncotarget 2017, 8, 68381–68392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, R.A.; Owen, G.I. Glucose Transporters: Expression, Regulation and Cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef] [PubMed]
- McQuade, D.T.; Plutschack, M.B.; Seeberger, P.H. Passive Fructose Transporters in Disease: A Molecular Overview of Their Structural Specificity. Org. Biomol. Chem. 2013, 11, 4909–4920. [Google Scholar] [CrossRef]
- Pujol-Gimenez, J.; de Heredia, F.P.; Idoate, M.A.; Airley, R.; Lostao, M.P.; Evans, A.R. Could GLUT12 Be a Potential Therapeutic Target in Cancer Treatment? A Preliminary Report. J. Cancer 2015, 6, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Nahrjou, N.; Ghosh, A.; Tanasova, M. Targeting of GLUT5 for Transporter-Mediated Drug-Delivery Is Contingent upon Substrate Hydrophilicity. Int. J. Mol. Sci. 2021, 22, 5073. [Google Scholar] [CrossRef] [PubMed]
- Begoyan, V.V.; Weseliński, Ł.J.; Xia, S.; Fedie, J.; Kannan, S.; Ferrier, A.; Rao, S.; Tanasova, M. Multicolor GLUT5-Permeable Fluorescent Probes for Fructose Transport Analysis. Chem. Commun. 2018, 54, 3855–3858. [Google Scholar] [CrossRef]
- Kannan, S.; Begoyan, V.V.; Fedie, J.R.; Xia, S.; Weseliński, Ł.J.; Tanasova, M.; Rao, S. Metabolism-Driven High-Throughput Cancer Identification with GLUT5-Specific Molecular Probes. Biosensors 2018, 8, E39. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.-C.; Kim, S.T.; Tang, R.; Yan, B.; Rotello, V.M. Insulin-Based Regulation of Glucose-Functionalized Nanoparticle Uptake in Muscle Cells. J. Mater. Chem. B 2014, 2, 4610–4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageyama, T.; Nakamura, M.; Matsuo, A.; Yamasaki, Y.; Takakura, Y.; Hashida, M.; Kanai, Y.; Naito, M.; Tsuruo, T.; Minato, N.; et al. The 4F2hc/LAT1 Complex Transports L-DOPA across the Blood-Brain Barrier. Brain Res. 2000, 879, 115–121. [Google Scholar] [CrossRef]
- Duelli, R.; Enerson, B.E.; Gerhart, D.Z.; Drewes, L.R. Expression of Large Amino Acid Transporter LAT1 in Rat Brain Endothelium. J. Cereb. Blood Flow Metab. 2000, 20, 1557–1562. [Google Scholar] [CrossRef] [Green Version]
- Asano, S.; Kameyama, M.; Oura, A.; Morisato, A.; Sakai, H.; Tabuchi, Y.; Chairoungdua, A.; Endou, H.; Kanai, Y. L-Type Amino Acid Transporter-1 Expressed in Human Astrocytomas, U343MGa. Biol. Pharm. Bull. 2007, 30, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Smith, Q.R. Carrier-Mediated Transport to Enhance Drug Delivery to Brain. Int. Congr. Ser. 2005, 1277, 63–74. [Google Scholar] [CrossRef]
- Puris, E.; Gynther, M.; Huttunen, J.; Auriola, S.; Huttunen, K.M. L-Type Amino Acid Transporter 1 Utilizing Prodrugs of Ferulic Acid Revealed Structural Features Supporting the Design of Prodrugs for Brain Delivery. Eur. J. Pharm. Sci. 2019, 129, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Huttunen, J.; Montaser, A.; Huttunen, K.M. Hemocompatible LAT1-Inhibitor Can Induce Apoptosis in Cancer Cells without Affecting Brain Amino Acid Homeostasis. Apoptosis 2020, 25, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Puris, E.; Peltokangas, S.; Auriola, S.; Kanninen, K.M.; Koistinaho, J.; Huttunen, K.M.; Ruponen, M.; Vellonen, K.-S. Alzheimer’s Disease Phenotype or Inflammatory Insult Does Not Alter Function of L-Type Amino Acid Transporter 1 in Mouse Blood-Brain Barrier and Primary Astrocytes. Pharm. Res. 2018, 36, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, Y.; Segawa, H.; Miyamoto, K.I.; Uchino, H.; Takeda, E.; Endou, H. Expression Cloning and Characterization of a Transporter for Large Neutral Amino Acids Activated by the Heavy Chain of 4F2 AntiGen. (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef] [Green Version]
- Montaser, A.B.; Järvinen, J.; Löffler, S.; Huttunen, J.; Auriola, S.; Lehtonen, M.; Jalkanen, A.; Huttunen, K.M. L-Type Amino Acid Transporter 1 Enables the Efficient Brain Delivery of Small-Sized Prodrug across the Blood-Brain Barrier and into Human and Mouse Brain Parenchymal Cells. ACS Chem. NeuroSci. 2020, 11, 4301–4315. [Google Scholar] [CrossRef]
- Häfliger, P.; Charles, R.-P. The L-Type Amino Acid Transporter LAT1-An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [Green Version]
- Cornford, E.M.; Young, D.; Paxton, J.W.; Finlay, G.J.; Wilson, W.R.; Pardridge, W.M. Melphalan Penetration of the Blood-Brain Barrier via the Neutral Amino Acid Transporter in Tumor-Bearing Brain. Cancer Res. 1992, 52, 138–143. [Google Scholar]
- Wang, Y.; Welty, D.F. The Simultaneous Estimation of the Influx and Efflux Blood-Brain Barrier Permeabilities of Gabapentin Using a Microdialysis-Pharmacokinetic Approach. Pharm. Res. 1996, 13, 398–403. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nishimura, T.; Higuchi, K.; Noguchi, S.; Tega, Y.; Kurosawa, T.; Deguchi, Y.; Tomi, M. Transport of Pregabalin Via L-Type Amino Acid Transporter 1 (SLC7A5) in Human Brain Capillary Endothelial Cell Line. Pharm. Res. 2018, 35, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puris, E.; Gynther, M.; Huttunen, J.; Petsalo, A.; Huttunen, K.M. L-Type Amino Acid Transporter 1 Utilizing Prodrugs: How to Achieve Effective Brain Delivery and Low Systemic Exposure of Drugs. J. Control. Release 2017, 261, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Peura, L.; Malmioja, K.; Huttunen, K.; Leppänen, J.; Hämäläinen, M.; Forsberg, M.M.; Gynther, M.; Rautio, J.; Laine, K. Design, Synthesis and Brain Uptake of LAT1-Targeted Amino Acid Prodrugs of Dopamine. Pharm. Res. 2013, 30, 2523–2537. [Google Scholar] [CrossRef]
- Peura, L.; Malmioja, K.; Laine, K.; Leppänen, J.; Gynther, M.; Isotalo, A.; Rautio, J. Large Amino Acid Transporter 1 (LAT1) Prodrugs of Valproic Acid: New Prodrug Design Ideas for Central Nervous System Delivery. Mol. Pharm. 2011, 8, 1857–1866. [Google Scholar] [CrossRef]
- Bonina, F.P.; Arenare, L.; Palagiano, F.; Saija, A.; Nava, F.; Trombetta, D.; de Caprariis, P. Synthesis, Stability, and Pharmacological Evaluation of Nipecotic Acid Prodrugs. J. Pharm. Sci. 1999, 88, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Walker, I.; Nicholls, D.; Irwin, W.J.; Freeman, S. Drug Delivery via Active Transport at the Blood-Brain Barrier: Affinity of a Prodrug of Phosphonoformate for the Large Amino Acid Transporter. Int. J. Pharm. 1994, 104, 157–167. [Google Scholar] [CrossRef]
- Huttunen, J.; Gynther, M.; Huttunen, K.M. Targeted Efflux Transporter Inhibitors—A Solution to Improve Poor Cellular Accumulation of Anti-Cancer Agents. Int. J. Pharm. 2018, 550, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type Amino Acid Transporter 1 as a Target for Drug Delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef]
- Hong, S.; Fang, Z.; Jung, H.-Y.; Yoon, J.-H.; Hong, S.-S.; Maeng, H.-J. Synthesis of Gemcitabine-Threonine Amide Prodrug Effective on Pancreatic Cancer Cells with Improved Pharmacokinetic Properties. Molecules 2018, 23, E2608. [Google Scholar] [CrossRef] [Green Version]
- Hokari, M.; Wu, H.Q.; Schwarcz, R.; Smith, Q.R. Facilitated Brain Uptake of 4-Chlorokynurenine and Conversion to 7-Chlorokynurenic Acid. Neuroreport 1996, 8, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Uchino, H.; Kanai, Y.; Kim, D.K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.W.; Endou, H. Transport of Amino Acid-Related Compounds Mediated by L-Type Amino Acid Transporter 1 (LAT1): Insights into the Mechanisms of Substrate Recognition. Mol. Pharmacol. 2002, 61, 729–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gynther, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Mannila, A.; Nevalainen, T.; Savolainen, J.; Järvinen, T.; Rautio, J. Large Neutral Amino Acid Transporter Enables Brain Drug Delivery via Prodrugs. J. Med. Chem. 2008, 51, 932–936. [Google Scholar] [CrossRef]
- Li, L.; Di, X.; Zhang, S.; Kan, Q.; Liu, H.; Lu, T.; Wang, Y.; Fu, Q.; Sun, J.; He, Z. Large Amino Acid Transporter 1 Mediated Glutamate Modified Docetaxel-Loaded Liposomes for Glioma Targeting. Colloids Surf. B Biointerfaces 2016, 141, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Ylikangas, H.; Peura, L.; Malmioja, K.; Leppänen, J.; Laine, K.; Poso, A.; Lahtela-Kakkonen, M.; Rautio, J. Structure-Activity Relationship Study of Compounds Binding to Large Amino Acid Transporter 1 (LAT1) Based on Pharmacophore Modeling and in Situ Rat Brain Perfusion. Eur. J. Pharm. Sci. 2013, 48, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Ylikangas, H.; Malmioja, K.; Peura, L.; Gynther, M.; Nwachukwu, E.O.; Leppänen, J.; Laine, K.; Rautio, J.; Lahtela-Kakkonen, M.; Huttunen, K.M.; et al. Quantitative Insight into the Design of Compounds Recognized by the L-Type Amino Acid Transporter 1 (LAT1). ChemMedChem 2014, 9, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, K.; Järvinen, J.; Laitinen, T.; Reinisalo, M.; Honkakoski, P.; Poso, A.; Huttunen, K.M.; Rautio, J. Structural Features Affecting the Interactions and Transportability of LAT1-Targeted Phenylalanine Drug Conjugates. Mol. Pharm. 2023, 20, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Kinne, A.; Schülein, R.; Krause, G. Primary and Secondary Thyroid Hormone Transporters. Thyroid Res. 2011, 4 (Suppl. 1), S7. [Google Scholar] [CrossRef] [Green Version]
- Huttunen, K.M.; Huttunen, J.; Aufderhaar, I.; Gynther, M.; Denny, W.A.; Spicer, J.A. L-Type Amino Acid Transporter 1 (Lat1)-Mediated Targeted Delivery of Perforin Inhibitors. Int. J. Pharm. 2016, 498, 205–216. [Google Scholar] [CrossRef]
- van de Looij, S.M.; Hebels, E.R.; Viola, M.; Hembury, M.; Oliveira, S.; Vermonden, T. Gold Nanoclusters: Imaging, Therapy, and Theranostic Roles in Biomedical Applications. Bioconjugate Chem. 2022, 33, 4–23. [Google Scholar] [CrossRef]
- Chen, H.; Li, B.; Ren, X.; Li, S.; Ma, Y.; Cui, S.; Gu, Y. Multifunctional Near-Infrared-Emitting Nano-Conjugates Based on Gold Clusters for Tumor Imaging and Therapy. Biomaterials 2012, 33, 8461–8476. [Google Scholar] [CrossRef]
- Bhunia, S.; Vangala, V.; Bhattacharya, D.; Ravuri, H.G.; Kuncha, M.; Chakravarty, S.; Sistla, R.; Chaudhuri, A. Large Amino Acid Transporter 1 Selective Liposomes of L-DOPA Functionalized Amphiphile for Combating Glioblastoma. Mol. Pharm. 2017, 14, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Venu, Y.; Bhattacharya, D.; Kompella, S.D.; Madhusudana, K.; Chakravarty, S.; Ramakrishna, S.; Chaudhuri, A. Combating Established Mouse Glioblastoma through Nicotinylated-Liposomes-Mediated Targeted Chemotherapy in Combination with Dendritic-Cell-Based Genetic Immunization. Adv. Biosyst 2017, 1, e1600009. [Google Scholar] [CrossRef]
- Kharya, P.; Jain, A.; Gulbake, A.; Shilpi, S.; Jain, A.; Hurkat, P.; Majumdar, S.; Jain, S.K. Phenylalanine-Coupled Solid Lipid Nanoparticles for Brain Tumor Targeting. J. Nanopart Res. 2013, 15, 2022. [Google Scholar] [CrossRef]
- Rautio, J.; Gynther, M.; Laine, K. LAT1-Mediated Prodrug Uptake: A Way to Breach the Blood-Brain Barrier? Ther. Deliv. 2013, 4, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Di, X.; Wu, M.; Sun, Z.; Zhong, L.; Wang, Y.; Fu, Q.; Kan, Q.; Sun, J.; He, Z. Targeting Tumor Highly-Expressed LAT1 Transporter with Amino Acid-Modified Nanoparticles: Toward a Novel Active Targeting Strategy in Breast Cancer Therapy. Nanomedicine 2017, 13, 987–998. [Google Scholar] [CrossRef]
- Maekawa-Matsuura, M.; Fujieda, K.; Maekawa, Y.; Nishimura, T.; Nagase, K.; Kanazawa, H. LAT1-Targeting Thermoresponsive Liposomes for Effective Cellular Uptake by Cancer Cells. ACS Omega 2019, 4, 6443–6451. [Google Scholar] [CrossRef] [Green Version]
- Sloan, J.L.; Mager, S. Cloning and Functional Expression of a Human Na(+) and Cl(-)-Dependent Neutral and Cationic Amino Acid Transporter B(0+). J. Biol. Chem. 1999, 274, 23740–23745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.M.H.; Howard, A.; Walters, J.R.F.; Ganapathy, V.; Thwaites, D.T. Taurine Uptake across the Human Intestinal Brush-Border Membrane Is via Two Transporters: H+-Coupled PAT1 (SLC36A1) and Na+- and Cl(-)-Dependent TauT (SLC6A6). J. Physiol. 2009, 587, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Ruffin, M.; Mercier, J.; Calmel, C.; Mésinèle, J.; Bigot, J.; Sutanto, E.N.; Kicic, A.; Corvol, H.; Guillot, L. Update on SLC6A14 in Lung and Gastrointestinal Physiology and Physiopathology: Focus on Cystic Fibrosis. Cell Mol. Life Sci. 2020, 77, 3311–3323. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Miyauchi, S.; Martindale, R.G.; Herdman, A.V.; Podolsky, R.; Miyake, K.; Mager, S.; Prasad, P.D.; Ganapathy, M.E.; Ganapathy, V. Upregulation of the Amino Acid Transporter ATB0,+ (SLC6A14) in Colorectal Cancer and Metastasis in Humans. Biochim. Biophys. Acta 2005, 1741, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Prasad, P.D.; Ghamande, S.; Moore-Martin, P.; Herdman, A.V.; Martindale, R.G.; Podolsky, R.; Mager, S.; Ganapathy, M.E.; Ganapathy, V. Up-Regulation of the Amino Acid Transporter ATB(0,+) (SLC6A14) in Carcinoma of the Cervix. Gynecol. Oncol. 2006, 100, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, S.; Umapathy, N.S.; Thangaraju, M.; Hatanaka, T.; Itagaki, S.; Munn, D.H.; Prasad, P.D.; Ganapathy, V. Interaction of Tryptophan Derivatives with SLC6A14 (ATB0,+) Reveals the Potential of the Transporter as a Drug Target for Cancer Chemotherapy. BioChem. J. 2008, 414, 343–355. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Prasad, P.D.; Ganapathy, V. The Amino Acid Transporter SLC6A14 in Cancer and Its Potential Use in Chemotherapy. Asian J. Pharm. Sci. 2014, 9, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, T.; Haramura, M.; Fei, Y.-J.; Miyauchi, S.; Bridges, C.C.; Ganapathy, P.S.; Smith, S.B.; Ganapathy, V.; Ganapathy, M.E. Transport of Amino Acid-Based Prodrugs by the Na+- and Cl(-) -Coupled Amino Acid Transporter ATB0,+ and Expression of the Transporter in Tissues Amenable for Drug Delivery. J. Pharmacol. Exp. Ther. 2004, 308, 1138–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.-Y.; Shim, W.-S.; Chang, J.-E.; Chong, S.; Kim, D.-D.; Chung, S.-J.; Shim, C.-K. Enhanced Intracellular Accumulation of a Non-Nucleoside Anti-Cancer Agent via Increased Uptake of Its Valine Ester Prodrug through Amino Acid Transporters. Xenobiotica 2012, 42, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.R.; Prasad, P.D.; Umapathy, N.S.; Ganapathy, V.; Shekhawat, P.S. Transport of Butyryl-L-Carnitine, a Potential Prodrug, via the Carnitine Transporter OCTN2 and the Amino Acid Transporter ATB(0,+). Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1046–G1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Q.; Yang, B.; Tao, W.; Li, J.; Kou, L.; Lian, H.; Che, X.; He, Z.; Sun, J. ATB0,+ Transporter-Mediated Targeting Delivery to Human Lung Cancer Cells via Aspartate-Modified Docetaxel-Loading Stealth Liposomes. Biomater. Sci. 2017, 5, 295–304. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S.K. L-Valine Appended PLGA Nanoparticles for Oral Insulin Delivery. Acta Diabetol. 2015, 52, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chi, D.; Wu, X.; Wang, Y.; Lin, X.; Xu, Z.; Liu, H.; Sun, J.; He, Z.; Wang, Y. Tyrosine Modified Irinotecan-Loaded Liposomes Capable of Simultaneously Targeting LAT1 and ATB0,+ for Efficient Tumor Therapy. J. Control. Release 2019, 316, 22–33. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The Role of ASCT2 in Cancer: A Review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef]
- Wahi, K.; Holst, J. ASCT2: A Potential Cancer Drug Target. Expert Opin. Ther. Targets 2019, 23, 555–558. [Google Scholar] [CrossRef]
- Lopes, C.; Pereira, C.; Medeiros, R. ASCT2 and LAT1 Contribution to the Hallmarks of Cancer: From a Molecular Perspective to Clinical Translation. Cancers 2021, 13, E203. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino Acid Transporters Revisited: New Views in Health and Disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 Controls Glutamine Uptake and Tumour Growth in Triple-Negative Basal-like Breast Cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Arriza, J.L.; Kavanaugh, M.P.; Fairman, W.A.; Wu, Y.N.; Murdoch, G.H.; North, R.A.; Amara, S.G. Cloning and Expression of a Human Neutral Amino Acid Transporter with Structural Similarity to the Glutamate Transporter Gene Family. J. Biol. Chem. 1993, 268, 15329–15332. [Google Scholar] [CrossRef]
- Bröer, A.; Wagner, C.; Lang, F.; Bröer, S. Neutral Amino Acid Transporter ASCT2 Displays Substrate-Induced Na+ Exchange and a Substrate-Gated Anion Conductance. BioChem. J. 2000, 346 Pt 3, 705–710. [Google Scholar] [CrossRef]
- Kanai, Y.; Hediger, M.A. The Glutamate/Neutral Amino Acid Transporter Family SLC1: Molecular, Physiological and Pharmacological Aspects. Pflug. Arch. 2004, 447, 469–479. [Google Scholar] [CrossRef]
- Kanai, Y.; Clémençon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M.A. The SLC1 High-Affinity Glutamate and Neutral Amino Acid Transporter Family. Mol. Aspects Med. 2013, 34, 108–120. [Google Scholar] [CrossRef]
- Ganapathy, V.; Thangaraju, M.; Prasad, P.D. Nutrient Transporters in Cancer: Relevance to Warburg Hypothesis and Beyond. Pharmacol. Ther. 2009, 121, 29–40. [Google Scholar] [CrossRef]
- Lieberman, B.P.; Ploessl, K.; Wang, L.; Qu, W.; Zha, Z.; Wise, D.R.; Chodosh, L.A.; Belka, G.; Thompson, C.B.; Kung, H.F. PET Imaging of Glutaminolysis in Tumors by 18F-(2S,4R)4-Fluoroglutamine. J. Nucl. Med. 2011, 52, 1947–1955. [Google Scholar] [CrossRef] [Green Version]
- Ploessl, K.; Wang, L.; Lieberman, B.P.; Qu, W.; Kung, H.F. Comparative Evaluation of 18F-Labeled Glutamic Acid and Glutamine as Tumor Metabolic Imaging Agents. J. Nucl. Med. 2012, 53, 1616–1624. [Google Scholar] [CrossRef] [Green Version]
- Nanni, C.; Schiavina, R.; Brunocilla, E.; Boschi, S.; Borghesi, M.; Zanoni, L.; Pettinato, C.; Martorana, G.; Fanti, S. 18F-Fluciclovine PET/CT for the Detection of Prostate Cancer Relapse: A Comparison to 11C-Choline PET/CT. Clin. Nucl. Med. 2015, 40, e386–e391. [Google Scholar] [CrossRef]
- Odewole, O.A.; Tade, F.I.; Nieh, P.T.; Savir-Baruch, B.; Jani, A.B.; Master, V.A.; Rossi, P.J.; Halkar, R.K.; Osunkoya, A.O.; Akin-Akintayo, O.; et al. Recurrent Prostate Cancer Detection with Anti-3-[(18)F]FACBC PET/CT: Comparison with CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Okudaira, H.; Oka, S.; Ono, M.; Nakanishi, T.; Schuster, D.M.; Kobayashi, M.; Goodman, M.M.; Tamai, I.; Kawai, K.; Shirakami, Y. Accumulation of Trans-1-Amino-3-[(18)F]Fluorocyclobutanecarboxylic Acid in Prostate Cancer Due to Androgen-Induced Expression of Amino Acid Transporters. Mol. Imaging Biol. 2014, 16, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Ravera, M.; Gabano, E.; Tinello, S.; Zanellato, I.; Osella, D. May Glutamine Addiction Drive the Delivery of Antitumor Cisplatin-Based Pt(IV) Prodrugs? J. InOrg. BioChem. 2017, 167, 27–35. [Google Scholar] [CrossRef]
- Zhou, P.; Liang, X.; Zhou, C.; Qin, J.; Hou, C.; Zhu, Z.; Zhang, W.; Wang, S.; Zhong, D. Glutamine-β-Cyclodextrin for Targeted Doxorubicin Delivery to Triple-Negative Breast Cancer Tumors via the Transporter ASCT2. J. Mater. Chem. B 2019, 7, 5363–5375. [Google Scholar] [CrossRef]
- Wang, C.; Wu, J.; Wang, Z.; Yang, Z.; Li, Z.; Deng, H.; Li, L.; Peng, X.; Feng, M. Glutamine Addiction Activates Polyglutamine-Based Nanocarriers Delivering Therapeutic SiRNAs to Orthotopic Lung Tumor Mediated by Glutamine Transporter SLC1A5. Biomaterials 2018, 183, 77–92. [Google Scholar] [CrossRef]
- Nakamura, E.; Sato, M.; Yang, H.; Miyagawa, F.; Harasaki, M.; Tomita, K.; Matsuoka, S.; Noma, A.; Iwai, K.; Minato, N. 4F2 (CD98) Heavy Chain Is Associated Covalently with an Amino Acid Transporter and Controls Intracellular Trafficking and Membrane Topology of 4F2 Heterodimer. J. Biol. Chem. 1999, 274, 3009–3016. [Google Scholar] [CrossRef] [Green Version]
- Devés, R.; Boyd, C.A. Surface AntiGen. CD98(4F2): Not a Single Membrane Protein, but a Family of Proteins with Multiple Functions. J. Membr. Biol. 2000, 173, 165–177. [Google Scholar] [CrossRef]
- Palacín, M.; Nunes, V.; Font-Llitjós, M.; Jiménez-Vidal, M.; Fort, J.; Gasol, E.; Pineda, M.; Feliubadaló, L.; Chillarón, J.; Zorzano, A. The Genetics of Heteromeric Amino Acid Transporters. Physiology 2005, 20, 112–124. [Google Scholar] [CrossRef]
- Yan, Y.; Vasudevan, S.; Nguyen, H.T.T.; Merlin, D. Intestinal Epithelial CD98: An Oligomeric and Multifunctional Protein. Biochim. BioPhys. Acta 2008, 1780, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.T.; Dalmasso, G.; Torkvist, L.; Halfvarson, J.; Yan, Y.; Laroui, H.; Shmerling, D.; Tallone, T.; D’Amato, M.; Sitaraman, S.V.; et al. CD98 Expression Modulates Intestinal Homeostasis, Inflammation, and Colitis-Associated Cancer in Mice. J. Clin. Invest. 2011, 121, 1733–1747. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Viennois, E.; Chen, Q.; Wang, L.; Han, M.K.; Zhang, Y.; Zhang, Z.; Kang, Y.; Wan, Y.; Merlin, D. Silencing of Intestinal Glycoprotein CD98 by Orally Targeted Nanoparticles Enhances Chemosensitization of Colon Cancer. ACS Nano 2018, 12, 5253–5265. [Google Scholar] [CrossRef]
- Xiao, B.; Laroui, H.; Viennois, E.; Ayyadurai, S.; Charania, M.A.; Zhang, Y.; Zhang, Z.; Baker, M.T.; Zhang, B.; Gewirtz, A.T.; et al. Nanoparticles with Surface Antibody against CD98 and Carrying CD98 Small Interfering RNA Reduce Colitis in Mice. Gastroenterology 2014, 146, 1289–1300.e1-19. [Google Scholar] [CrossRef] [Green Version]
- Berczeller, A. Bile Acid Derivatives of Aryl Sulfonamides. U.S. Patent 2441129, 1948. [Google Scholar]
- Kramer, W.; Wess, G. Bile Acid Transport Systems as Pharmaceutical Targets. Eur. J. Clin. Invest. 1996, 26, 715–732. [Google Scholar] [CrossRef]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting Bile-Acid Signalling for Metabolic Diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef] [PubMed]
- Holm, R.; Müllertz, A.; Mu, H. Bile Salts and Their Importance for Drug Absorption. Int. J. Pharm. 2013, 453, 44–55. [Google Scholar] [CrossRef]
- Boegh, M.; Nielsen, H.M. Mucus as a Barrier to Drug Delivery—Understanding and Mimicking the Barrier Properties. Basic Clin. Pharmacol. Toxicol. 2015, 116, 179–186. [Google Scholar] [CrossRef]
- Ko, C.-W.; Qu, J.; Black, D.D.; Tso, P. Regulation of Intestinal Lipid Metabolism: Current Concepts and Relevance to Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef]
- Yáñez, J.A.; Wang, S.W.J.; Knemeyer, I.W.; Wirth, M.A.; Alton, K.B. Intestinal Lymphatic Transport for Drug Delivery. Adv. Drug Deliv. Rev. 2011, 63, 923–942. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [CrossRef]
- Tso, P.; Balint, J.A. Formation and Transport of Chylomicrons by Enterocytes to the Lymphatics. Am. J. Physiol. 1986, 250, G715–G726. [Google Scholar] [CrossRef] [PubMed]
- Rahmany, S.; Jialal, I. Biochemistry, Chylomicron. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J.H. Lipid-Based Delivery Systems and Intestinal Lymphatic Drug Transport: A Mechanistic Update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, Y.; Qi, J.; Wu, W. An Update on Oral Drug Delivery via Intestinal Lymphatic Transport. Acta Pharm. Sin. B 2021, 11, 2449–2468. [Google Scholar] [CrossRef] [PubMed]
- Managuli, R.S.; Raut, S.Y.; Reddy, M.S.; Mutalik, S. Targeting the Intestinal Lymphatic System: A Versatile Path for Enhanced Oral Bioavailability of Drugs. Expert Opin. Drug Deliv. 2018, 15, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.; Zhang, J.; Kuolee, R.; Patel, G.B.; Chen, W. Intestinal M Cells: The Fallible Sentinels? World J. Gastroenterol. 2007, 13, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acid Metabolism and Signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, P.A. Role of the Intestinal Bile Acid Transporters in Bile Acid and Drug Disposition. Handb. Exp. Pharmacol. 2011, 169–203. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, K.M.; Sugiyama, Y. Membrane Transporters and Drug Response. Goodman Gilman Pharmacol. Basis Ther. 2006, 11, 41–70. [Google Scholar]
- Mizuno, N.; Niwa, T.; Yotsumoto, Y.; Sugiyama, Y. Impact of Drug Transporter Studies on Drug Discovery and Development. Pharmacol. Rev. 2003, 55, 425–461. [Google Scholar] [CrossRef]
- Kramer, W.; Schneider, S. 3-Diazirine-Derivatives of Bile Salts for Photoaffinity Labeling. J. Lipid Res. 1989, 30, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Kramer, W.; Wess, G.; Neckermann, G.; Schubert, G.; Fink, J.; Girbig, F.; Gutjahr, U.; Kowalewski, S.; Baringhaus, K.H.; Böger, G. Intestinal Absorption of PeptiDes. by Coupling to Bile Acids. J. Biol. Chem. 1994, 269, 10621–10627. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Dawson, P. The Sodium Bile Salt Cotransport Family SLC10. Pflug. Arch. 2004, 447, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Anwer, M.S.; Stieger, B. Sodium-Dependent Bile Salt Transporters of the SLC10A Transporter Family: More than Solute Transporters. Pflug. Arch. 2014, 466, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Li, Q.; Guo, D.; Shu, Y.; Polli, J.E. Synthesis and Evaluation of Bile Acid-Ribavirin Conjugates as Prodrugs to Target the Liver. J. Pharm. Sci. 2015, 104, 2864–2876. [Google Scholar] [CrossRef] [Green Version]
- Tolle-Sander, S.; Lentz, K.A.; Maeda, D.Y.; Coop, A.; Polli, J.E. Increased Acyclovir Oral Bioavailability via a Bile Acid Conjugate. Mol. Pharm. 2004, 1, 40–48. [Google Scholar] [CrossRef]
- Vivian, D.; Polli, J.E. Synthesis and in Vitro Evaluation of Bile Acid Prodrugs of Floxuridine to Target the Liver. Int. J. Pharm. 2014, 475, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, D.; Shang, L.; He, Z.; Sun, J. Transporter-Targeted Cholic Acid-Cytarabine Conjugates for Improved Oral Absorption. Int. J. Pharm. 2016, 511, 161–169. [Google Scholar] [CrossRef]
- Lee, Y.; Nam, J.H.; Shin, H.C.; Byun, Y. Conjugation of Low-Molecular-Weight Heparin and Deoxycholic Acid for the Development of a New Oral Anticoagulant Agent. Circulation 2001, 104, 3116–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kim, S.K.; Lee, D.Y.; Lee, S.; Kim, C.-Y.; Shin, H.-C.; Moon, H.T.; Byun, Y. Efficacy of Orally Active Chemical Conjugate of Low Molecular Weight Heparin and Deoxycholic Acid in Rats, Mice and Monkeys. J. Control. Release 2006, 111, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Lee, D.Y.; Kim, C.Y.; Nam, J.H.; Moon, H.T.; Byun, Y. A Newly Developed Oral Heparin Derivative for Deep Vein Thrombosis: Non-Human Primate Study. J. Control. Release 2007, 123, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, K.; Kumar, T.S.; Lee, J.; Kim, S.K.; Lee, D.Y.; Lee, Y.; Byun, Y. Synthesis and Biological Properties of Insulin-Deoxycholic Acid Chemical Conjugates. Bioconjug. Chem. 2005, 16, 615–620. [Google Scholar] [CrossRef]
- Lu, X.; Wu, H.; Liang, Y.; Zhang, Z.; Lv, H. Redox-Responsive Prodrug for Improving Oral Bioavailability of Paclitaxel through Bile Acid Transporter-Mediated Pathway. Int. J. Pharm. 2021, 600, 120496. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Feng, F.; Meng, F.; Deng, C.; Feijen, J.; Zhong, Z. Glutathione-Responsive Nano-Vehicles as a Promising Platform for Targeted Intracellular Drug and Gene Delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yang, J.; Guan, J.; Yang, B.; Zhang, S.; Sun, M.; Yang, R.; Zhang, T.; Zhang, R.; Kan, Q.; et al. In Vivo Tailor-Made Protein Corona of a Prodrug-Based Nanoassembly Fabricated by Redox Dual-Sensitive Paclitaxel Prodrug for the Superselective Treatment of Breast Cancer. Biomater. Sci. 2018, 6, 2360–2374. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Z.; Cao, Z.; Zhou, W.; Zhang, Y.; Chen, Q.; Lu, Y.; Chen, X.; Guo, Q.; Li, C.; et al. Endogenous Albumin-Mediated Delivery of Redox-Responsive Paclitaxel-Loaded Micelles for Targeted Cancer Therapy. Biomaterials 2018, 183, 243–257. [Google Scholar] [CrossRef]
- Swaan, P.W.; Szoka, F.C.; Oie, S. Molecular Modeling of the Intestinal Bile Acid Carrier: A Comparative Molecular Field Analysis Study. J. Comput. Aided Mol. Des. 1997, 11, 581–588. [Google Scholar] [CrossRef]
- Bhat, L.; Jandeleit, B.; Dias, T.M.; Moors, T.L.; Gallop, M.A. Synthesis and Biological Evaluation of Novel Steroidal Pyrazoles as Substrates for Bile Acid Transporters. BioOrg. Med. Chem. Lett. 2005, 15, 85–87. [Google Scholar] [CrossRef]
- Kramer, W. Transporters, Trojan Horses and Therapeutics: Suitability of Bile Acid and Peptide Transporters for Drug Delivery. Biol. Chem. 2011, 392, 77–94. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Wring, S.A.; Polli, J.E. Interaction of Native Bile Acids with Human Apical Sodium-Dependent Bile Acid Transporter (HASBT): Influence of Steroidal Hydroxylation Pattern and C-24 Conjugation. Pharm. Res. 2006, 23, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, A.; Wring, S.A.; Coop, A.; Polli, J.E. Influence of Charge and Steric Bulk in the C-24 Region on the Interaction of Bile Acids with Human Apical Sodium-Dependent Bile Acid Transporter. Mol. Pharm. 2006, 3, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Pan, Y.; Acharya, C.; Swaan, P.W.; Polli, J.E. Structural Requirements of the ASBT by 3D-QSAR Analysis Using Aminopyridine Conjugates of Chenodeoxycholic Acid. Bioconjug. Chem. 2010, 21, 2038–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, N.-J.; Iwata, S.; Cameron, A.D.; Drew, D. Crystal Structure of a Bacterial Homologue of the Bile Acid Sodium Symporter ASBT. Nature 2011, 478, 408–411. [Google Scholar] [CrossRef]
- Al-Hilal, T.A.; Park, J.; Alam, F.; Chung, S.W.; Park, J.W.; Kim, K.; Kwon, I.C.; Kim, I.-S.; Kim, S.Y.; Byun, Y. Oligomeric Bile Acid-Mediated Oral Delivery of Low Molecular Weight Heparin. J. Control. Release 2014, 175, 17–24. [Google Scholar] [CrossRef]
- Fan, W.; Xia, D.; Zhu, Q.; Li, X.; He, S.; Zhu, C.; Guo, S.; Hovgaard, L.; Yang, M.; Gan, Y. Functional Nanoparticles Exploit the Bile Acid Pathway to Overcome Multiple Barriers of the Intestinal Epithelium for Oral Insulin Delivery. Biomaterials 2018, 151, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, C.; Li, J.; Li, J.; Li, J.; Sun, T.; Wang, L.; Wu, H.; Yu, G. Chitosan-Based Nanomaterials for Drug Delivery. Molecules 2018, 23, E2661. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Rad, J.; Quispe, C.; Butnariu, M.; Rotariu, L.S.; Sytar, O.; Sestito, S.; Rapposelli, S.; Akram, M.; Iqbal, M.; Krishna, A.; et al. Chitosan Nanoparticles as a Promising Tool in Nanomedicine with Particular Emphasis on Oncological Treatment. Cancer Cell Int. 2021, 21, 318. [Google Scholar] [CrossRef]
- Zhang, Z.; Cai, H.; Liu, Z.; Yao, P. Effective Enhancement of Hypoglycemic Effect of Insulin by Liver-Targeted Nanoparticles Containing Cholic Acid-Modified Chitosan Derivative. Mol. Pharm. 2016, 13, 2433–2442. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.; Xu, G.; Yao, P. Liver-Targeted Delivery of Insulin-Loaded Nanoparticles via Enterohepatic Circulation of Bile Acids. Drug Deliv. 2018, 25, 1224–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Bin, W.; Tu, B.; Li, X.; Wang, W.; Liao, S.; Sun, C. A Delivery System for Oral Administration of Proteins/Peptides Through Bile Acid Transport Channels. J. Pharm. Sci. 2019, 108, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, K.; Ganguly, K.; Kulkarni, A.R.; Rudzinski, W.E.; Krauss, L.; Nadagouda, M.N.; Aminabhavi, T.M. Oral Insulin Delivery Using Deoxycholic Acid Conjugated PEGylated Polyhydroxybutyrate Co-Polymeric Nanoparticles. Nanomedicine 2015, 10, 1569–1583. [Google Scholar] [CrossRef]
- Kemp, M.M.; Linhardt, R.J. Heparin-Based Nanoparticles. Wiley Interdiscip. Rev. NanoMed. NanoBiotechnol. 2010, 2, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Khatun, Z.; Nurunnabi, M.; Reeck, G.R.; Cho, K.J.; Lee, Y.-K. Oral Delivery of Taurocholic Acid Linked Heparin-Docetaxel Conjugates for Cancer Therapy. J. Control. Release 2013, 170, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Khatun, Z.; Nurunnabi, M.; Cho, K.J.; Byun, Y.; Bae, Y.H.; Lee, Y. Oral Absorption Mechanism and Anti-Angiogenesis Effect of Taurocholic Acid-Linked Heparin-Docetaxel Conjugates. J. Control. Release 2014, 177, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, D.; Xu, J.; Wang, J.; Wei, Q.; Xiong, S. Bile Acid Transporter Mediated STC/Soluplus Self-Assembled Hybrid Nanoparticles for Enhancing the Oral Drug Bioavailability. Int. J. Pharm. 2020, 579, 119120. [Google Scholar] [CrossRef]
- Kang, S.H.; Revuri, V.; Lee, S.-J.; Cho, S.; Park, I.-K.; Cho, K.J.; Bae, W.K.; Lee, Y.-K. Oral SiRNA Delivery to Treat Colorectal Liver Metastases. ACS Nano 2017, 11, 10417–10429. [Google Scholar] [CrossRef]
- Suzuki, K.; Kim, K.S.; Bae, Y.H. Long-Term Oral Administration of Exendin-4 to Control Type 2 Diabetes in a Rat Model. J. Control. Release 2019, 294, 259–267. [Google Scholar] [CrossRef]
- Nałęcz, K.A. Solute Carriers in the Blood-Brain Barier: Safety in Abundance. NeuroChem. Res. 2017, 42, 795–809. [Google Scholar] [CrossRef]
- Inazu, M. Functional Expression of Choline Transporters in the Blood-Brain Barrier. Nutrients 2019, 11, E2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, T.; Haga, T.; Kanai, Y.; Endou, H.; Ishihara, T.; Katsura, I. Identification and Characterization of the High-Affinity Choline Transporter. Nat. NeuroSci. 2000, 3, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.; George, R.L.; Bridges, C.C.; Prasad, P.D.; Ganapathy, V. Transport of Choline and Its Relationship to the Expression of the Organic Cation Transporters in a Rat Brain Microvessel Endothelial Cell Line (RBE4). Biochim. BioPhys. Acta 2001, 1512, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwao, B.; Yara, M.; Hara, N.; Kawai, Y.; Yamanaka, T.; Nishihara, H.; Inoue, T.; Inazu, M. Functional Expression of Choline Transporter Like-Protein 1 (CTL1) and CTL2 in Human Brain Microvascular Endothelial Cells. Neurochem. Int. 2016, 93, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Inazu, M.; Takeda, H.; Matsumiya, T. Molecular and Functional Characterization of an Na+-Independent Choline Transporter in Rat Astrocytes. J. NeuroChem. 2005, 94, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.D.; Lockman, P.R. The Blood-Brain Barrier Choline Transporter as a Brain Drug Delivery Vector. Life Sci. 2003, 73, 1609–1615. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Lockman, P.R.; McAfee, J.H.; Fitzpatrick, K.T.; Van der Schyf, C.J.; Allen, D.D. Molecular Modeling Studies on the Active Binding Site of the Blood-Brain Barrier Choline Transporter. BioOrg. Med. Chem. Lett. 2004, 14, 3085–3092. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Lockman, P.R.; Nguyen, T.H.; Van der Schyf, C.J.; Crooks, P.A.; Dwoskin, L.P.; Allen, D.D. 3D-QSAR Study of Bis-Azaaromatic Quaternary Ammonium Analogs at the Blood-Brain Barrier Choline Transporter. BioOrg. Med. Chem. 2005, 13, 4253–4261. [Google Scholar] [CrossRef]
- Zheng, G.; Zhang, Z.; Lockman, P.R.; Geldenhuys, W.J.; Allen, D.D.; Dwoskin, L.P.; Crooks, P.A. Bis-Azaaromatic Quaternary Ammonium Salts as Ligands for the Blood-Brain Barrier Choline Transporter. BioOrg. Med. Chem. Lett. 2010, 20, 3208–3210. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhou, L.; Ye, D.; Huang, S.; Shao, K.; Huang, R.; Han, L.; Liu, Y.; Liu, S.; Ye, L.; et al. Choline-Derivate-Modified Nanoparticles for Brain-Targeting Gene Delivery. Adv. Mater. 2011, 23, 4516–4520. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, S.; Shao, K.; Liu, Y.; An, S.; Kuang, Y.; Guo, Y.; Ma, H.; Wang, X.; Jiang, C. A Choline Derivate-Modified Nanoprobe for Glioma Diagnosis Using MRI. Sci. Rep. 2013, 3, 1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Guo, Y.; Kuang, Y.; An, S.; Ma, H.; Jiang, C. Choline Transporter-Targeting and Co-Delivery System for Glioma Therapy. Biomaterials 2013, 34, 9142–9148. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, H.; Zhang, Y.; Jiang, X.; Guo, Y.; An, S.; Ma, H.; He, X.; Jiang, C. Choline Derivate-Modified Doxorubicin Loaded Micelle for Glioma Therapy. ACS Appl. Mater. Interfaces 2015, 7, 21589–21601. [Google Scholar] [CrossRef]
- Bürzle, M.; Suzuki, Y.; Ackermann, D.; Miyazaki, H.; Maeda, N.; Clémençon, B.; Burrier, R.; Hediger, M.A. The Sodium-Dependent Ascorbic Acid Transporter Family SLC23. Mol. Aspects Med. 2013, 34, 436–454. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A Family of Mammalian Na+-Dependent L-Ascorbic Acid Transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Ulloa, V.; Saldivia, N.; Ferrada, L.; Salazar, K.; Martínez, F.; Silva-Alvarez, C.; Magdalena, R.; Oviedo, M.J.; Montecinos, H.; Torres-Vergara, P.; et al. Basal Sodium-Dependent Vitamin C Transporter 2 Polarization in Choroid Plexus Explant Cells in Normal or Scorbutic Conditions. Sci. Rep. 2019, 9, 14422. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, S.; Pappalardo, J.S.; Sawant, R.R.; Musacchio, T.; Rockwell, K.; Caliceti, P.; Torchilin, V.P. Targeting Glioma Cells in Vitro with Ascorbate-Conjugated Pharmaceutical Nanocarriers. Bioconjug. Chem. 2009, 20, 2348–2355. [Google Scholar] [CrossRef]
- Cioffi, N.; Losito, I.; Terzano, R.; Zambonin, C.G. An Electrospray Ionization Ion Trap Mass Spectrometric (ESI-MS-MSn) Study of Dehydroascorbic Acid Hydrolysis at Neutral PH. Analyst 2000, 125, 2244–2248. [Google Scholar] [CrossRef]
- Miyata, H.; Toyoda, Y.; Takada, T.; Hiragi, T.; Kubota, Y.; Shigesawa, R.; Koyama, R.; Ikegaya, Y.; Suzuki, H. Identification of an Exporter That Regulates Vitamin C Supply from Blood to the Brain. iScience 2022, 25, 103642. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L. Autoradiographic Studies on the Distribution of C14-Labelled Ascorbic Acid and Dehydroascorbic Acid. Acta Physiol. Scand. 1966, 70, 1–83. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Welch, R.W.; Garraffo, H.M.; Ge, P.; Lu, S.F.; Crossman, A.T.; Kirk, K.L.; Levine, M. Specificity of Ascorbate Analogs for Ascorbate Transport. Synthesis and Detection of [(125)I]6-Deoxy-6-Iodo-L-Ascorbic Acid and Characterization of Its Ascorbate-Specific Transport Properties. J. Biol. Chem. 1999, 274, 23215–23222. [Google Scholar] [CrossRef] [Green Version]
- Corpe, C.P.; Lee, J.-H.; Kwon, O.; Eck, P.; Narayanan, J.; Kirk, K.L.; Levine, M. 6-Bromo-6-Deoxy-L-Ascorbic Acid: An Ascorbate Analog Specific for Na+-Dependent Vitamin C Transporter but Not Glucose Transporter Pathways. J. Biol. Chem. 2005, 280, 5211–5220. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Kino, T.; Yamamoto, F.; Kaneshiro, T.; Mukai, T.; Maeda, M. Ascorbate Analogs for Use in Medical Imaging: Synthesis and Radical Scavenging Activity of 5-O-(4’-Iodobenzyl)-L-Ascorbic Acid. Chem. Pharm. Bull. 2007, 55, 1700–1703. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, F.; Sasaki, S.; Maeda, M. Positron Labeled Antioxidants: Synthesis and Tissue Biodistribution of 6-Deoxy-6-[18F]Fluoro-L-Ascorbic Acid. Int. J. Rad. Appl. Instrum. A 1992, 43, 633–639. [Google Scholar] [CrossRef]
- Yamamoto, F.; Kuwano, E.; Kaneshiro, T.; Sasaki, S.; Maeda, M. 125I-Labeled 2-O- and 3-O-m-Iodobenzyl, and 6-O-m-Iodophenyl Derivatives of L-Ascorbic Acid: Synthesis and Preliminary Tissue Distribution. J. Label. Compd. Radiopharm. 2003, 46, 737–750. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Scaglianti, M.; Vitali, F.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; Prasad, P.; et al. Transporter-Mediated Effects of Diclofenamic Acid and Its Ascorbyl pro-Drug in the in Vivo Neurotropic Activity of Ascorbyl Nipecotic Acid Conjugate. J. Pharm. Sci. 2004, 93, 78–85. [Google Scholar] [CrossRef]
- Manfredini, S.; Pavan, B.; Vertuani, S.; Scaglianti, M.; Compagnone, D.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; Prasad, P.; et al. Design, Synthesis and Activity of Ascorbic Acid Prodrugs of Nipecotic, Kynurenic and Diclophenamic Acids, Liable to Increase Neurotropic Activity. J. Med. Chem. 2002, 45, 559–562. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Pavan, B.; Vertuani, S.; Vitali, F.; Scaglianti, M.; Bortolotti, F.; Biondi, C.; Scatturin, A.; Tanganelli, S.; Ferraro, L.; et al. Ascorbic and 6-Br-Ascorbic Acid Conjugates as a Tool to Increase the Therapeutic Effects of Potentially Central Active Drugs. Eur. J. Pharm. Sci. 2005, 24, 259–269. [Google Scholar] [CrossRef]
- Quéléver, G.; Kachidian, P.; Melon, C.; Garino, C.; Laras, Y.; Pietrancosta, N.; Sheha, M.; Louis Kraus, J. Enhanced Delivery of Gamma-Secretase Inhibitor DAPT into the Brain via an Ascorbic Acid Mediated Strategy. Org. Biomol. Chem. 2005, 3, 2450–2457. [Google Scholar] [CrossRef]
- Li, L.; Tuo, J.; Xie, Y.; Huang, M.; Huang, M.; Pi, R.; Hu, H. Preparation, Transportation Mechanisms and Brain-Targeting Evaluation in Vivo of a Chemical Delivery System Exploiting the Blood-Cerebrospinal Fluid Barrier. J. Drug Target. 2014, 22, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Gajbhiye, K.R.; Gajbhiye, V.; Siddiqui, I.A.; Pilla, S.; Soni, V. Ascorbic Acid Tethered Polymeric Nanoparticles Enable Efficient Brain Delivery of Galantamine: An in Vitro-in Vivo Study. Sci. Rep. 2017, 7, 11086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, P.D.; Wang, H.; Kekuda, R.; Fujita, T.; Fei, Y.J.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V. Cloning and Functional Expression of a CDNA Encoding a Mammalian Sodium-Dependent Vitamin Transporter Mediating the Uptake of Pantothenate, Biotin, and Lipoate. J. Biol. Chem. 1998, 273, 7501–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, P.D.; Wang, H.; Huang, W.; Fei, Y.J.; Leibach, F.H.; Devoe, L.D.; Ganapathy, V. Molecular and Functional Characterization of the Intestinal Na+-Dependent Multivitamin Transporter. Arch. BioChem. BioPhys. 1999, 366, 95–106. [Google Scholar] [CrossRef]
- Chen, L.L.; Frankel, A.D.; Harder, J.L.; Fawell, S.; Barsoum, J.; Pepinsky, B. Increased Cellular Uptake of the Human Immunodeficiency Virus-1 Tat Protein after Modification with Biotin. Anal. BioChem. 1995, 227, 168–175. [Google Scholar] [CrossRef]
- Choudhury, I.; Wang, J.; Rabson, A.B.; Stein, S.; Pooyan, S.; Stein, S.; Leibowitz, M.J. Inhibition of HIV-1 Replication by a Tat RNA-Binding Domain Peptide Analog. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 17, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, I.; Wang, J.; Stein, S.; Rabson, A.; Leibowitz, M.J. Translational Effects of Peptide Antagonists of Tat Protein of Human Immunodeficiency Virus Type 1. J. Gen. Virol. 1999, 80 Pt 3, 777–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, S.; Pooyan, S.; Stein, S.; Prasad, P.D.; Wang, J.; Leibowitz, M.J.; Ganapathy, V.; Sinko, P.J. Targeting the Sodium-Dependent Multivitamin Transporter (SMVT) for Improving the Oral Absorption Properties of a Retro-Inverso Tat Nonapeptide. Pharm. Res. 2001, 18, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Qiu, B.; Pooyan, S.; Zhang, G.; Stein, S.; Leibowitz, M.J.; Sinko, P.J. Targeted PEG-Based Bioconjugates Enhance the Cellular Uptake and Transport of a HIV-1 TAT Nonapeptide. J. Control. Release 2001, 77, 199–212. [Google Scholar] [CrossRef]
- Minko, T.; Paranjpe, P.V.; Qiu, B.; Lalloo, A.; Won, R.; Stein, S.; Sinko, P.J. Enhancing the Anticancer Efficacy of Camptothecin Using Biotinylated Poly(Ethylene Glycol) Conjugates in Sensitive and Multidrug-Resistant Human Ovarian Carcinoma Cells. Cancer Chemother. Pharmacol. 2002, 50, 143–150. [Google Scholar] [CrossRef]
- Vadlapudi, A.D.; Vadlapatla, R.K.; Kwatra, D.; Earla, R.; Samanta, S.K.; Pal, D.; Mitra, A.K. Targeted Lipid Based Drug Conjugates: A Novel Strategy for Drug Delivery. Int. J. Pharm. 2012, 434, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Vadlapudi, A.D.; Vadlapatla, R.K.; Earla, R.; Sirimulla, S.; Bailey, J.B.; Pal, D.; Mitra, A.K. Novel Biotinylated Lipid Prodrugs of Acyclovir for the Treatment of Herpetic Keratitis (HK): Transporter Recognition, Tissue Stability and Antiviral Activity. Pharm. Res. 2013, 30, 2063–2076. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, J.; Lu, Y.; He, W.; Li, X.; Wu, W. Biotinylated Liposomes as Potential Carriers for the Oral Delivery of Insulin. Nanomedicine 2014, 10, 167–176. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, X.; Ye, Y.; Zhang, T.; Wang, H.; Ma, Z.; Wu, B. Nanostructured Lipid Carriers Used for Oral Delivery of Oridonin: An Effect of Ligand Modification on Absorption. Int. J. Pharm. 2015, 479, 391–398. [Google Scholar] [CrossRef]
- Russell-Jones, G.; McTavish, K.; McEwan, J.; Rice, J.; Nowotnik, D. Vitamin-Mediated Targeting as a Potential Mechanism to Increase Drug Uptake by Tumours. J. InOrg. BioChem. 2004, 98, 1625–1633. [Google Scholar] [CrossRef]
- Russell-Jones, G.; McTavish, K.; McEwan, J. Preliminary Studies on the Selective Accumulation of Vitamin-Targeted Polymers within Tumors. J. Drug Target. 2011, 19, 133–139. [Google Scholar] [CrossRef]
- Maiti, S.; Paira, P. Biotin Conjugated Organic Molecules and Proteins for Cancer Therapy: A Review. Eur. J. Med. Chem. 2018, 145, 206–223. [Google Scholar] [CrossRef]
- Na, K.; Bum Lee, T.; Park, K.-H.; Shin, E.K.; Lee, Y.-B.; Choi, H.-K. Self-Assembled Nanoparticles of Hydrophobically-Modified Polysaccharide Bearing Vitamin H as a Targeted Anti-Cancer Drug Delivery System. Eur. J. Pharm. Sci. 2003, 18, 165–173. [Google Scholar] [CrossRef]
- Krueger, A.; Stegk, J.; Liang, Y.; Lu, L.; Jarre, G. Biotinylated Nanodiamond: Simple and Efficient Functionalization of Detonation Diamond. Langmuir 2008, 24, 4200–4204. [Google Scholar] [CrossRef]
- Yang, W.; Cheng, Y.; Xu, T.; Wang, X.; Wen, L.-P. Targeting Cancer Cells with Biotin-Dendrimer Conjugates. Eur. J. Med. Chem. 2009, 44, 862–868. [Google Scholar] [CrossRef]
- Yellepeddi, V.K.; Kumar, A.; Palakurthi, S. Biotinylated Poly(Amido)Amine (PAMAM) Dendrimers as Carriers for Drug Delivery to Ovarian Cancer Cells in Vitro. Anticancer. Res. 2009, 29, 2933–2943. [Google Scholar]
- Yellepeddi, V.K.; Kumar, A.; Maher, D.M.; Chauhan, S.C.; Vangara, K.K.; Palakurthi, S. Biotinylated PAMAM. Dendrimers for Intracellular Delivery of Cisplatin to Ovarian Cancer: Role of SMVT. Anticancer. Res. 2011, 31, 897–906. [Google Scholar]
- Kim, J.H.; Li, Y.; Kim, M.S.; Kang, S.W.; Jeong, J.H.; Lee, D.S. Synthesis and Evaluation of Biotin-Conjugated PH-Responsive Polymeric Micelles as Drug Carriers. Int. J. Pharm. 2012, 427, 435–442. [Google Scholar] [CrossRef]
- Aleandri, S.; Bandera, D.; Mezzenga, R.; Landau, E.M. Biotinylated Cubosomes: A Versatile Tool for Active Targeting and Codelivery of Paclitaxel and a Fluorescein-Based Lipid Dye. Langmuir 2015, 31, 12770–12776. [Google Scholar] [CrossRef]
- Morral-Ruíz, G.; Melgar-Lesmes, P.; López-Vicente, A.; Solans, C.; García-Celma, M.J. Biotinylated Polyurethane-Urea Nanoparticles for Targeted Theranostics in Human Hepatocellular Carcinoma. Nano Res. 2015, 8, 1729–1745. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.R.; Jain, N.K. Biotinylated Methotrexate Loaded Erythrocytes for Enhanced Liver Uptake. “A Study on the Rat.” Int. J. Pharm. 2002, 231, 145–153. [Google Scholar] [CrossRef]
- Pandey, S.; Garg, P.; Lim, K.T.; Kim, J.; Choung, Y.-H.; Choi, Y.-J.; Choung, P.-H.; Cho, C.-S.; Chung, J.H. The Efficiency of Membrane Transport of Vitamin B6 Coupled to Poly(Ester Amine) Gene Transporter and Transfection in Cancer Cells. Biomaterials 2013, 34, 3716–3728. [Google Scholar] [CrossRef]
- Hellmann, H.; Mooney, S. Vitamin B6: A Molecule for Human Health? Molecules 2010, 15, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Parsa, S.; Ortega-Molina, A.; Ying, H.-Y.; Jiang, M.; Teater, M.; Wang, J.; Zhao, C.; Reznik, E.; Pasion, J.P.; Kuo, D.; et al. The Serine Hydroxymethyltransferase-2 (SHMT2) Initiates Lymphoma Development through Epigenetic Tumor Suppressor Silencing. Nat. Cancer 2020, 1, 653–664. [Google Scholar] [CrossRef]
- Yamashiro, T.; Yasujima, T.; Said, H.M.; Yuasa, H. PH-Dependent Pyridoxine Transport by SLC19A2 and SLC19A3: Implications for Absorption in Acidic Microclimates. J. Biol. Chem. 2020, 295, 16998–17008. [Google Scholar] [CrossRef]
- Visentin, M.; Diop-Bove, N.; Zhao, R.; Goldman, I.D. The Intestinal Absorption of Folates. Annu Rev. Physiol. 2014, 76, 251–274. [Google Scholar] [CrossRef] [Green Version]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an Intestinal Folate Transporter and the Molecular Basis for Hereditary Folate Malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Aluri, S.; Goldman, I.D. The Proton-Coupled Folate Transporter (PCFT-SLC46A1) and the Syndrome of Systemic and Cerebral Folate Deficiency of Infancy: Hereditary Folate Malabsorption. Mol. Aspects Med. 2017, 53, 57–72. [Google Scholar] [CrossRef] [Green Version]
- Grapp, M.; Wrede, A.; Schweizer, M.; Hüwel, S.; Galla, H.-J.; Snaidero, N.; Simons, M.; Bückers, J.; Low, P.S.; Urlaub, H.; et al. Choroid Plexus Transcytosis and Exosome Shuttling Deliver Folate into Brain Parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Min, S.H.; Wang, Y.; Campanella, E.; Low, P.S.; Goldman, I.D. A Role for the Proton-Coupled Folate Transporter (PCFT-SLC46A1) in Folate Receptor-Mediated Endocytosis. J. Biol. Chem. 2009, 284, 4267–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Zhang, F.; Yang, C.; Wang, L.; Sung, J.; Garg, P.; Zhang, M.; Merlin, D. Oral Targeted Delivery by Nanoparticles Enhances Efficacy of an Hsp90 Inhibitor by Reducing Systemic Exposure in Murine Models of Colitis and Colitis-Associated Cancer. J. Crohns Colitis 2020, 14, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Yu, M.; Wang, A.; Qiu, Y.; Fan, W.; Hovgaard, L.; Yang, M.; Li, Y.; Wang, R.; et al. The Upregulated Intestinal Folate Transporters Direct the Uptake of Ligand-Modified Nanoparticles for Enhanced Oral Insulin Delivery. Acta Pharm. Sin. B 2022, 12, 1460–1472. [Google Scholar] [CrossRef]
- Kamen, B.A.; Smith, A.K. A Review of Folate Receptor Alpha Cycling and 5-Methyltetrahydrofolate Accumulation with an Emphasis on Cell Models in Vitro. Adv. Drug Deliv. Rev. 2004, 56, 1085–1097. [Google Scholar] [CrossRef]
- Zheng, Y.; Cai, Z.; Song, X.; Chen, Q.; Bi, Y.; Li, Y.; Hou, S. Preparation and Characterization of Folate Conjugated N-Trimethyl Chitosan Nanoparticles as Protein Carrier Targeting Folate Receptor: In Vitro Studies. J. Drug Target. 2009, 17, 294–303. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Ma, D.; Tang, H.; Tan, L.; Xie, Q.; Yao, S. Biocompatible Multi-Walled Carbon Nanotube-Chitosan-Folic Acid Nanoparticle Hybrids as GFP Gene Delivery Materials. Colloids Surf. B Biointerfaces 2013, 111, 224–231. [Google Scholar] [CrossRef]
- Kuplennik, N.; Lang, K.; Steinfeld, R.; Sosnik, A. Folate Receptor α-Modified Nanoparticles for Targeting of the Central Nervous System. ACS Appl. Mater. Interfaces 2019, 11, 39633–39647. [Google Scholar] [CrossRef]
- Kur, E.; Mecklenburg, N.; Cabrera, R.M.; Willnow, T.E.; Hammes, A. LRP2 Mediates Folate Uptake in the Developing Neural Tube. J. Cell Sci. 2014, 127, 2261–2268. [Google Scholar] [CrossRef] [Green Version]
- Varma, M.V.; Ambler, C.M.; Ullah, M.; Rotter, C.J.; Sun, H.; Litchfield, J.; Fenner, K.S.; El-Kattan, A.F. Targeting Intestinal Transporters for Optimizing Oral Drug Absorption. Curr. Drug Metab. 2010, 11, 730–742. [Google Scholar] [CrossRef]
- Dahan, A.; Khamis, M.; Agbaria, R.; Karaman, R. Targeted Prodrugs in Oral Drug Delivery: The Modern Molecular Biopharmaceutical Approach. Expert Opin. Drug Deliv. 2012, 9, 1001–1013. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, J.; Sun, Y.; Wang, Y.; He, Z. Prodrug Design Targeting Intestinal PepT1 for Improved Oral Absorption: Design and Performance. Curr. Drug Metab. 2013, 14, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Zimmermann, E.M.; Ben-Shabat, S. Modern Prodrug Design for Targeted Oral Drug Delivery. Molecules 2014, 19, 16489–16505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, L.M.; Orr, G.F.; de Miranda, P.; Bumette, T.; Krenitsky, T.A. Amino Acid Ester Prodrugs of Acyclovir. Antivir. Chem. Chemother. 1992, 3, 157–164. [Google Scholar] [CrossRef]
- Weller, S.; Blum, M.R.; Doucette, M.; Burnette, T.; Cederberg, D.M.; de Miranda, P.; Smiley, M.L. Pharmacokinetics of the Acyclovir Pro-Drug Valaciclovir after Escalating Single- and Multiple-Dose Administration to Normal Volunteers. Clin. Pharmacol. Ther. 1993, 54, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.J.; Kanai, Y.; Nussberger, S.; Ganapathy, V.; Leibach, F.H.; Romero, M.F.; Singh, S.K.; Boron, W.F.; Hediger, M.A. Expression Cloning of a Mammalian Proton-Coupled Oligopeptide Transporter. Nature 1994, 368, 563–566. [Google Scholar] [CrossRef]
- Han, H.; de Vrueh, R.L.; Rhie, J.K.; Covitz, K.M.; Smith, P.L.; Lee, C.P.; Oh, D.M.; Sadée, W.; Amidon, G.L. 5’-Amino Acid Esters of Antiviral Nucleosides, Acyclovir, and AZT Are Absorbed by the Intestinal PEPT1 Peptide Transporter. Pharm. Res. 1998, 15, 1154–1159. [Google Scholar] [CrossRef]
- Smith, D.E.; Clémençon, B.; Hediger, M.A. Proton-Coupled Oligopeptide Transporter Family SLC15: Physiological, Pharmacological and Pathological Implications. Mol. Aspects Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liang, R.; Ramamoorthy, S.; Fei, Y.J.; Ganapathy, M.E.; Hediger, M.A.; Ganapathy, V.; Leibach, F.H. Molecular Cloning of PEPT 2, a New Member of the H+/Peptide Cotransporter Family, from Human Kidney. Biochim. BioPhys. Acta 1995, 1235, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Berger, U.V.; Hediger, M.A. Distribution of Peptide Transporter PEPT2 MRNA in the Rat Nervous System. ANat. Embryol. 1999, 199, 439–449. [Google Scholar] [CrossRef]
- Verri, T.; Barca, A.; Pisani, P.; Piccinni, B.; Storelli, C.; Romano, A. Di- and Tripeptide Transport in Vertebrates: The Contribution of Teleost Fish Models. J. Comp. Physiol. B 2017, 187, 395–462. [Google Scholar] [CrossRef]
- Nakamura, N.; Lill, J.R.; Phung, Q.; Jiang, Z.; Bakalarski, C.; de Mazière, A.; Klumperman, J.; Schlatter, M.; Delamarre, L.; Mellman, I. Endosomes Are Specialized Platforms for Bacterial Sensing and NOD2 Signalling. Nature 2014, 509, 240–244. [Google Scholar] [CrossRef]
- Song, F.; Yi, Y.; Li, C.; Hu, Y.; Wang, J.; Smith, D.E.; Jiang, H. Regulation and Biological Role of the Peptide/Histidine Transporter SLC15A3 in Toll-like Receptor-Mediated Inflammatory Responses in Macrophage. Cell Death Dis. 2018, 9, 770. [Google Scholar] [CrossRef] [Green Version]
- Rimann, I.; Gonzalez-Quintial, R.; Baccala, R.; Kiosses, W.B.; Teijaro, J.R.; Parker, C.G.; Li, X.; Beutler, B.; Kono, D.H.; Theofilopoulos, A.N. The Solute Carrier SLC15A4 Is Required for Optimal Trafficking of Nucleic Acid-Sensing TLRs and Ligands to Endolysosomes. Proc. Natl. Acad. Sci. USA 2022, 119, e2200544119. [Google Scholar] [CrossRef]
- Anand, B.S.; Patel, J.; Mitra, A.K. Interactions of the Dipeptide Ester Prodrugs of Acyclovir with the Intestinal Oligopeptide Transporter: Competitive Inhibition of Glycylsarcosine Transport in Human Intestinal Cell Line-Caco-2. J. Pharmacol. Exp. Ther. 2003, 304, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Anand, B.S.; Katragadda, S.; Mitra, A.K. Pharmacokinetics of Novel Dipeptide Ester Prodrugs of Acyclovir after Oral Administration: Intestinal Absorption and Liver Metabolism. J. Pharmacol. Exp. Ther. 2004, 311, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, A.E.; Friedrichsen, G.M.; Sørensen, A.H.; Andersen, R.; Nielsen, C.U.; Brodin, B.; Begtrup, M.; Frokjaer, S.; Steffansen, B. Prodrugs of Purine and Pyrimidine Analogues for the Intestinal Di/Tri-Peptide Transporter PepT1: Affinity for HPepT1 in Caco-2 Cells, Drug Release in Aqueous Media and in Vitro Metabolism. J. Control. Release 2003, 86, 279–292. [Google Scholar] [CrossRef]
- Thomsen, A.E.; Christensen, M.S.; Bagger, M.A.; Steffansen, B. Acyclovir Prodrug for the Intestinal Di/Tri-Peptide Transporter PEPT1: Comparison of in Vivo Bioavailability in Rats and Transport in Caco-2 Cells. Eur. J. Pharm. Sci. 2004, 23, 319–325. [Google Scholar] [CrossRef]
- Sugawara, M.; Huang, W.; Fei, Y.J.; Leibach, F.H.; Ganapathy, V.; Ganapathy, M.E. Transport of Valganciclovir, a Ganciclovir Prodrug, via Peptide Transporters PEPT1 and PEPT2. J. Pharm. Sci. 2000, 89, 781–789. [Google Scholar] [CrossRef]
- Li, F.; Hong, L.; Mau, C.-I.; Chan, R.; Hendricks, T.; Dvorak, C.; Yee, C.; Harris, J.; Alfredson, T. Transport of Levovirin Prodrugs in the Human Intestinal Caco-2 Cell Line. J. Pharm. Sci. 2006, 95, 1318–1325. [Google Scholar] [CrossRef]
- Gupta, D.; Varghese Gupta, S.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.-D.; Amidon, G.L. Increasing Oral Absorption of Polar Neuraminidase Inhibitors: A Prodrug Transporter Approach Applied to Oseltamivir Analogue. Mol. Pharm. 2013, 10, 512–522. [Google Scholar] [CrossRef]
- Incecayir, T.; Sun, J.; Tsume, Y.; Xu, H.; Gose, T.; Nakanishi, T.; Tamai, I.; Hilfinger, J.; Lipka, E.; Amidon, G.L. Carrier-Mediated Prodrug Uptake to Improve the Oral Bioavailability of Polar Drugs: An Application to an Oseltamivir Analogue. J. Pharm. Sci. 2016, 105, 925–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.V.; Gupta, D.; Sun, J.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.-D.; Amidon, G.L. Enhancing the Intestinal Membrane Permeability of Zanamivir: A Carrier Mediated Prodrug Approach. Mol. Pharm. 2011, 8, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Gan, W.; Lei, M.; Jiang, W.; Cheng, M.; He, J.; Sun, Q.; Liu, W.; Hu, L.; Jin, Y. PEPT1-Mediated Prodrug Strategy for Oral Delivery of Peramivir. Asian J. Pharm. Sci. 2018, 13, 555–565. [Google Scholar] [CrossRef]
- Mandal, A.; Pal, D.; Mitra, A.K. Circumvention of P-Gp and MRP2 Mediated Efflux of Lopinavir by a Histidine Based Dipeptide Prodrug. Int. J. Pharm. 2016, 512, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.W.; Sala-Rabanal, M.; Krylov, I.S.; Serpi, M.; Kashemirov, B.A.; McKenna, C.E. Serine Side Chain-Linked Peptidomimetic Conjugates of Cyclic HPMPC and HPMPA: Synthesis and Interaction with HPEPT1. Mol. Pharm. 2010, 7, 2349–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Maag, H.; Alfredson, T. Prodrugs of Nucleoside Analogues for Improved Oral Absorption and Tissue Targeting. J. Pharm. Sci. 2008, 97, 1109–1134. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, J.; Shi, S.; Jing, Y.; Yin, S.; Chen, Y.; Li, G.; Xu, Y.; He, Z. Synthesis, Transport and Pharmacokinetics of 5’-Amino Acid Ester Prodrugs of 1-Beta-D-Arabinofuranosylcytosine. Mol. Pharm. 2009, 6, 315–325. [Google Scholar] [CrossRef]
- Li, Y.; Yang, M.; Zhao, Y.; Li, L.; Xu, W. Preparation and in Vitro Evaluation of Amphiphilic Paclitaxel Small Molecule Prodrugs and Enhancement of Oral Absorption. Eur. J. Med. Chem. 2021, 215, 113276. [Google Scholar] [CrossRef]
- Vig, B.S.; Lorenzi, P.J.; Mittal, S.; Landowski, C.P.; Shin, H.-C.; Mosberg, H.I.; Hilfinger, J.M.; Amidon, G.L. Amino Acid Ester Prodrugs of Floxuridine: Synthesis and Effects of Structure, Stereochemistry, and Site of Esterification on the Rate of Hydrolysis. Pharm. Res. 2003, 20, 1381–1388. [Google Scholar] [CrossRef]
- Landowski, C.P.; Vig, B.S.; Song, X.; Amidon, G.L. Targeted Delivery to PEPT1-Overexpressing Cells: Acidic, Basic, and Secondary Floxuridine Amino Acid Ester Prodrugs. Mol. Cancer Ther. 2005, 4, 659–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landowski, C.P.; Song, X.; Lorenzi, P.L.; Hilfinger, J.M.; Amidon, G.L. Floxuridine Amino Acid Ester Prodrugs: Enhancing Caco-2 Permeability and Resistance to Glycosidic Bond Metabolism. Pharm. Res. 2005, 22, 1510–1518. [Google Scholar] [CrossRef]
- Tsume, Y.; Hilfinger, J.M.; Amidon, G.L. Enhanced Cancer Cell Growth Inhibition by Dipeptide Prodrugs of Floxuridine: Increased Transporter Affinity and Metabolic Stability. Mol. Pharm. 2008, 5, 717–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsume, Y.; Vig, B.S.; Sun, J.; Landowski, C.P.; Hilfinger, J.M.; Ramachandran, C.; Amidon, G.L. Enhanced Absorption and Growth Inhibition with Amino Acid Monoester Prodrugs of Floxuridine by Targeting HPEPT1 Transporters. Molecules 2008, 13, 1441–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsume, Y.; Hilfinger, J.M.; Amidon, G.L. Potential of Amino Acid/Dipeptide Monoester Prodrugs of Floxuridine in Facilitating Enhanced Delivery of Active Drug to Interior Sites of Tumors: A Two-Tier Monolayer in Vitro Study. Pharm. Res. 2011, 28, 2575–2588. [Google Scholar] [CrossRef]
- Song, X.; Lorenzi, P.L.; Landowski, C.P.; Vig, B.S.; Hilfinger, J.M.; Amidon, G.L. Amino Acid Ester Prodrugs of the Anticancer Agent Gemcitabine: Synthesis, Bioconversion, Metabolic Bioevasion, and HPEPT1-Mediated Transport. Mol. Pharm. 2005, 2, 157–167. [Google Scholar] [CrossRef]
- Mittal, S.; Song, X.; Vig, B.S.; Landowski, C.P.; Kim, I.; Hilfinger, J.M.; Amidon, G.L. Prolidase, a Potential Enzyme Target for Melanoma: Design of Proline-Containing Dipeptide-like Prodrugs. Mol. Pharm. 2005, 2, 37–46. [Google Scholar] [CrossRef]
- Yuri, T.; Kono, Y.; Okada, T.; Terada, T.; Miyauchi, S.; Fujita, T. Transport Characteristics of 5-Aminosalicylic Acid Derivatives Conjugated with Amino Acids via Human H+-Coupled Oligopeptide Transporter PEPT1. Biol. Pharm. Bull. 2020, 43, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Foley, D.; Bailey, P.; Pieri, M.; Meredith, D. Targeting Ketone Drugs towards Transport by the Intestinal Peptide Transporter, PepT1. Org. BioMol. Chem. 2009, 7, 1064–1067. [Google Scholar] [CrossRef] [Green Version]
- Foley, D.; Pieri, M.; Pettecrew, R.; Price, R.; Miles, S.; Miles, S.; Lam, H.K.; Bailey, P.; Meredith, D. The in Vitro Transport of Model Thiodipeptide Prodrugs Designed to Target the Intestinal Oligopeptide Transporter, PepT1. Org. BioMol. Chem. 2009, 7, 3652–3656. [Google Scholar] [CrossRef]
- Cao, F.; Jia, J.; Yin, Z.; Gao, Y.; Sha, L.; Lai, Y.; Ping, Q.; Zhang, Y. Ethylene Glycol-Linked Amino Acid Diester Prodrugs of Oleanolic Acid for PepT1-Mediated Transport: Synthesis, Intestinal Permeability and Pharmacokinetics. Mol. Pharm. 2012, 9, 2127–2135. [Google Scholar] [CrossRef]
- Cao, F.; Gao, Y.; Wang, M.; Fang, L.; Ping, Q. Propylene Glycol-Linked Amino Acid/Dipeptide Diester Prodrugs of Oleanolic Acid for PepT1-Mediated Transport: Synthesis, Intestinal Permeability, and Pharmacokinetics. Mol. Pharm. 2013, 10, 1378–1387. [Google Scholar] [CrossRef]
- Fang, L.; Wang, M.; Gou, S.; Liu, X.; Zhang, H.; Cao, F. Combination of Amino Acid/Dipeptide with Nitric Oxide Donating Oleanolic Acid Derivatives as PepT1 Targeting Antitumor Prodrugs. J. Med. Chem. 2014, 57, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Gilzad Kohan, H.; Kaur, K.; Jamali, F. Synthesis and Characterization of a New Peptide Prodrug of Glucosamine with Enhanced Gut Permeability. PLoS One 2015, 10, e0126786. [Google Scholar] [CrossRef]
- Tsuji, A.; Tamai, I.; Nakanishi, M.; Amidon, G.L. Mechanism of Absorption of the Dipeptide Alpha-Methyldopa-Phe in Intestinal Brush-Border Membrane Vesicles. Pharm. Res. 1990, 7, 308–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.P.; Lu, H.H.; Lee, J.S.; Cheng, C.Y.; Mah, J.R.; Ku, C.Y.; Hsu, W.; Yen, C.F.; Lin, C.J.; Kuo, H.S. Intestinal Absorption Studies on Peptide Mimetic Alpha-Methyldopa Prodrugs. J. Pharm. Pharmacol. 1996, 48, 270–276. [Google Scholar] [CrossRef]
- Tamai, I.; Nakanishi, T.; Nakahara, H.; Sai, Y.; Ganapathy, V.; Leibach, F.H.; Tsuji, A. Improvement of L-Dopa Absorption by Dipeptidyl Derivation, Utilizing Peptide Transporter PepT1. J. Pharm. Sci. 1998, 87, 1542–1546. [Google Scholar] [CrossRef]
- Wang, C.-L.; Fan, Y.-B.; Lu, H.-H.; Tsai, T.-H.; Tsai, M.-C.; Wang, H.-P. Evidence of D-Phenylglycine as Delivering Tool for Improving L-Dopa Absorption. J. BioMed. Sci. 2010, 17, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya, M.; Tanaka, K.; Tsuchida, Y.; Muto, Y.; Koketsu, M.; Watanabe, K. Increased Bioavailability of Tricin-Amino Acid Derivatives via a Prodrug Approach. J. Med. Chem. 2011, 54, 1529–1536. [Google Scholar] [CrossRef]
- Azzolini, M.; Mattarei, A.; La Spina, M.; Fanin, M.; Chiodarelli, G.; Romio, M.; Zoratti, M.; Paradisi, C.; Biasutto, L. New Natural Amino Acid-Bearing Prodrugs Boost Pterostilbene’s Oral Pharmacokinetic and Distribution Profile. Eur. J. Pharm. BioPharm. 2017, 115, 149–158. [Google Scholar] [CrossRef]
- Ezra, A.; Golomb, G. Administration Routes and Delivery Systems of Bisphosphonates for the Treatment of Bone Resorption. Adv. Drug Deliv. Rev. 2000, 42, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Ezra, A.; Hoffman, A.; Breuer, E.; Alferiev, I.S.; Mönkkönen, J.; El Hanany-Rozen, N.; Weiss, G.; Stepensky, D.; Gati, I.; Cohen, H.; et al. A Peptide Prodrug Approach for Improving Bisphosphonate Oral Absorption. J. Med. Chem. 2000, 43, 3641–3652. [Google Scholar] [CrossRef]
- Foley, D.W.; Pathak, R.B.; Phillips, T.R.; Wilson, G.L.; Bailey, P.D.; Pieri, M.; Senan, A.; Meredith, D. Thiodipeptides Targeting the Intestinal Oligopeptide Transporter as a General Approach to Improving Oral Drug Delivery. Eur. J. Med. Chem. 2018, 156, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, L.; Luo, T.; Zhou, J.; Sun, L.; Xu, Y. Synthesis and Evaluation of a Dipeptide-Drug Conjugate Library as Substrates for PEPT1. ACS Comb. Sci. 2012, 14, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Minhas, G.S.; Newstead, S. Structural Basis for Prodrug Recognition by the SLC15 Family of Proton-Coupled Peptide Transporters. Proc. Natl. Acad. Sci. USA 2019, 116, 804–809. [Google Scholar] [CrossRef] [Green Version]
- Ural-Blimke, Y.; Flayhan, A.; Strauss, J.; Rantos, V.; Bartels, K.; Nielsen, R.; Pardon, E.; Steyaert, J.; Kosinski, J.; Quistgaard, E.M.; et al. Structure of Prototypic Peptide Transporter DtpA from E. Coli in Complex with Valganciclovir ProviDes. Insights into Drug Binding of Human PepT1. J. Am. Chem. Soc. 2019, 141, 2404–2412. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.L.; Deme, J.C.; Wu, Z.; Kuteyi, G.; Huo, J.; Owens, R.J.; Biggin, P.C.; Lea, S.M.; Newstead, S. Cryo-EM Structure of PepT2 Reveals Structural Basis for Proton-Coupled Peptide and Prodrug Transport in Mammals. Sci. Adv. 2021, 7, eabh3355. [Google Scholar] [CrossRef]
- Lyons, J.A.; Parker, J.L.; Solcan, N.; Brinth, A.; Li, D.; Shah, S.T.A.; Caffrey, M.; Newstead, S. Structural Basis for Polyspecificity in the POT Family of Proton-Coupled Oligopeptide Transporters. EMBO Rep. 2014, 15, 886–893. [Google Scholar] [CrossRef]
- Martinez Molledo, M.; Quistgaard, E.M.; Flayhan, A.; Pieprzyk, J.; Löw, C. Multispecific Substrate Recognition in a Proton-Dependent Oligopeptide Transporter. Structure 2018, 26, 467–476.e4. [Google Scholar] [CrossRef] [Green Version]
- Samsudin, F.; Parker, J.L.; Sansom, M.S.P.; Newstead, S.; Fowler, P.W. Accurate Prediction of Ligand Affinities for a Proton-Dependent Oligopeptide Transporter. Cell Chem. Biol. 2016, 23, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Meredith, D.; Temple, C.S.; Guha, N.; Sword, C.J.; Boyd, C.A.; Collier, I.D.; Morgan, K.M.; Bailey, P.D. Modified Amino Acids and Peptides as Substrates for the Intestinal Peptide Transporter PepT1. Eur. J. BioChem. 2000, 267, 3723–3728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, F.; Will, J.; Amasheh, S.; Clauss, W.; Ahlbrecht, H.; Daniel, H. Minimal Molecular Determinants of Substrates for Recognition by the Intestinal Peptide Transporter. J. Biol. Chem. 1998, 273, 23211–23218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minhas, G.S.; Newstead, S. Recent Advances in Understanding Prodrug Transport through the SLC15 Family of Proton-Coupled Transporters. BioChem. Soc. Trans 2020, 48, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Våbenø, J.; Lejon, T.; Nielsen, C.U.; Steffansen, B.; Chen, W.; Ouyang, H.; Borchardt, R.T.; Luthman, K. Phe-Gly Dipeptidomimetics Designed for the Di-/Tripeptide Transporters PEPT1 and PEPT2: Synthesis and Biological Investigations. J. Med. Chem. 2004, 47, 1060–1069. [Google Scholar] [CrossRef]
- Bueno, A.B.; Collado, I.; de Dios, A.; Domínguez, C.; Martín, J.A.; Martín, L.M.; Martínez-Grau, M.A.; Montero, C.; Pedregal, C.; Catlow, J.; et al. Dipeptides as Effective Prodrugs of the Unnatural Amino Acid (+)-2-Aminobicyclo[3.1.0]Hexane-2,6-Dicarboxylic Acid (LY354740), a Selective Group II Metabotropic Glutamate Receptor Agonist. J. Med. Chem. 2005, 48, 5305–5320. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Tian, C.; Wang, M.; Huang, D.; Wei, W.; Liu, Y.; Li, L.; Sun, B.; Kou, L.; Kan, Q.; et al. Dipeptide-Modified Nanoparticles to Facilitate Oral Docetaxel Delivery: New Insights into PepT1-Mediated Targeting Strategy. Drug Deliv. 2018, 25, 1403–1413. [Google Scholar] [CrossRef]
- Gourdon, B.; Chemin, C.; Moreau, A.; Arnauld, T.; Baumy, P.; Cisternino, S.; Péan, J.-M.; Declèves, X. Functionalized PLA-PEG Nanoparticles Targeting Intestinal Transporter PepT1 for Oral Delivery of Acyclovir. Int. J. Pharm. 2017, 529, 357–370. [Google Scholar] [CrossRef]
- Durán, J.M.; Peral, M.J.; Calonge, M.L.; Ilundáin, A.A. Functional Characterization of Intestinal L-Carnitine Transport. J. Membr. Biol. 2002, 185, 65–74. [Google Scholar] [CrossRef]
- Kato, Y.; Sugiura, M.; Sugiura, T.; Wakayama, T.; Kubo, Y.; Kobayashi, D.; Sai, Y.; Tamai, I.; Iseki, S.; Tsuji, A. Organic Cation/Carnitine Transporter OCTN2 (Slc22a5) Is Responsible for Carnitine Transport across Apical Membranes of Small Intestinal Epithelial Cells in Mouse. Mol. Pharmacol. 2006, 70, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. OCTN Cation Transporters in Health and Disease: Role as Drug Targets and Assay Development. J. BioMol. Screen 2013, 18, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Hatanaka, T.; Huang, W.; Prasad, P.D.; Leibach, F.H.; Ganapathy, M.E.; Ganapathy, V. Na+- and Cl--Coupled Active Transport of Carnitine by the Amino Acid Transporter ATB(0,+) from Mouse Colon Expressed in HRPE Cells and Xenopus Oocytes. J. Physiol. 2001, 532, 297–304. [Google Scholar] [CrossRef]
- Taylor, P.M. Absorbing Competition for Carnitine. J. Physiol. 2001, 532, 283. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, H.; Zhao, D.; Ding, D.; Sun, M.; Kou, L.; Luo, C.; Zhang, D.; Yi, X.; Dong, J.; et al. Combination of L-Carnitine with Lipophilic Linkage-Donating Gemcitabine Derivatives as Intestinal Novel Organic Cation Transporter 2-Targeting Oral Prodrugs. J. Med. Chem. 2017, 60, 2552–2561. [Google Scholar] [CrossRef]
- Mo, J.; Shi, S.; Zhang, Q.; Gong, T.; Sun, X.; Zhang, Z. Synthesis, Transport and Mechanism of a Type I Prodrug: L-Carnitine Ester of Prednisolone. Mol. Pharm. 2011, 8, 1629–1640. [Google Scholar] [CrossRef]
- Nakamura, T.; Nakanishi, T.; Haruta, T.; Shirasaka, Y.; Keogh, J.P.; Tamai, I. Transport of Ipratropium, an Anti-Chronic Obstructive Pulmonary Disease Drug, Is Mediated by Organic Cation/Carnitine Transporters in Human Bronchial Epithelial Cells: Implications for Carrier-Mediated Pulmonary Absorption. Mol. Pharm. 2010, 7, 187–195. [Google Scholar] [CrossRef]
- Kou, L.; Yao, Q.; Sun, M.; Wu, C.; Wang, J.; Luo, Q.; Wang, G.; Du, Y.; Fu, Q.; Wang, J.; et al. Cotransporting Ion Is a Trigger for Cellular Endocytosis of Transporter-Targeting Nanoparticles: A Case Study of High-Efficiency SLC22A5 (OCTN2)-Mediated Carnitine-Conjugated Nanoparticles for Oral Delivery of Therapeutic Drugs. Adv. Healthc. Mater. 2017, 6, 1700165. [Google Scholar] [CrossRef] [PubMed]
- Kido, Y.; Tamai, I.; Ohnari, A.; Sai, Y.; Kagami, T.; Nezu, J.; Nikaido, H.; Hashimoto, N.; Asano, M.; Tsuji, A. Functional Relevance of Carnitine Transporter OCTN2 to Brain Distribution of L-Carnitine and Acetyl-L-Carnitine across the Blood-Brain Barrier. J. NeuroChem. 2001, 79, 959–969. [Google Scholar] [CrossRef]
- Inano, A.; Sai, Y.; Nikaido, H.; Hasimoto, N.; Asano, M.; Tsuji, A.; Tamai, I. Acetyl-L-Carnitine Permeability across the Blood-Brain Barrier and Involvement of Carnitine Transporter OCTN2. BioPharm. Drug Dispos. 2003, 24, 357–365. [Google Scholar] [CrossRef]
- Berezowski, V.; Miecz, D.; Marszałek, M.; Bröer, A.; Bröer, S.; Cecchelli, R.; Nałecz, K.A. Involvement of OCTN2 and B0,+ in the Transport of Carnitine through an in Vitro Model of the Blood-Brain Barrier. J. NeuroChem. 2004, 91, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Miecz, D.; Januszewicz, E.; Czeredys, M.; Hinton, B.T.; Berezowski, V.; Cecchelli, R.; Nałecz, K.A. Localization of Organic Cation/Carnitine Transporter (OCTN2) in Cells Forming the Blood-Brain Barrier. J. NeuroChem. 2008, 104, 113–123. [Google Scholar] [CrossRef]
- Napolitano, C.; Scaglianti, M.; Scalambra, E.; Manfredini, S.; Ferraro, L.; Beggiato, S.; Vertuani, S. Carnitine Conjugate of Nipecotic Acid: A New Example of Dual Prodrug. Molecules 2009, 14, 3268–3274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, L.; Yao, Q.; Sivaprakasam, S.; Luo, Q.; Sun, Y.; Fu, Q.; He, Z.; Sun, J.; Ganapathy, V. Dual Targeting of L-Carnitine-Conjugated Nanoparticles to OCTN2 and ATB0,+ to Deliver Chemotherapeutic Agents for Colon Cancer Therapy. Drug Deliv. 2017, 24, 1338–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenbuch, B.; Stieger, B. The SLCO (Former SLC21) Superfamily of Transporters. Mol. Aspects Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, N.F.; Figg, W.D.; Sparreboom, A. Role of the Liver-Specific Transporters OATP1B1 and OATP1B3 in Governing Drug Elimination. Expert Opin. Drug Metab. Toxicol. 2005, 1, 429–445. [Google Scholar] [CrossRef]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The Organic Anion and Cation Transporters of the SLCO and SLC22A Gene Superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [Green Version]
- Pfefferkorn, J.A.; Guzman-Perez, A.; Litchfield, J.; Aiello, R.; Treadway, J.L.; Pettersen, J.; Minich, M.L.; Filipski, K.J.; Jones, C.S.; Tu, M.; et al. Discovery of (S)-6-(3-Cyclopentyl-2-(4-(Trifluoromethyl)-1H-Imidazol-1-Yl)Propanamido)Nicotinic Acid as a Hepatoselective Glucokinase Activator Clinical Candidate for Treating Type 2 Diabetes Mellitus. J. Med. Chem. 2012, 55, 1318–1333. [Google Scholar] [CrossRef]
- Thilagavathi, R.; Hosseini-Zare, M.S.; Malini, M.; Selvam, C. A Comprehensive Review on Glucokinase Activators: Promising Agents for the Treatment of Type 2 Diabetes. Chem. Biol. Drug Des. 2022, 99, 247–263. [Google Scholar] [CrossRef]
- Pfefferkorn, J.A. Strategies for the Design of Hepatoselective Glucokinase Activators to Treat Type 2 Diabetes. Expert Opin. Drug Discov. 2013, 8, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Oballa, R.M.; Belair, L.; Black, W.C.; Bleasby, K.; Chan, C.C.; Desroches, C.; Du, X.; Gordon, R.; Guay, J.; Guiral, S.; et al. Development of a Liver-Targeted Stearoyl-CoA Desaturase (SCD) Inhibitor (MK-8245) to Establish a Therapeutic Window for the Treatment of Diabetes and Dyslipidemia. J. Med. Chem. 2011, 54, 5082–5096. [Google Scholar] [CrossRef]
- Zhang, Z.; Dales, N.A.; Winther, M.D. Opportunities and Challenges in Developing Stearoyl-Coenzyme A Desaturase-1 Inhibitors as Novel Therapeutics for Human Disease. J. Med. Chem. 2014, 57, 5039–5056. [Google Scholar] [CrossRef]
- Powell, D.A.; Black, W.C.; Bleasby, K.; Chan, C.-C.; Deschenes, D.; Gagnon, M.; Gordon, R.; Guay, J.; Guiral, S.; Hafey, M.J.; et al. Nicotinic Acids: Liver-Targeted SCD Inhibitors with Preclinical Anti-Diabetic Efficacy. BioOrg. Med. Chem. Lett. 2011, 21, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.K.; Saksena, S.; Alrefai, W.A.; Sarwar, Z.; Goldstein, J.L.; Carroll, R.E.; Ramaswamy, K.; Dudeja, P.K. Expression and Membrane Localization of MCT Isoforms along the Length of the Human Intestine. Am. J. Physiol. Cell Physiol. 2005, 289, C846–C852. [Google Scholar] [CrossRef] [Green Version]
- Kirat, D.; Inoue, H.; Iwano, H.; Hirayama, K.; Yokota, H.; Taniyama, H.; Kato, S. Monocarboxylate Transporter 1 Gene Expression in the Ovine Gastrointestinal Tract. Vet. J. 2006, 171, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, T.; Takebe, K.; Kato, I.; Karaki, S.-I.; Kuwahara, A. Cellular Expression of Monocarboxylate Transporters (MCT) in the Digestive Tract of the Mouse, Rat, and Humans, with Special Reference to Slc5a8. BioMed. Res. 2006, 27, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhao, L.; Jiang, Q.; Sun, Y.; Zhao, D.; Sun, M.; He, Z.; Sun, J.; Wang, Y. Intestinal OCTN2- and MCT1-Targeted Drug Delivery to Improve Oral Bioavailability. Asian J. Pharm. Sci. 2020, 15, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate Transporters in the Brain and in Cancer. Biochim. BioPhys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cundy, K.C.; Branch, R.; Chernov-Rogan, T.; Dias, T.; Estrada, T.; Hold, K.; Koller, K.; Liu, X.; Mann, A.; Panuwat, M.; et al. XP13512 [(+/-)-1-([(Alpha-Isobutanoyloxyethoxy)Carbonyl] Aminomethyl)-1-Cyclohexane Acetic Acid], a Novel Gabapentin Prodrug: I. Design, Synthesis, Enzymatic Conversion to Gabapentin, and Transport by Intestinal Solute Transporters. J. Pharmacol. Exp. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Sukbuntherng, J.; Luo, W.; Chen, D.; Vu, A.; Tovera, J.; Cundy, K.C. Pharmacokinetics and Tolerability of Single Escalating Doses of Gabapentin Enacarbil: A Randomized-Sequence, Double-Blind, Placebo-Controlled Crossover Study in Healthy Volunteers. Clin. Ther. 2009, 31, 1776–1786. [Google Scholar] [CrossRef]
- Kushida, C.A.; Walters, A.S.; Becker, P.; Thein, S.G.; Perkins, A.T.; Roth, T.; Canafax, D.; Barrett, R.W. XP021 Study Group A Randomized, Double-Blind, Placebo-Controlled, Crossover Study of XP13512/GSK1838262 in the Treatment of Patients with Primary Restless Legs Syndrome. Sleep 2009, 32, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Kushida, C.A.; Becker, P.M.; Ellenbogen, A.L.; Canafax, D.M.; Barrett, R.W. XP052 Study Group Randomized, Double-Blind, Placebo-Controlled Study of XP13512/GSK1838262 in Patients with RLS. Neurology 2009, 72, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, D.; Wang, G.; Jiang, Q.; Guo, M.; Kan, Q.; He, Z.; Sun, J. A Novel Oral Prodrug-Targeting Transporter MCT 1: 5-Fluorouracil-Dicarboxylate Monoester Conjugates. Asian J. Pharm. Sci. 2019, 14, 631–639. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.; Chen, H.; Sun, Y.; Sun, M.; Liu, X.; Jian, W.; He, Z.; Sun, J. A Facile Di-Acid Mono-Amidation Strategy to Prepare Cyclization-Activating Mono-Carboxylate Transporter 1-Targeting Gemcitabine Prodrugs for Enhanced Oral Delivery. Int. J. Pharm. 2020, 573, 118718. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of Monocarboxylate Transporters in Human Cancers: State of the Art. J. Bioenerg. BioMembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Xu, P.; Zhao, H.; Chen, Q.; Chen, D.; Hu, H.; Zhao, X.; Qiao, M. Effect of Surface Ligand Density on Cytotoxicity and Pharmacokinetic Profile of Docetaxel Loaded Liposomes. Asian J. Pharm. Sci. 2016, 11, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Kawano, K.; Maitani, Y. Effects of Polyethylene Glycol Spacer Length and Ligand Density on Folate Receptor Targeting of Liposomal Doxorubicin in Vitro. J. Drug Deliv. 2011, 2011, 160967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Liu, M.; Shan, W.; Zhu, X.; Li, L.; Zhang, Z.; Huang, Y. Bioinspired Butyrate-Functionalized Nanovehicles for Targeted Oral Delivery of Biomacromolecular Drugs. J. Control. Release 2017, 262, 273–283. [Google Scholar] [CrossRef]
- Gerhart, D.Z.; Enerson, B.E.; Zhdankina, O.Y.; Leino, R.L.; Drewes, L.R. Expression of Monocarboxylate Transporter MCT1 by Brain Endothelium and Glia in Adult and Suckling Rats. Am. J. Physiol. 1997, 273, E207–E213. [Google Scholar] [CrossRef]
- Kido, Y.; Tamai, I.; Okamoto, M.; Suzuki, F.; Tsuji, A. Functional Clarification of MCT1-Mediated Transport of Monocarboxylic Acids at the Blood-Brain Barrier Using in Vitro Cultured Cells and in Vivo BUI Studies. Pharm. Res. 2000, 17, 55–62. [Google Scholar] [CrossRef]
- Venishetty, V.K.; Samala, R.; Komuravelli, R.; Kuncha, M.; Sistla, R.; Diwan, P.V. β-Hydroxybutyric Acid Grafted Solid Lipid Nanoparticles: A Novel Strategy to Improve Drug Delivery to Brain. Nanomedicine 2013, 9, 388–397. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. Pluronic Block Copolymers as Novel Polymer Therapeutics for Drug and Gene Delivery. J. Control. Release 2002, 82, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Manickam, D.S.; Brynskikh, A.; Kabanov, A.V. Agile Delivery of Protein Therapeutics to CNS. J. Control. Release 2014, 190, 637–663. [Google Scholar] [CrossRef] [Green Version]
- Batrakova, E.V.; Zhang, Y.; Li, Y.; Li, S.; Vinogradov, S.V.; Persidsky, Y.; Alakhov, V.Y.; Miller, D.W.; Kabanov, A.V. Effects of Pluronic P85 on GLUT1 and MCT1 Transporters in the Blood-Brain Barrier. Pharm. Res. 2004, 21, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Bröer, A.; Brookes, N.; Ganapathy, V.; Dimmer, K.S.; Wagner, C.A.; Lang, F.; Bröer, S. The Astroglial ASCT2 Amino Acid Transporter as a Mediator of Glutamine Efflux. J. NeuroChem. 1999, 73, 2184–2194. [Google Scholar] [PubMed]
- Gliddon, C.M.; Shao, Z.; LeMaistre, J.L.; Anderson, C.M. Cellular Distribution of the Neutral Amino Acid Transporter Subtype ASCT2 in Mouse Brain. J. NeuroChem. 2009, 108, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Lane, M.D. Expression of a Novel Insulin-Activated Amino Acid Transporter Gene during Differentiation of 3T3-L1 Preadipocytes into Adipocytes. BioChem. BioPhys. Res. Commun. 1995, 208, 1008–1015. [Google Scholar] [CrossRef]
- Utsunomiya-Tate, N.; Endou, H.; Kanai, Y. Cloning and Functional Characterization of a System ASC-like Na+-Dependent Neutral Amino Acid Transporter. J. Biol. Chem. 1996, 271, 14883–14890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avissar, N.E.; Ryan, C.K.; Ganapathy, V.; Sax, H.C. Na(+)-Dependent Neutral Amino Acid Transporter ATB(0) Is a Rabbit Epithelial Cell Brush-Border Protein. Am. J. Physiol. Cell Physiol. 2001, 281, C963–C971. [Google Scholar] [CrossRef]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and MTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, M.; Hinkle, P.C. Reconstitution and Purification of the D-Glucose Transporter from Human Erythrocytes. J. Biol. Chem. 1977, 252, 7384–7390. [Google Scholar] [CrossRef]
- Takata, K.; Kasahara, T.; Kasahara, M.; Ezaki, O.; Hirano, H. Erythrocyte/HepG2-Type Glucose Transporter Is Concentrated in Cells of Blood-Tissue Barriers. BioChem. BioPhys. Res. Commun. 1990, 173, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Boado, R.J.; Farrell, C.R. Brain-Type Glucose Transporter (GLUT-1) Is Selectively Localized to the Blood-Brain Barrier. Studies with Quantitative Western Blotting and in Situ Hybridization. J. Biol. Chem. 1990, 265, 18035–18040. [Google Scholar] [CrossRef]
- Frolova, A.I.; Moley, K.H. Quantitative Analysis of Glucose Transporter MRNAs in Endometrial Stromal Cells Reveals Critical Role of GLUT1 in Uterine Receptivity. Endocrinology 2011, 152, 2123–2128. [Google Scholar] [CrossRef] [Green Version]
- Tal, M.; Schneider, D.L.; Thorens, B.; Lodish, H.F. Restricted Expression of the Erythroid/Brain Glucose Transporter Isoform to Perivenous Hepatocytes in Rats. Modulation by Glucose. J. Clin. Invest. 1990, 86, 986–992. [Google Scholar] [CrossRef] [Green Version]
- Mueckler, M.; Caruso, C.; Baldwin, S.A.; Panico, M.; Blench, I.; Morris, H.R.; Allard, W.J.; Lienhard, G.E.; Lodish, H.F. Sequence and Structure of a Human Glucose Transporter. Science 1985, 229, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian Facilitative Hexose Transporters Mediate the Transport of Dehydroascorbic Acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose Transporter Isoforms GLUT1 and GLUT3 Transport Dehydroascorbic Acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef] [Green Version]
- Uldry, M.; Ibberson, M.; Hosokawa, M.; Thorens, B. GLUT2 Is a High Affinity Glucosamine Transporter. FEBS Lett. 2002, 524, 199–203. [Google Scholar] [CrossRef] [PubMed]
- James, D.E.; Strube, M.; Mueckler, M. Molecular Cloning and Characterization of an Insulin-Regulatable Glucose Transporter. Nature 1989, 338, 83–87. [Google Scholar] [CrossRef]
- Fukumoto, H.; Kayano, T.; Buse, J.B.; Edwards, Y.; Pilch, P.F.; Bell, G.I.; Seino, S. Cloning and Characterization of the Major Insulin-Responsive Glucose Transporter Expressed in Human Skeletal Muscle and Other Insulin-Responsive Tissues. J. Biol. Chem. 1989, 264, 7776–7779. [Google Scholar] [CrossRef]
- Birnbaum, M.J. Identification of a Novel Gene Encoding an Insulin-Responsive Glucose Transporter Protein. Cell 1989, 57, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kayano, T.; Burant, C.F.; Fukumoto, H.; Gould, G.W.; Fan, Y.S.; Eddy, R.L.; Byers, M.G.; Shows, T.B.; Seino, S.; Bell, G.I. Human Facilitative Glucose Transporters. Isolation, Functional Characterization, and Gene Localization of CDNAs Encoding an Isoform (GLUT5) Expressed in Small Intestine, Kidney, Muscle, and Adipose Tissue and an Unusual Glucose Transporter Pseudogene-like Sequence (GLUT6). J. Biol. Chem. 1990, 265, 13276–13282. [Google Scholar] [PubMed]
- Burant, C.F.; Takeda, J.; Brot-Laroche, E.; Bell, G.I.; Davidson, N.O. Fructose Transporter in Human Spermatozoa and Small Intestine Is GLUT5. J. Biol. Chem. 1992, 267, 14523–14526. [Google Scholar] [CrossRef] [PubMed]
- Mantych, G.J.; James, D.E.; Devaskar, S.U. Jejunal/Kidney Glucose Transporter Isoform (Glut-5) Is Expressed in the Human Blood-Brain Barrier. Endocrinology 1993, 132, 35–40. [Google Scholar] [CrossRef]
- Douard, V.; Ferraris, R.P. Regulation of the Fructose Transporter GLUT5 in Health and Disease. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E227–E237. [Google Scholar] [CrossRef] [Green Version]
- Tatibouët, A.; Yang, J.; Morin, C.; Holman, G.D. Synthesis and Evaluation of Fructose Analogues as Inhibitors of the D-Fructose Transporter GLUT5. BioOrg. Med. Chem. 2000, 8, 1825–1833. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Fei, Y.J.; Xia, H.; Yang-Feng, T.L.; Leibach, F.H.; Devoe, L.D.; Ganapathy, V.; Prasad, P.D. Human Placental Na+-Dependent Multivitamin Transporter. Cloning, Functional Expression, Gene Structure, and Chromosomal Localization. J. Biol. Chem. 1999, 274, 14875–14883. [Google Scholar] [CrossRef] [Green Version]
- Baur, B.; Baumgartner, E.R. Biotin and Biocytin Uptake into Cultured Primary Calf Brain Microvessel Endothelial Cells of the Blood-Brain Barrier. Brain Res. 2000, 858, 348–355. [Google Scholar] [CrossRef]
- Uchida, Y.; Ito, K.; Ohtsuki, S.; Kubo, Y.; Suzuki, T.; Terasaki, T. Major Involvement of Na(+) -Dependent Multivitamin Transporter (SLC5A6/SMVT) in Uptake of Biotin and Pantothenic Acid by Human Brain Capillary Endothelial Cells. J. NeuroChem. 2015, 134, 97–112. [Google Scholar] [CrossRef]
- Ohkura, Y.; Akanuma, S.; Tachikawa, M.; Hosoya, K. Blood-to-Retina Transport of Biotin via Na+-Dependent Multivitamin Transporter (SMVT) at the Inner Blood-Retinal Barrier. Exp. Eye Res. 2010, 91, 387–392. [Google Scholar] [CrossRef]
- de Carvalho, F.D.; Quick, M. Surprising Substrate Versatility in SLC5A6: Na+-Coupled I- Transport by the Human Na+/Multivitamin Transporter (HSMVT). J. Biol. Chem. 2011, 286, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, J.L.; Grubb, B.R.; Mager, S. Expression of the Amino Acid Transporter ATB 0+ in Lung: Possible Role in Luminal Protein Removal. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L39–L49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotoli, B.M.; Bussolati, O.; Sala, R.; Gazzola, G.C.; Dall’Asta, V. The Transport of Cationic Amino Acids in Human Airway Cells: Expression of System Y+L Activity and Transepithelial Delivery of NOS Inhibitors. FASEB J. 2005, 19, 810–812. [Google Scholar] [CrossRef]
- Uchiyama, T.; Fujita, T.; Gukasyan, H.J.; Kim, K.-J.; Borok, Z.; Crandall, E.D.; Lee, V.H.L. Functional Characterization and Cloning of Amino Acid Transporter B(0,+) (ATB(0,+)) in Primary Cultured Rat Pneumocytes. J. Cell Physiol. 2008, 214, 645–654. [Google Scholar] [CrossRef]
- Su, T.Z.; Lunney, E.; Campbell, G.; Oxender, D.L. Transport of Gabapentin, a Gamma-Amino Acid Drug, by System l Alpha-Amino Acid Transporters: A Comparative Study in Astrocytes, Synaptosomes, and CHO Cells. J. NeuroChem. 1995, 64, 2125–2131. [Google Scholar] [CrossRef] [PubMed]
- Papin-Michault, C.; Bonnetaud, C.; Dufour, M.; Almairac, F.; Coutts, M.; Patouraux, S.; Virolle, T.; Darcourt, J.; Burel-Vandenbos, F. Study of LAT1 Expression in Brain Metastases: Towards a Better Understanding of the Results of Positron Emission Tomography Using Amino Acid Tracers. PLoS One 2016, 11, e0157139. [Google Scholar] [CrossRef] [Green Version]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.; Fukasawa, Y.; et al. Human L-Type Amino Acid Transporter 1 (LAT1): Characterization of Function and Expression in Tumor Cell Lines. Biochim. BioPhys. Acta 2001, 1514, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Verrey, F. System L: Heteromeric Exchangers of Large, Neutral Amino Acids Involved in Directional Transport. Pflug. Arch. 2003, 445, 529–533. [Google Scholar] [CrossRef]
- Tomi, M.; Mori, M.; Tachikawa, M.; Katayama, K.; Terasaki, T.; Hosoya, K. L-Type Amino Acid Transporter 1-Mediated L-Leucine Transport at the Inner Blood-Retinal Barrier. Invest. OphthalMol. Vis. Sci. 2005, 46, 2522–2530. [Google Scholar] [CrossRef] [Green Version]
- Kudo, Y.; Boyd, C.A. Characterisation of L-Tryptophan Transporters in Human Placenta: A Comparison of Brush Border and Basal Membrane Vesicles. J. Physiol. 2001, 531, 405–416. [Google Scholar] [CrossRef]
- Ritchie, J.W.; Taylor, P.M. Role of the System L Permease LAT1 in Amino Acid and Iodothyronine Transport in Placenta. BioChem. J. 2001, 356, 719–725. [Google Scholar] [CrossRef]
- Nakada, N.; Mikami, T.; Hana, K.; Ichinoe, M.; Yanagisawa, N.; Yoshida, T.; Endou, H.; Okayasu, I. Unique and Selective Expression of L-Amino Acid Transporter 1 in Human Tissue as Well as Being an Aspect of Oncofetal Protein. Histol. Histopathol. 2014, 29, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Vumma, R.; Wiesel, F.-A.; Flyckt, L.; Bjerkenstedt, L.; Venizelos, N. Functional Characterization of Tyrosine Transport in Fibroblast Cells from Healthy Controls. NeuroSci. Lett. 2008, 434, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.R.; Oh, Y.-J.; Kang, S.W.; Lee, E.B.; Lee, W.-W. Role of SLC7A5 in Metabolic Reprogramming of Human Monocyte/Macrophage Immune Responses. Front. Immunol. 2018, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, D.; Doi, H.; Fukushima, K.; Katsura, K.; Ogawa, N.; Sekiguchi, S.; Fujimori, K.; Sato, A.; Satomi, S.; Ishida, K.; et al. Glutamate Exocrine Dynamics Augmented by Plasma Glutamine and the Distribution of Amino Acid Transporters of the Rat Pancreas. J. Physiol. Pharmacol. 2010, 61, 265–271. [Google Scholar] [PubMed]
- Zhou, Y.; Waanders, L.F.; Holmseth, S.; Guo, C.; Berger, U.V.; Li, Y.; Lehre, A.-C.; Lehre, K.P.; Danbolt, N.C. Proteome Analysis and Conditional Deletion of the EAAT2 Glutamate Transporter Provide Evidence against a Role of EAAT2 in Pancreatic Insulin Secretion in Mice. J. Biol. Chem. 2014, 289, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Huttunen, K.M.; Gynther, M.; Huttunen, J.; Puris, E.; Spicer, J.A.; Denny, W.A. A Selective and Slowly Reversible Inhibitor of L-Type Amino Acid Transporter 1 (LAT1) Potentiates Antiproliferative Drug Efficacy in Cancer Cells. J. Med. Chem. 2016, 59, 5740–5751. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Meier, P.J. Molecular Cloning, Chromosomal Localization, and Functional Characterization of a Human Liver Na+/Bile Acid Cotransporter. J. Clin. Invest. 1994, 93, 1326–1331. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kim, K.H.; Lee, J.A.; Namkung, W.; Sun, A.-Q.; Ananthanarayanan, M.; Suchy, F.J.; Shin, D.M.; Muallem, S.; Lee, M.G. Transporter-Mediated Bile Acid Uptake Causes Ca2+-Dependent Cell Death in Rat Pancreatic Acinar Cells. Gastroenterology 2002, 122, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Kullak-Ublick, G.A.; Beuers, U.; Paumgartner, G. Molecular and Functional Characterization of Bile Acid Transport in Human Hepatoblastoma HepG2 Cells. Hepatology 1996, 23, 1053–1060. [Google Scholar] [CrossRef]
- Schroeder, A.; Eckhardt, U.; Stieger, B.; Tynes, R.; Schteingart, C.D.; Hofmann, A.F.; Meier, P.J.; Hagenbuch, B. Substrate Specificity of the Rat Liver Na(+)-Bile Salt Cotransporter in Xenopus LaeVis Oocytes and in CHO Cells. Am. J. Physiol. 1998, 274, G370–G375. [Google Scholar] [CrossRef] [PubMed]
- Craddock, A.L.; Love, M.W.; Daniel, R.W.; Kirby, L.C.; Walters, H.C.; Wong, M.H.; Dawson, P.A. Expression and Transport Properties of the Human Ileal and Renal Sodium-Dependent Bile Acid Transporter. Am. J. Physiol. 1998, 274, G157–G169. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H.; Oelkers, P.; Craddock, A.L.; Dawson, P.A. Expression Cloning and Characterization of the Hamster Ileal Sodium-Dependent Bile Acid Transporter. J. Biol. Chem. 1994, 269, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Shneider, B.L.; Dawson, P.A.; Christie, D.M.; Hardikar, W.; Wong, M.H.; Suchy, F.J. Cloning and Molecular Characterization of the Ontogeny of a Rat Ileal Sodium-Dependent Bile Acid Transporter. J. Clin. Invest. 1995, 95, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Christie, D.M.; Dawson, P.A.; Thevananther, S.; Shneider, B.L. Comparative Analysis of the Ontogeny of a Sodium-Dependent Bile Acid Transporter in Rat Kidney and Ileum. Am. J. Physiol. 1996, 271, G377–G385. [Google Scholar] [CrossRef]
- Alpini, G.; Glaser, S.S.; Rodgers, R.; Phinizy, J.L.; Robertson, W.E.; Lasater, J.; Caligiuri, A.; Tretjak, Z.; LeSage, G.D. Functional Expression of the Apical Na+-Dependent Bile Acid Transporter in Large but Not Small Rat Cholangiocytes. Gastroenterology 1997, 113, 1734–1740. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Pham, L.; Tietz, P.; Marinelli, R.A.; deGroen, P.C.; Levine, S.; Dawson, P.A.; LaRusso, N.F. Rat Cholangiocytes Absorb Bile Acids at Their Apical Domain via the Ileal Sodium-Dependent Bile Acid Transporter. J. Clin. Invest. 1997, 100, 2714–2721. [Google Scholar] [CrossRef]
- Chignard, N.; Mergey, M.; Veissière, D.; Parc, R.; Capeau, J.; Poupon, R.; Paul, A.; Housset, C. Bile Acid Transport and Regulating Functions in the Human Biliary Epithelium. Hepatology 2001, 33, 496–503. [Google Scholar] [CrossRef]
- Alrefai, W.A.; Sarwar, Z.; Tyagi, S.; Saksena, S.; Dudeja, P.K.; Gill, R.K. Cholesterol Modulates Human Intestinal Sodium-Dependent Bile Acid Transporter. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G978–G985. [Google Scholar] [CrossRef] [Green Version]
- Freeman, T.C.; Bentsen, B.S.; Thwaites, D.T.; Simmons, N.L. H+/Di-Tripeptide Transporter (PepT1) Expression in the Rabbit Intestine. Pflug. Arch. 1995, 430, 394–400. [Google Scholar] [CrossRef]
- Ogihara, H.; Saito, H.; Shin, B.C.; Terado, T.; Takenoshita, S.; Nagamachi, Y.; Inui, K.; Takata, K. Immuno-Localization of H+/Peptide Cotransporter in Rat Digestive Tract. BioChem. BioPhys. Res. Commun. 1996, 220, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Smith, D.E.; Yang, T.; Huang, Y.G.; Schnermann, J.B.; Brosius, F.C. Localization of PEPT1 and PEPT2 Proton-Coupled Oligopeptide Transporter MRNA and Protein in Rat Kidney. Am. J. Physiol. 1999, 276, F658–F665. [Google Scholar] [CrossRef]
- Bockman, D.E.; Ganapathy, V.; Oblak, T.G.; Leibach, F.H. Localization of Peptide Transporter in Nuclei and Lysosomes of the Pancreas. Int. J. Pancreatol. 1997, 22, 221–225. [Google Scholar] [CrossRef]
- Knütter, I.; Rubio-Aliaga, I.; Boll, M.; Hause, G.; Daniel, H.; Neubert, K.; Brandsch, M. H+-Peptide Cotransport in the Human Bile Duct Epithelium Cell Line SK-ChA-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G222–G229. [Google Scholar] [CrossRef] [Green Version]
- Charrier, L.; Driss, A.; Yan, Y.; Nduati, V.; Klapproth, J.-M.; Sitaraman, S.V.; Merlin, D. HPepT1 Mediates Bacterial Tripeptide FMLP Uptake in Human Monocytes. Lab Invest. 2006, 86, 490–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T.; Kishida, T.; Wada, M.; Okada, N.; Yamamoto, A.; Leibach, F.H.; Ganapathy, V. Functional Characterization of Brain Peptide Transporter in Rat Cerebral Cortex: Identification of the High-Affinity Type H+/Peptide Transporter PEPT2. Brain Res. 2004, 997, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; Döring, F.; Nickolaus, M.; Daniel, H.; Fischer, A. Expression of PEPT2 Peptide Transporter MRNA and Protein in Glial Cells of Rat Dorsal Root Ganglia. NeuroSci. Lett. 2001, 304, 181–184. [Google Scholar] [CrossRef]
- Rühl, A.; Hoppe, S.; Frey, I.; Daniel, H.; Schemann, M. Functional Expression of the Peptide Transporter PEPT2 in the Mammalian Enteric Nervous System. J. Comp. Neurol 2005, 490, 1–11. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Nickolaus, M.; Springer, J.; Döring, F.; Daniel, H.; Fischer, A. Localization of the Peptide Transporter PEPT2 in the Lung: Implications for Pulmonary Oligopeptide Uptake. Am. J. Pathol. 2001, 158, 707–714. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Döring, F.; Theis, S.; Nickolaus, M.; Fischer, A.; Daniel, H. Peptide Transport in the Mammary Gland: Expression and Distribution of PEPT2 MRNA and Protein. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1172–E1179. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; King, N. Demonstration of Functional Dipeptide Transport with Expression of PEPT2 in Guinea Pig Cardiomyocytes. Pflug. Arch. 2007, 453, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, Y.; Tan, F.; Fang, D.; Hu, Y.; Smith, D.E.; Jiang, H. Functional and Molecular Expression of the Proton-Coupled Oligopeptide Transporters in Spleen and Macrophages from Mouse and Human. Mol. Pharm. 2013, 10, 1409–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.Z.; Zhu, T.; Smith, D.E.; Hediger, M.A. Stoichiometry and Kinetics of the High-Affinity H+-Coupled Peptide Transporter PepT2. J. Biol. Chem. 1999, 274, 2773–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Lu, K. Substrates of the Human Oligopeptide Transporter HPEPT2. BioSci. Trends 2015, 9, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.M.; Goldstein, J.L.; Brown, M.S. CDNA Cloning of MEV, a Mutant Protein That Facilitates Cellular Uptake of Mevalonate, and Identification of the PoInt. Mutation Responsible for Its Gain of Function. J. Biol. Chem. 1992, 267, 23113–23121. [Google Scholar] [CrossRef]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular Characterization of a Membrane Transporter for Lactate, Pyruvate, and Other Monocarboxylates: Implications for the Cori Cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef]
- Pierre, K.; Pellerin, L.; Debernardi, R.; Riederer, B.M.; Magistretti, P.J. Cell-Specific Localization of Monocarboxylate Transporters, MCT1 and MCT2, in the Adult Mouse Brain Revealed by Double Immunohistochemical Labeling and Confocal Microscopy. Neuroscience 2000, 100, 617–627. [Google Scholar] [CrossRef]
- Gerhart, D.Z.; Leino, R.L.; Drewes, L.R. Distribution of Monocarboxylate Transporters MCT1 and MCT2 in Rat Retina. Neuroscience 1999, 92, 367–375. [Google Scholar] [CrossRef]
- Takebe, K.; Nio-Kobayashi, J.; Takahashi-Iwanaga, H.; Yajima, T.; Iwanaga, T. Cellular Expression of a Monocarboxylate Transporter (MCT1) in the Mammary Gland and Sebaceous Gland of Mice. HistoChem. Cell Biol. 2009, 131, 401–409. [Google Scholar] [CrossRef]
- Ritzhaupt, A.; Wood, I.S.; Ellis, A.; Hosie, K.B.; Shirazi-Beechey, S.P. Identification and Characterization of a Monocarboxylate Transporter (MCT1) in Pig and Human Colon: Its Potential to Transport L-Lactate as Well as Butyrate. J. Physiol. 1998, 513 Pt 3, 719–732. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Price, N.T. The Proton-Linked Monocarboxylate Transporter (MCT) Family: Structure, Function and Regulation. Biochem. J. 1999, 343 Pt 2, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Ritzhaupt, A.; Ellis, A.; Hosie, K.B.; Shirazi-Beechey, S.P. The Characterization of Butyrate Transport across Pig and Human Colonic Luminal Membrane. J. Physiol. 1998, 507 Pt 3, 819–830. [Google Scholar] [CrossRef]
- Hadjiagapiou, C.; Schmidt, L.; Dudeja, P.K.; Layden, T.J.; Ramaswamy, K. Mechanism(s) of Butyrate Transport in Caco-2 Cells: Role of Monocarboxylate Transporter 1. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G775–G780. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Flatley, R.M.; Matherly, L.H. The Human Reduced Folate Carrier Gene Is Ubiquitously and Differentially Expressed in Normal Human Tissues: Identification of Seven Non-Coding Exons and Characterization of a Novel Promoter. BioChem. J. 2002, 367, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, R.; Russell, R.G.; Goldman, I.D. Localization of the Murine Reduced Folate Carrier as Assessed by Immunohistochemical Analysis. Biochim. BioPhys. Acta 2001, 1513, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Dixon, K.H.; Lanpher, B.C.; Chiu, J.; Kelley, K.; Cowan, K.H. A Novel CDNA RestoRes. Reduced Folate Carrier Activity and Methotrexate Sensitivity to Transport Deficient Cells. J. Biol. Chem. 1994, 269, 17–20. [Google Scholar] [CrossRef]
- Prasad, P.D.; Ramamoorthy, S.; Leibach, F.H.; Ganapathy, V. Molecular Cloning of the Human Placental Folate Transporter. BioChem. BioPhys. Res. Commun. 1995, 206, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Kakyo, M.; Tokui, T.; Nakagomi, R.; Nishio, T.; Nakai, D.; Nomura, H.; Unno, M.; Suzuki, M.; Naitoh, T.; et al. Identification of a Novel Gene Family Encoding Human Liver-Specific Organic Anion Transporter LST-1. J. Biol. Chem. 1999, 274, 17159–17163. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.K.; Hammond, C.L.; Ballatori, N. Intracellular Glutathione Regulates Taurocholate Transport in HepG2 Cells. Toxicol. Appl. Pharmacol. 2001, 174, 207–215. [Google Scholar] [CrossRef]
- Cui, Y.; König, J.; Leier, I.; Buchholz, U.; Keppler, D. Hepatic Uptake of Bilirubin and Its Conjugates by the Human Organic Anion Transporter SLC21A6. J. Biol. Chem. 2001, 276, 9626–9630. [Google Scholar] [CrossRef] [Green Version]
- Kullak-Ublick, G.A.; Ismair, M.G.; Stieger, B.; Landmann, L.; Huber, R.; Pizzagalli, F.; Fattinger, K.; Meier, P.J.; Hagenbuch, B. Organic Anion-Transporting Polypeptide B (OATP-B) and Its Functional Comparison with Three Other OATPs of Human Liver. Gastroenterology 2001, 120, 525–533. [Google Scholar] [CrossRef]
- Maeda, K.; Kambara, M.; Tian, Y.; Hofmann, A.F.; Sugiyama, Y. Uptake of Ursodeoxycholate and Its Conjugates by Human Hepatocytes: Role of Na(+)-Taurocholate Cotransporting Polypeptide (NTCP), Organic Anion Transporting Polypeptide (OATP) 1B1 (OATP-C), and Oatp1B3 (OATP8). Mol. Pharm. 2006, 3, 70–77. [Google Scholar] [CrossRef]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and Genomic Organization of a New Hepatocellular Organic Anion Transporting Polypeptide. J. Biol. Chem. 2000, 275, 23161–23168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briz, O.; Romero, M.R.; Martinez-Becerra, P.; Macias, R.I.R.; Perez, M.J.; Jimenez, F.; San Martin, F.G.; Marin, J.J.G. OATP8/1B3-Mediated Cotransport of Bile Acids and Glutathione: An Export Pathway for Organic Anions from Hepatocytes? J. Biol. Chem. 2006, 281, 30326–30335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briz, O.; Serrano, M.A.; MacIas, R.I.R.; Gonzalez-Gallego, J.; Marin, J.J.G. Role of Organic Anion-Transporting Polypeptides, OATP-A, OATP-C and OATP-8, in the Human Placenta-Maternal Liver Tandem Excretory Pathway for Foetal Bilirubin. BioChem. J. 2003, 371, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, T.; Kusuhara, H.; Utsunomiya-Tate, N.; Tsuda, M.; Sugiyama, Y.; Kanai, Y.; Endou, H. Molecular Cloning and Characterization of High-Affinity Carnitine Transporter from Rat Intestine. BioChem. BioPhys. Res. Commun. 1998, 251, 586–591. [Google Scholar] [CrossRef]
- Wu, X.; Prasad, P.D.; Leibach, F.H.; Ganapathy, V. CDNA Sequence, Transport Function, and Genomic Organization of Human OCTN2, a New Member of the Organic Cation Transporter Family. BioChem. BioPhys. Res. Commun. 1998, 246, 589–595. [Google Scholar] [CrossRef]
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Molecular and Functional Identification of Sodium Ion-Dependent, High Affinity Human Carnitine Transporter OCTN2. J. Biol. Chem. 1998, 273, 20378–20382. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.A.; Lükewille, U.; Kaltenbach, S.; Moschen, I.; Bröer, A.; Risler, T.; Bröer, S.; Lang, F. Functional and Pharmacological Characterization of Human Na(+)-Carnitine Cotransporter HOCTN2. Am. J. Physiol. Renal Physiol. 2000, 279, F584–F591. [Google Scholar] [CrossRef]
- Schömig, E.; Spitzenberger, F.; Engelhardt, M.; Martel, F.; Ording, N.; Gründemann, D. Molecular Cloning and Characterization of Two Novel Transport Proteins from Rat Kidney. FEBS Lett. 1998, 425, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Huang, W.; Prasad, P.D.; Seth, P.; Rajan, D.P.; Leibach, F.H.; Chen, J.; Conway, S.J.; Ganapathy, V. Functional Characteristics and Tissue Distribution Pattern of Organic Cation Transporter 2 (OCTN2), an Organic Cation/Carnitine Transporter. J. Pharmacol. Exp. Ther. 1999, 290, 1482–1492. [Google Scholar]
- Tachikawa, M.; Takeda, Y.; Tomi, M.; Hosoya, K. Involvement of OCTN2 in the Transport of Acetyl-L-Carnitine across the Inner Blood-Retinal Barrier. Invest. OphthalMol. Vis. Sci. 2010, 51, 430–436. [Google Scholar] [CrossRef]
- Grube, M.; Meyer Zu Schwabedissen, H.; Draber, K.; Präger, D.; Möritz, K.-U.; Linnemann, K.; Fusch, C.; Jedlitschky, G.; Kroemer, H.K. Expression, Localization, and Function of the Carnitine Transporter Octn2 (Slc22a5) in Human Placenta. Drug Metab. Dispos. 2005, 33, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Alcorn, J.; Lu, X.; Moscow, J.A.; McNamara, P.J. Transporter Gene Expression in Lactating and Nonlactating Human Mammary Epithelial Cells Using Real-Time Reverse Transcription-Polymerase Chain Reaction. J. Pharmacol. Exp. Ther. 2002, 303, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, R.; Tamai, I.; Yabuuchi, H.; Nezu, J.I.; Oku, A.; Sai, Y.; Shimane, M.; Tsuji, A. Na(+)-Dependent Carnitine Transport by Organic Cation Transporter (OCTN2): Its Pharmacological and Toxicological Relevance. J. Pharmacol. Exp. Ther. 1999, 291, 778–784. [Google Scholar]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human Vitamin C (L-Ascorbic Acid) Transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.; Rankin, G.D.; Bicer, E.M.; Roos-Engstrand, E.; Pourazar, J.; Blomberg, A.; Mudway, I.S.; Behndig, A.F. Identification of Vitamin C Transporters in the Human Airways: A Cross-Sectional in Vivo Study. BMJ. Open 2015, 5, e006979. [Google Scholar] [CrossRef] [Green Version]
- Steiling, H.; Longet, K.; Moodycliffe, A.; Mansourian, R.; Bertschy, E.; Smola, H.; Mauch, C.; Williamson, G. Sodium-Dependent Vitamin C Transporter Isoforms in Skin: Distribution, Kinetics, and Effect of UVB-Induced Oxidative Stress. Free Radic. Biol. Med. 2007, 43, 752–762. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, L.; Thumser, A.E.; Sharp, P. Decreased Expression of the Vitamin C Transporter SVCT1 by Ascorbic Acid in a Human Intestinal Epithelial Cell Line. Br J. Nutr. 2002, 87, 97–100. [Google Scholar] [CrossRef]
- O’Regan, S.; Traiffort, E.; Ruat, M.; Cha, N.; Compaore, D.; Meunier, F.M. An Electric Lobe Suppressor for a Yeast Choline Transport Mutation Belongs to a New Family of Transporter-like Proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 1835–1840. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Tie, A.; Tarnopolsky, M.; Bakovic, M. Genomic Organization, Promoter Activity, and Expression of the Human Choline Transporter-like Protein 1. Physiol. Genom. 2006, 26, 76–90. [Google Scholar] [CrossRef]
- Wille, S.; Szekeres, A.; Majdic, O.; Prager, E.; Staffler, G.; Stöckl, J.; Kunthalert, D.; Prieschl, E.E.; Baumruker, T.; Burtscher, H.; et al. Characterization of CDw92 as a Member of the Choline Transporter-like Protein Family Regulated Specifically on Dendritic Cells. J. Immunol. 2001, 167, 5795–5804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Wagner, L.; Poloumienko, A.; Bakovic, M. Identification and Expression of a Mouse Muscle-Specific CTL1 Gene. Gene 2004, 341, 305–312. [Google Scholar] [CrossRef]
- Michel, V.; Bakovic, M. The Solute Carrier 44A1 Is a Mitochondrial Protein and Mediates Choline Transport. FASEB J. 2009, 23, 2749–2758. [Google Scholar] [CrossRef]
- O’Regan, S.; Meunier, F.-M. Selection and Characterization of the Choline Transport Mutation Suppressor from Torpedo Electric Lobe, CTL1. NeuroChem. Res. 2003, 28, 551–555. [Google Scholar] [CrossRef]
- Taylor, A.; Grapentine, S.; Ichhpuniani, J.; Bakovic, M. Choline Transporter-like Proteins 1 and 2 Are Newly Identified Plasma Membrane and Mitochondrial Ethanolamine Transporters. J. Biol. Chem. 2021, 296, 100604. [Google Scholar] [CrossRef]
- Kommareddi, P.K.; Nair, T.S.; Thang, L.V.; Galano, M.M.; Babu, E.; Ganapathy, V.; Kanazawa, T.; McHugh, J.B.; Carey, T.E. Isoforms, Expression, Glycosylation, and Tissue Distribution of CTL2/SLC44A2. Protein J. 2010, 29, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Nair, T.S.; Kozma, K.E.; Hoefling, N.L.; Kommareddi, P.K.; Ueda, Y.; Gong, T.-W.; Lomax, M.I.; Lansford, C.D.; Telian, S.A.; Satar, B.; et al. Identification and Characterization of Choline Transporter-like Protein 2, an Inner Ear Glycoprotein of 68 and 72 KDa That Is the Target of Antibody-Induced Hearing Loss. J. NeuroSci. 2004, 24, 1772–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traiffort, E.; Ruat, M.; O’Regan, S.; Meunier, F.M. Molecular Characterization of the Family of Choline Transporter-like Proteins and Their Splice Variants. J. NeuroChem. 2005, 92, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Qiu, A.; Min, S.H.; Jansen, M.; Malhotra, U.; Tsai, E.; Cabelof, D.C.; Matherly, L.H.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Rodent Intestinal Folate Transporters (SLC46A1): Secondary Structure, Functional Properties, and Response to Dietary Folate Restriction. Am. J. Physiol. Cell Physiol. 2007, 293, C1669–C1678. [Google Scholar] [CrossRef] [Green Version]
- Wollack, J.B.; Makori, B.; Ahlawat, S.; Koneru, R.; Picinich, S.C.; Smith, A.; Goldman, I.D.; Qiu, A.; Cole, P.D.; Glod, J.; et al. Characterization of Folate Uptake by Choroid Plexus Epithelial Cells in a Rat Primary Culture Model. J. NeuroChem. 2008, 104, 1494–1503. [Google Scholar] [CrossRef]
- Costa, C.P.; Barreiro, S.; Moreira, J.N.; Silva, R.; Almeida, H.; Sousa Lobo, J.M.; Silva, A.C. In Vitro Studies on Nasal Formulations of Nanostructured Lipid Carriers (NLC) and Solid Lipid Nanoparticles (SLN). Pharmaceuticals 2021, 14, 711. [Google Scholar] [CrossRef]
- Anand, U.; Parikh, A.; Ugwu, M.C.; Agu, R.U. Drug Transporters in the Nasal Epithelium: An Overview of Strategies in Targeted Drug Delivery. Future Med. Chem. 2014, 6, 1381–1397. [Google Scholar] [CrossRef]
- Al-Ghabeish, M.; Scheetz, T.; Assem, M.; Donovan, M.D. Microarray Determination of the Expression of Drug Transporters in Humans and Animal Species Used for the Investigation of Nasal Absorption. Mol. Pharm. 2015, 12, 2742–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponto, L.L.B.; Huang, J.; Walsh, S.A.; Acevedo, M.R.; Mundt, C.; Sunderland, J.; Donovan, M. Demonstration of Nucleoside Transporter Activity in the Nose-to-Brain Distribution of [18F]Fluorothymidine Using PET Imaging. AAPS J. 2017, 20, 16. [Google Scholar] [CrossRef]
- Borrajo, M.L.; Alonso, M.J. Using Nanotechnology to Deliver Biomolecules from Nose to Brain-Peptides, Proteins, Monoclonal Antibodies and RNA. Drug Deliv. Transl. Res. 2022, 12, 862–880. [Google Scholar] [CrossRef]
- Wei, Y.; Zhao, L. Passive Lung-Targeted Drug Delivery Systems via Intravenous Administration. Pharm. Dev. Technol. 2014, 19, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Abed, S.; Turner, R.; Serniuck, N.; Tat, V.; Naiel, S.; Hayat, A.; Mekhael, O.; Vierhout, M.; Ask, K.; Rullo, A.F. Cell-Specific Drug Targeting in the Lung. BioChem. Pharmacol. 2021, 190, 114577. [Google Scholar] [CrossRef]
- Nickel, S.; Clerkin, C.G.; Selo, M.A.; Ehrhardt, C. Transport Mechanisms at the Pulmonary Mucosa: Implications for Drug Delivery. Expert Opin. Drug Deliv. 2016, 13, 667–690. [Google Scholar] [CrossRef]
- Salomon, J.J.; Ehrhardt, C. Organic Cation Transporters in the Blood-Air Barrier: Expression and Implications for Pulmonary Drug Delivery. Ther. Deliv. 2012, 3, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Selo, M.A.; Sake, J.A.; Ehrhardt, C.; Salomon, J.J. Organic Cation Transporters in the Lung-Current and Emerging (Patho)Physiological and Pharmacological Concepts. Int. J. Mol. Sci. 2020, 21, 9168. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Lim, L.Y.; Zhang, Z.-R. L-Carnitine Ester of Prednisolone: Pharmacokinetic and Pharmacodynamic Evaluation of a Type I Prodrug. Int. J. Pharm. 2014, 475, 123–129. [Google Scholar] [CrossRef]
- Betters, J.L.; Yu, L. NPC1L1 and Cholesterol Transport. FEBS Lett. 2010, 584, 2740–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-Function of CD36 and Importance of Fatty Acid Signal Transduction in Fat Metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.M.; Stahl, A. SLC27 Fatty Acid Transport Proteins. Mol. Aspects Med. 2013, 34, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Spector, R.; Boose, B. Active Transport of Riboflavin by the Isolated Choroid Plexus in Vitro. J. Biol. Chem. 1979, 254, 10286–10289. [Google Scholar] [CrossRef]
- Yonezawa, A.; Inui, K. Novel Riboflavin Transporter Family RFVT/SLC52: Identification, Nomenclature, Functional Characterization and Genetic Diseases of RFVT/SLC52. Mol. Aspects Med. 2013, 34, 693–701. [Google Scholar] [CrossRef]
- O’Hagan, S.; Wright Muelas, M.; Day, P.J.; Lundberg, E.; Kell, D.B. GeneGini: Assessment via the Gini Coefficient of Reference “Housekeeping” Genes and Diverse Human Transporter Expression Profiles. Cell Syst. 2018, 6, 230–244.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, X.; Miao, T.; Chen, H.; Ni, J.; Han, L. Overcoming Mfsd2a-Mediated Low Transcytosis to Boost Nanoparticle Delivery to Brain for Chemotherapy of Brain Metastases. Adv. Healthc. Mater. 2021, 10, e2001997. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 Reductions Exacerbate Alzheimer’s Disease Vasculo-Neuronal Dysfunction and Degeneration. Nat. NeuroSci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [Green Version]
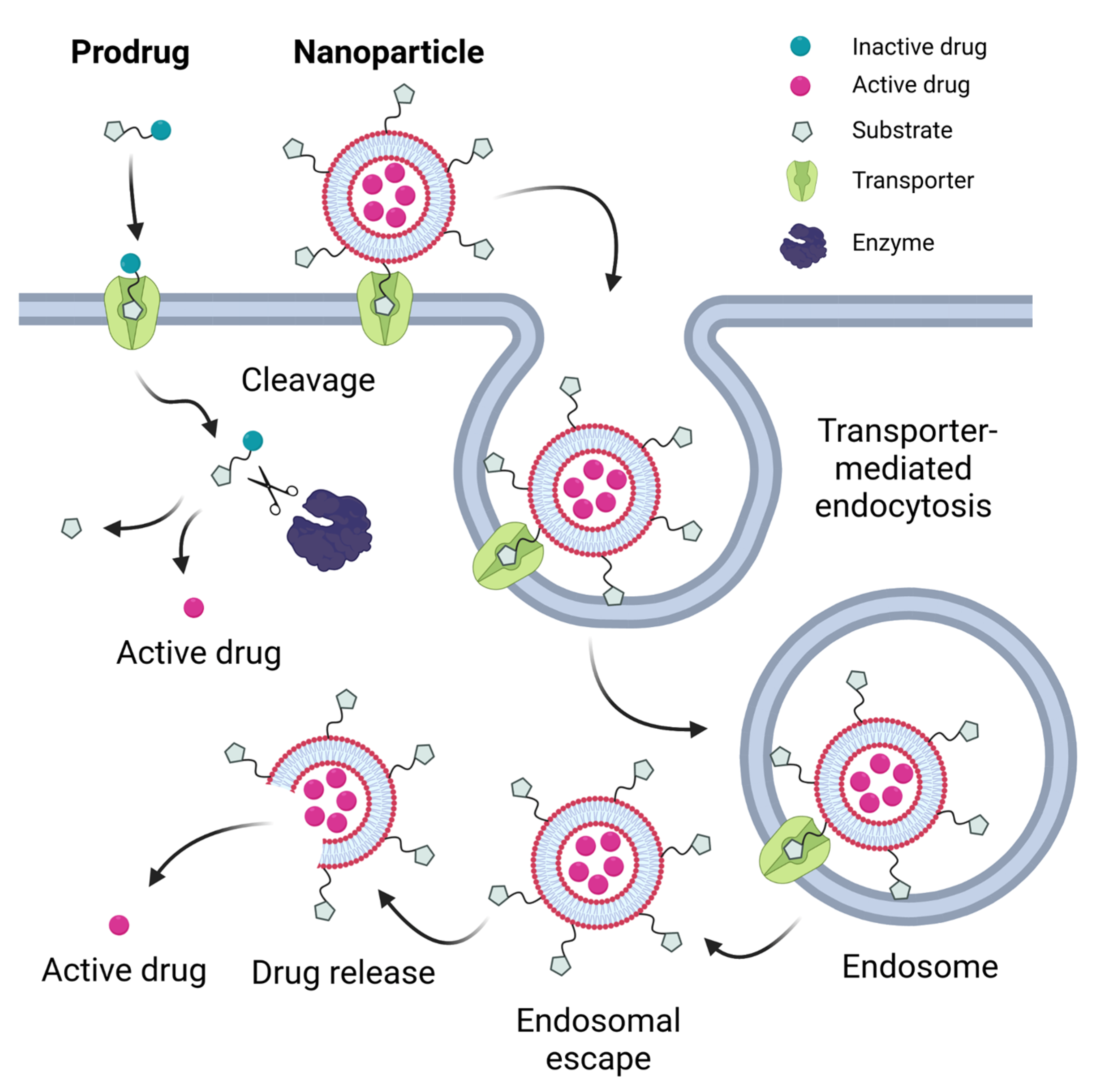

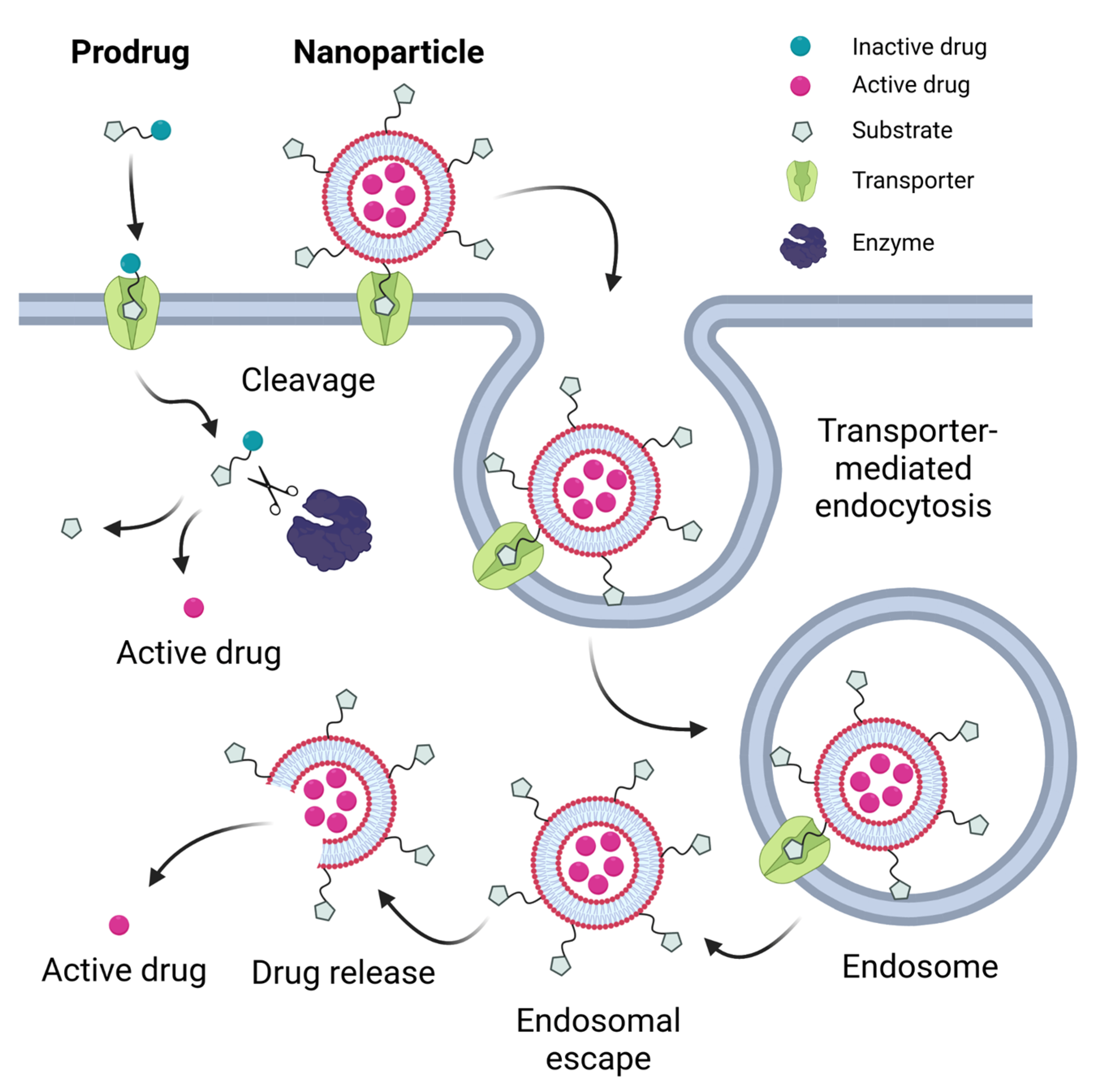{kind=link}
| Gene symbol/Protein Name | Tissue Expression | Barriers | Cell Lines | Substrates |
|---|---|---|---|---|
| SLC1A5/ASCT2 | brain [396,397], adipocytes [398,399], testis [399], colon, small intestine, pancreas, stomach [400], kidney [400], activated T cells [401], lung, skeletal muscle [399] | likely on the basolateral membrane of epithelial cells and responsible for taking up nutrients from the blood [399] | 9L [140,141], SF188 [140,141], A549 [145], A549/DPP [147], BT549 [146], DU145 [144], LNCaP [144], MDA-MB-231 [146], PC-3 [141] | L-Ala, L-Ser, L-Thr, L-Gln, L-Asn, L-Glu, L-Met, L-Leu, L-Gly, L-Val [399] |
| SLC2A1/GLUT1 | erythrocytes [402], eye, retina [403], brain microvessels [403,404], choroid plexus [403], peripheral nerves [403], placenta [403], uterus [405], liver [406] | BBB, placenta, BCSFB, blood–aqueous barrier, blood–retina barrier, blood–nerve barrier [403] | MDA-MB-231 [59,66], Bel-7402 [38], BMEC [70], BV2 [35], Caco-2 [35], hCMEC/D3 [70], HEp-2 [37], HRPE [55], human erythrocyte membranes [56], MCF-7 [66], Neuro-2a [69], RG-2 [36,61], SH-SY5Y [62], U-87 [63] | D-glucose [402,407], DHA [408,409], glucosamine [410] |
| SLC2A4/GLUT4 | adipose tissue, skeletal and cardiac muscle [411,412] | C1C12 (differentiated, with induction by insulin) [78] | D-glucose (2-deoxy-D-glucose) [413], glucosamine [410] | |
| SLC2A5/GLUT5 | small intestine [414], kidney [414], skeletal muscle [414], adipose tissue [414], testis, spermatozoa [415], brain [416] | intestinal [414,417], BBB [416] | MCF-7 [75,76,77], MCF10AneoT [77], MDA-MB-231 [75] | D-fructose [415,418] |
| SLC5A6/SMVT | heart, brain, placenta, lung, liver, skeletal muscle, kidney, pancreas [419], small intestine [243], brain microvessels [420,421] | intestinal [243], BBB [421], blood–retinal barrier [422] | Caco-2 [247,248,250,252], HeLa [259,263], HepG2 [257,264], HLCE-D36 [245,246], A2780/AD [249], A2780 [249], KB [259], MCF-7 [262], MDCK-MDR1 [250], MT2 [245], OVCAR-3 [260] | pantothenate, biotin [242], lipoate, iodide [423] |
| SLC6A14/ATB0,+ | lung, trachea, salivary gland, mammary gland, stomach, pituitary gland, uterus, prostate, testis [117,424], small intestine [118], colon [117,118,424] | blood–air barrier [117,425,426], intestinal [118,119] | MCF-7 [125,129,364], A549 [127], BxPC-3 [129], Caco-2 [364], CCD841 [364], HCT116 [364], HepG2 [30], HT29 [364], LS174T [364] | neutral and cationic amino acids (L-Ile, L-Leu, L-Met, L-Val, L-Ala, L-Gly, L-Ser, L-Cys, L-Asn, L-Thr, L-Gln, L-Phe, L-Trp, L-Tyr, L-His, L-Lys, L-Arg), β-alanine, 3,4-DOPA [117] |
| SLC7A5/LAT1 | brain [427,428], brain microvessels [79,80,86,429,430], retina [431], placenta [86,429,432,433], testis [86,429,434], ovary [434], colon [86,434], fibroblasts [435], bone marrow [429], lymph node [429], monocytes [436], macrophages [436], peripheral leukocytes [429], spleen [86], pancreas [434,437,438], thymus [429] | BBB [79,430], placenta [86,429], blood–retinal barrier [431,434], blood–testis barrier [434], blood–follicular barrier [434] | MCF-7 [97,108,110,439], ARPE-19 [83,92,97], C6 [103,113], GL261 [111,112], AsPC1 [99], Bel7402 [110], BxPC-3 [99], hCMEC/D3 [91], HeLa [116], MBEC4 [79], MDA-MB-231 [110], MIAPaCa-2 [99], PANC-1 [99], S180 [110] | large neutral L-amino acids (L-Leu, L-Ile, L-Phe, L-Met, L-Tyr, L-His, L-Trp, L-Val, D-Leu, D-Phe, D-Met, D-Leu) [86,429], T3, T4 [429] |
| SLC10A1/NTCP | liver [440], pancreas [441] | NTCP-transfected HEK293 [178,179], not expressed in HepG2 [442] | cholate, TC, GC, CDC, TCDC, GCDC, UDC, TUDC, LC, TLC, GLC, TLC, GLC, TDC [440], estrone-3-sulfate [443,444] | |
| SLC10A2/ASBT | small intestine [445,446], colon [446], kidney [447], bile duct [448,449], gallbladder [450] | intestinal [445,446] | Caco-2 [44,183,185,189,196,197,201,202,206,208,451] | cholate, TDC, TC, DC, TCDC, CDC, TUDC, UDC, GDC, GCDC, GUDC [444] |
| SLC15A1/PEPT1 | small intestine [288,452,453], colon [452], kidney [454], pancreas [455], bile duct [456], monocytes [457] | intestinal [288,452,453] | Caco-2 [289,297,298,299,300,301,302,303,304,305,306,307,310,313,314,315,316,318,319,320,321,322,324,329,332,334,335,346,347,348,349], AsPC-1 [315,316], Capan-2 [315,317], MDCK [307,331], A549 [325], MCF-7 [311] | di- and tripeptides [288,290,336] |
| SLC15A2/PEPT2 | kidney [454], brain [292,458], choroid plexus, retina [292], peripheral nervous system [292,459], enteric nervous system [460], lung [461], mammary gland [462], heart [463], spleen [464] | BCSFB [292] | SKPT [301,346], MDCK [307,331] | di- and tripeptides [465,466] |
| SLC16A1/MCT1 | bladder [467], brain [390,467,468], choroid plexus [469], stomach [467], intestine [467], small intestine [374,376,390,468], colon [374,376,467], erythrocytes [468], eye [468], retina [470], heart [390,467,468], kidney [390,467,468], liver [467,468], lung [467,468], mammary gland [471], muscle [467], skeletal muscle (red muscle) [390,468], ovary [467], placenta [471], spleen [467], testis [467,468], epididymis [468] | intestinal [374,376], BBB [390], BCSFB [469], blood–retinal barrier [470] | Caco-2 [40,380,384,385,389], 4T1 [40], bEnd [392], HT29-MTX-E12 [389], MDCK [380], PEAKrapid [380], U373 [392] | butyrate [472], L-lactate, pyruvate, acetoacetate, D,L-3-hydroxybutyrate, α-oxoisohexanoate, α-oxoisovalerate [473], acetate, propionate [474,475] |
| SLC19A1/RFC | expressed in 68 human tissues, highest levels in placenta, liver and peripheral blood leukocytes, also in heart [476], small intestine, colon, kidney and choroid plexus [477] | placenta [476] | A549 [278], Colon-26 [275], HeLa [279], MCF-7 [279], Raw 264.7 [275], SKOV3 [278] | 5-MTHF [478,479] |
| SLCO1B1/OATP1B1 | liver [480] | not expressed in HepG2 [481], primary rat hepatocytes have been routinely used [368,371,373] | DHEAS, estradiol-17β-glucuronide, estrone-3-sulfate, PG E2, TXB2, LTC4, LTE4, T4, T3, TC [480], bilirubin, MGB, BGB, cholate [482], GC [483], TUDC, GUDC [484] | |
| SLCO1B3/OATP1B3 | liver [485] | HepG2 [180] | DHEAS, estradiol-17β-glucuronide [485], estrone-3-sulfate, LTC4, T4, T3 [483], TC, GC [483,486], bilirubin [487], MGB [482], glutathione, cholate, TDC, TCDC [486], TUDC, GUDC [484] | |
| SLC22A5/OCTN2 | brain [488,489,490,491,492,493], brain capillary endothelial cells [359], spinal cord [490], retina [494], heart [489,490,491,492,493], salivary gland [492], small intestine [351,488,490,491,492], colon [488], kidney [488,489,490,491,492,493], liver [488,489,490,492], pancreas [489,490], trachea [490], lung [489,490,492], uterus [490], placenta [488,489,490,491,493,495], prostate [490], testis [488], skeletal muscle [488,489,490,491], striated muscle [492], adrenal gland [490,492], mammary gland [496], thymus [490], thyroid [490] | intestinal [351], BBB [359], blood–retinal barrier [494] | Caco-2 [355,358,364], BEAS-2B [356,357], RBE4 [361,362], BxPC-3 [355], CCD841 [364], hCEMC/D3 [45], HCT116 [364], HT29 [364], LS174T [364], MDA-MB-231 [364], MCF-7 [364], T98G [45] | L-carnitine, betaine [497] |
| SLC23A1/SVCT1 | kidney, liver, small intestine, colon, ovary, prostate, pancreas [225,498], lung [499], skin [500] | intestinal [501] | L-ascorbic acid [498] | |
| SLC23A2/SVCT2 | brain, spleen, prostate, testis, ovary, placenta, peripheral blood leukocytes [498], retina, small intestine, epididymis, brain, choroid plexus, pancreas, adrenal gland, gastric glands, spleen, thymus, testis [225], lung [225,499], skin [500] | BCSFB [225,226], blood–retinal barrier [225], intestinal [225,498] | HRPE [236,237,238], NIH/3T3 [227,241], CRL-1497 [232], C6 [227], F98 [227] | L-ascorbic acid [498] |
| SLC44A1/CTL1 | spinal cord, brain [502,503], lung [502,503], colon [502], peripheral blood monocytes and neutrophils, fibroblasts [504], brain microvessels [214], skeletal muscle, heart, testis [505], placenta, kidney, liver, small intestine, pancreas, spleen, ovary [503], mitochondria [214,506] | BBB [214], intestinal [502] | U-87 MG [221,222], brain capillary endothelial cells (BCECs) [220] | choline [502,505,507], ethanolamine [508] |
| SLC44A2/CTL2 | brain [214,509], inner ear [509,510], stomach [511], intestine [511], colon [509], kidney [509,511], heart [509,511], lung [509,511], muscle [509,511], tongue [509,511], liver [509], spleen [509], testis [511], mitochondria [214,506] | BBB [214] | U-87 MG [221,222], brain capillary endothelial cells (BCECs) [220] | choline [357,508,509], ethanolamine [508] |
| SLC46A1/PCFT | kidney, liver, placenta, small intestine, colon, spleen, brain, testis, skin, stomach [271,512], choroid plexus [513] | intestinal [271,512], BCSFB [513] | A549 [278], Caco-2 [276], Colon-26 [275], HeLa [279], MCF-7 [279], Raw 264.7 [275], SKOV3 [278] | folic acid, 5-MTHF [271] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyimesi, G.; Hediger, M.A. Transporter-Mediated Drug Delivery. Molecules 2023, 28, 1151. https://doi.org/10.3390/molecules28031151
Gyimesi G, Hediger MA. Transporter-Mediated Drug Delivery. Molecules. 2023; 28(3):1151. https://doi.org/10.3390/molecules28031151
Chicago/Turabian StyleGyimesi, Gergely, and Matthias A. Hediger. 2023. "Transporter-Mediated Drug Delivery" Molecules 28, no. 3: 1151. https://doi.org/10.3390/molecules28031151
APA StyleGyimesi, G., & Hediger, M. A. (2023). Transporter-Mediated Drug Delivery. Molecules, 28(3), 1151. https://doi.org/10.3390/molecules28031151